High Dose of Dietary Nicotinamide Riboside Induces Glucose Intolerance and White Adipose Tissue Dysfunction in Mice Fed a Mildly Obesogenic Diet


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Animals and Diets
2.2. Indirect Calorimetry and Metabolic Flexibility
2.3. Oral Glucose Tolerance Test
2.4. Sample Collection at Sacrifice
2.5. Plasma or Serum Parameters
2.6. Liver TG and NAD+ Metabolites
2.7. Gene Expression
2.8. Antibodies
2.9. Western Blot
2.10. Histology and Immunohistochemistry
2.11. Statistics
3. Results
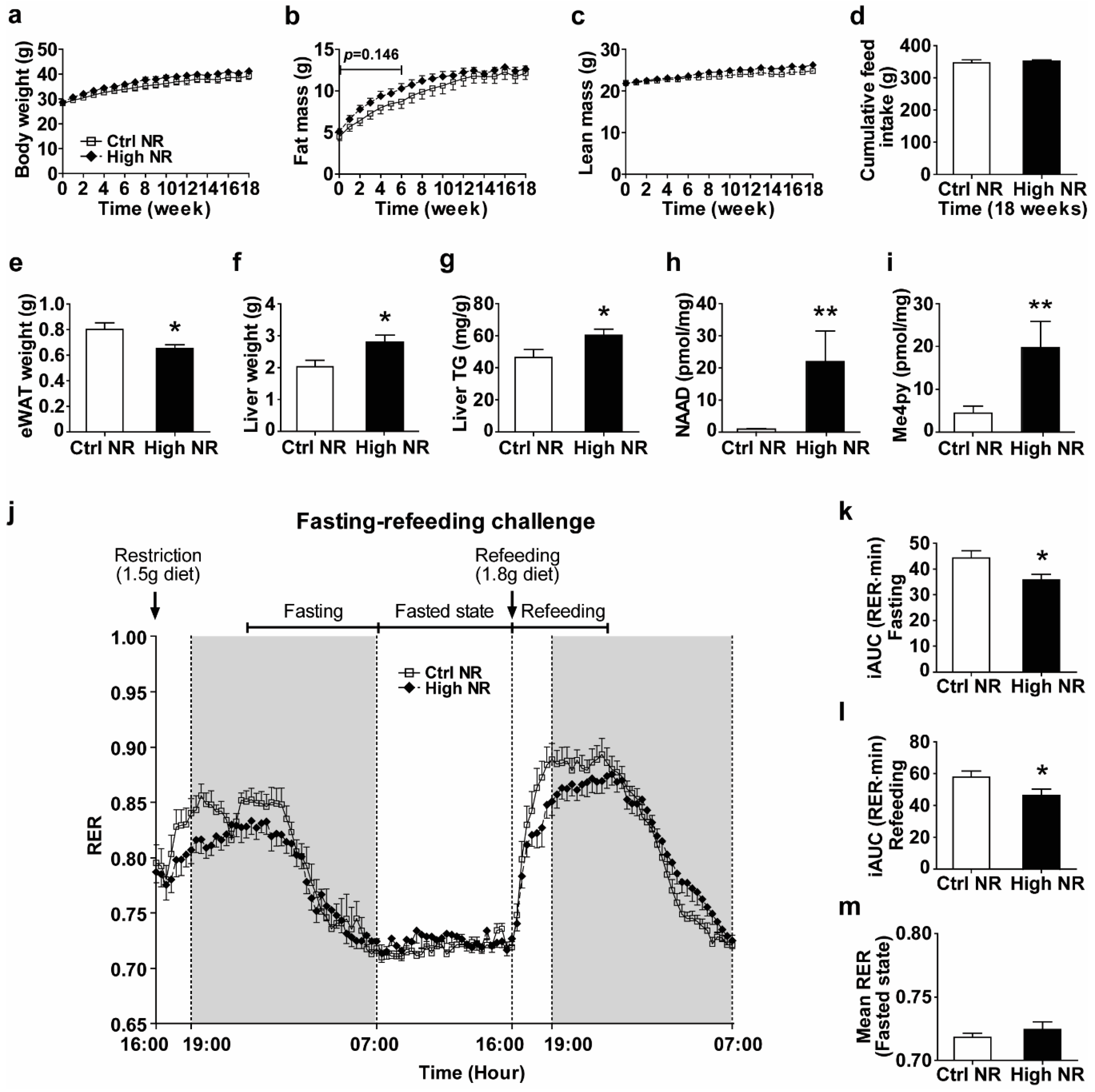
3.1. High Dose NR Reduces Metabolic Flexibility in HF Diet-Fed Male C57BI/6JRccHsd Mice
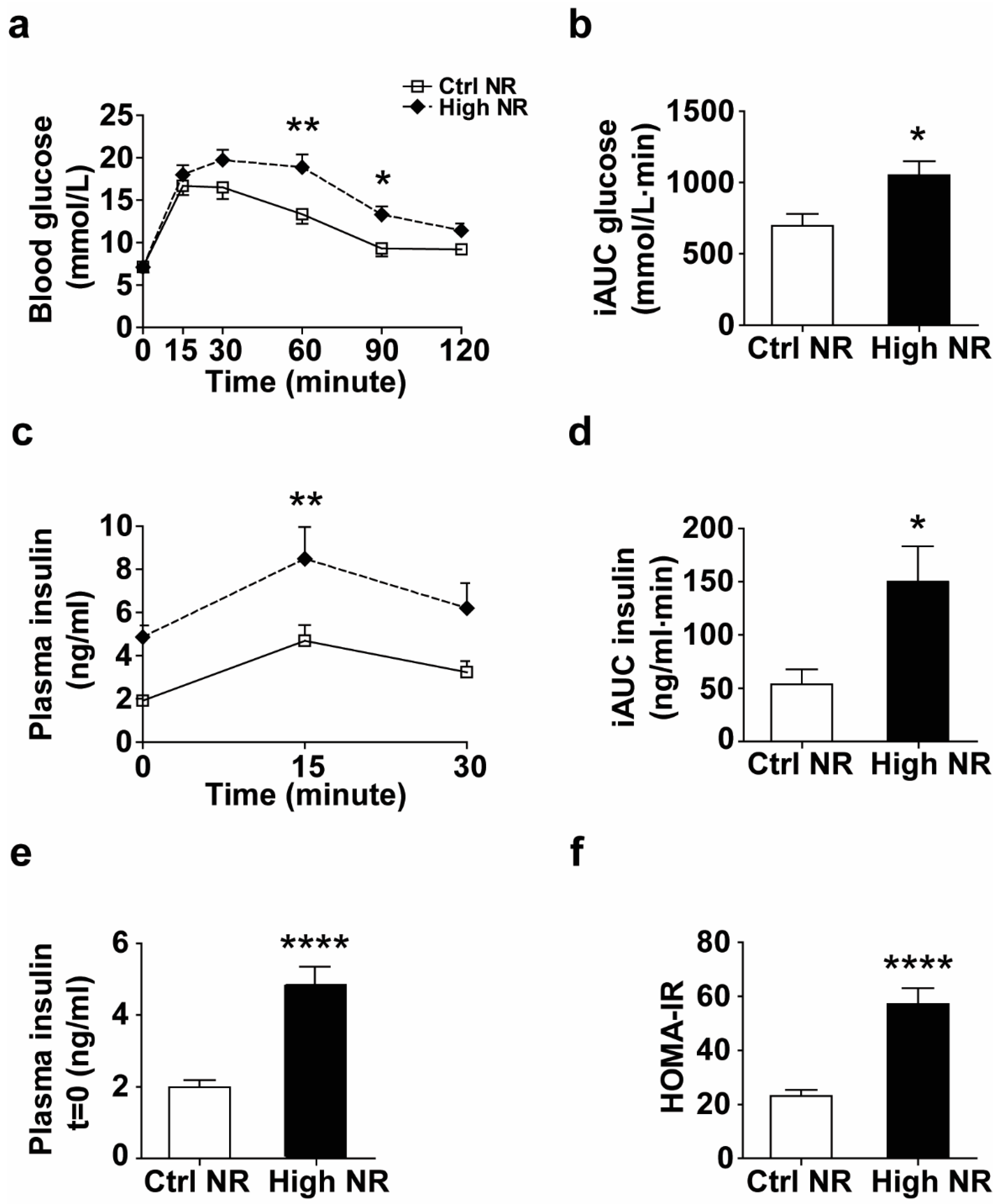
3.2. High NR-Fed Mice Have Lower Glucose Tolerance
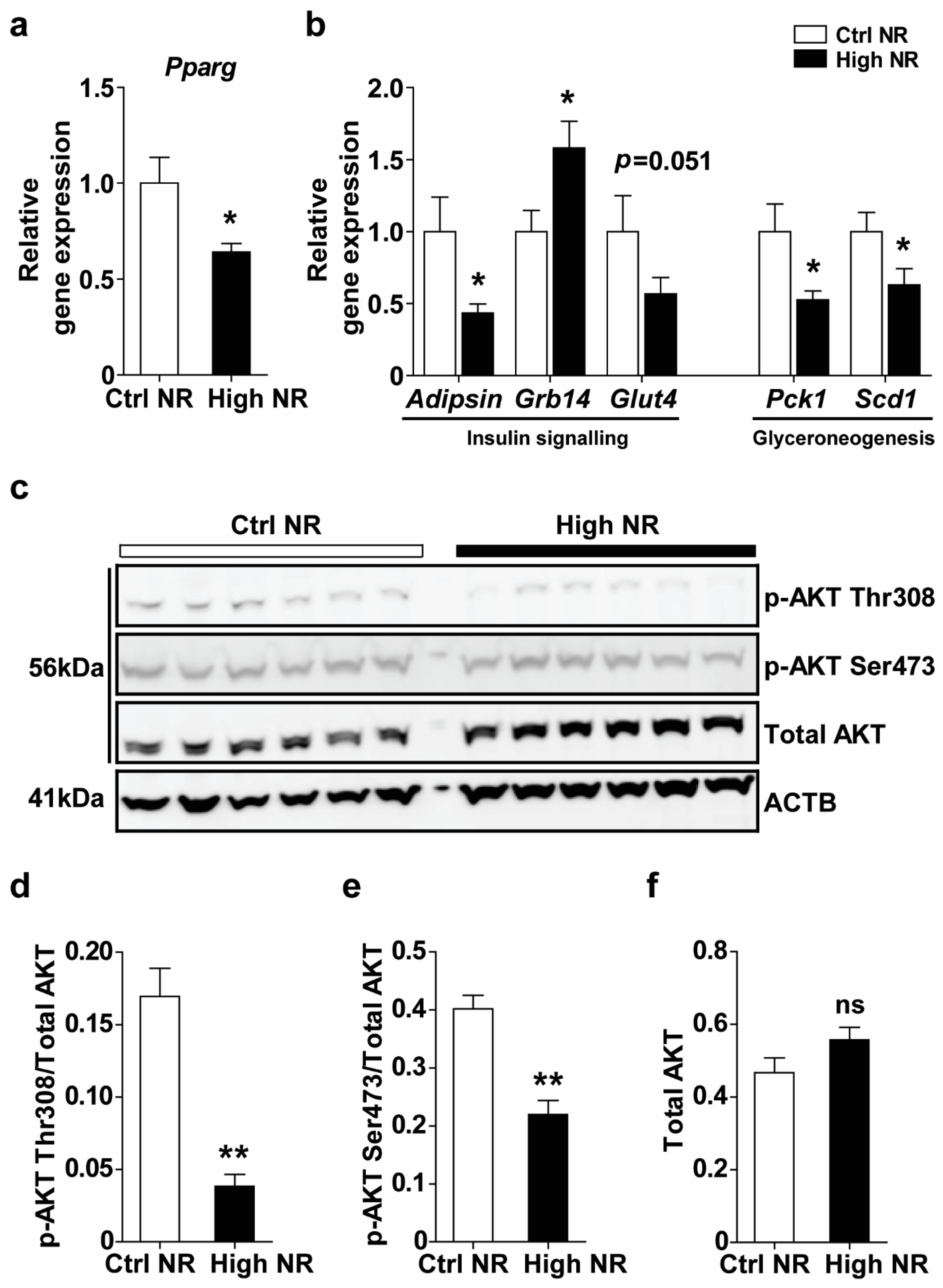
3.3. High NR Feeding Results in a Lowered Peroxisome Proliferator-Activated Receptor γ (PPARγ) Expression Signature in WAT
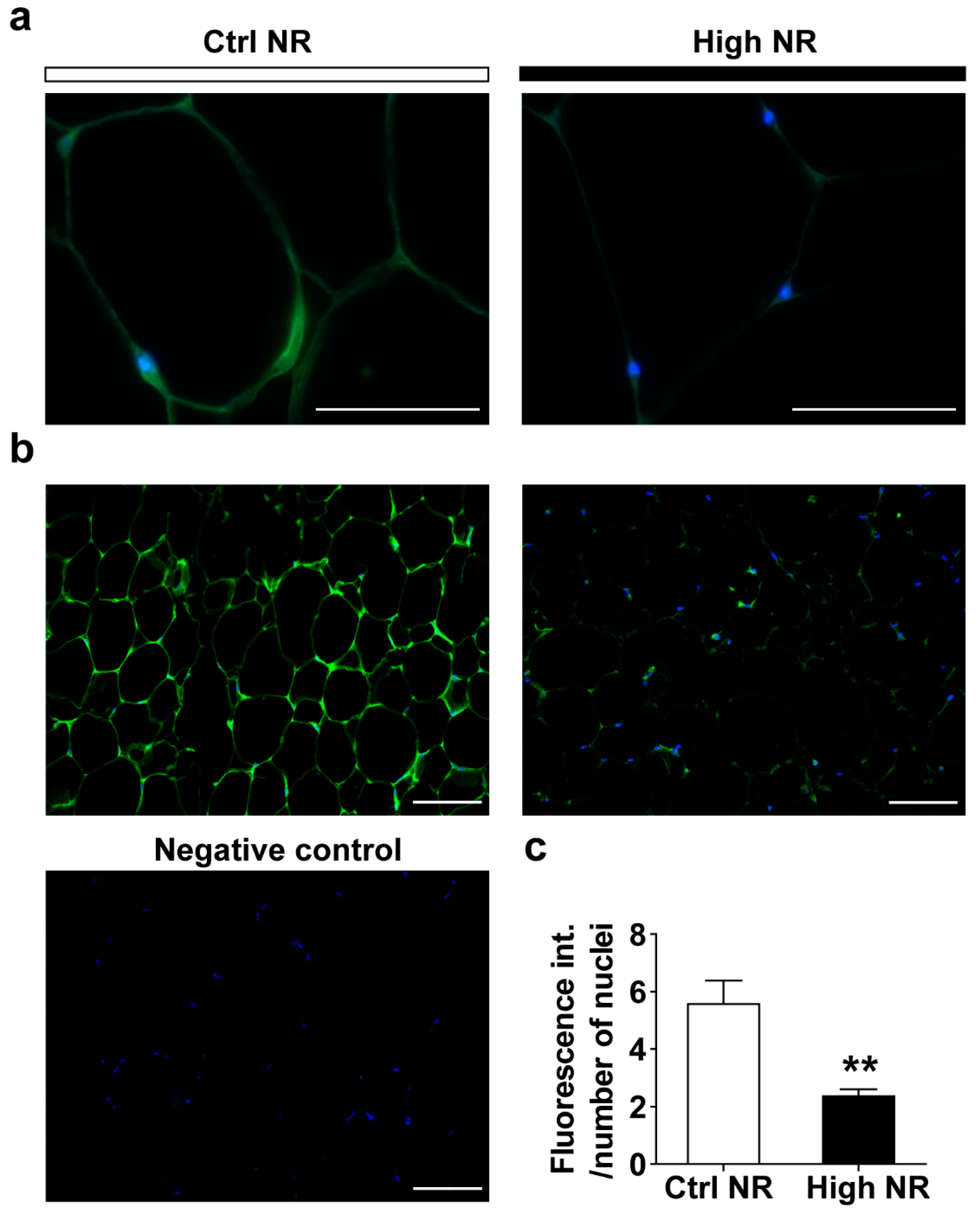
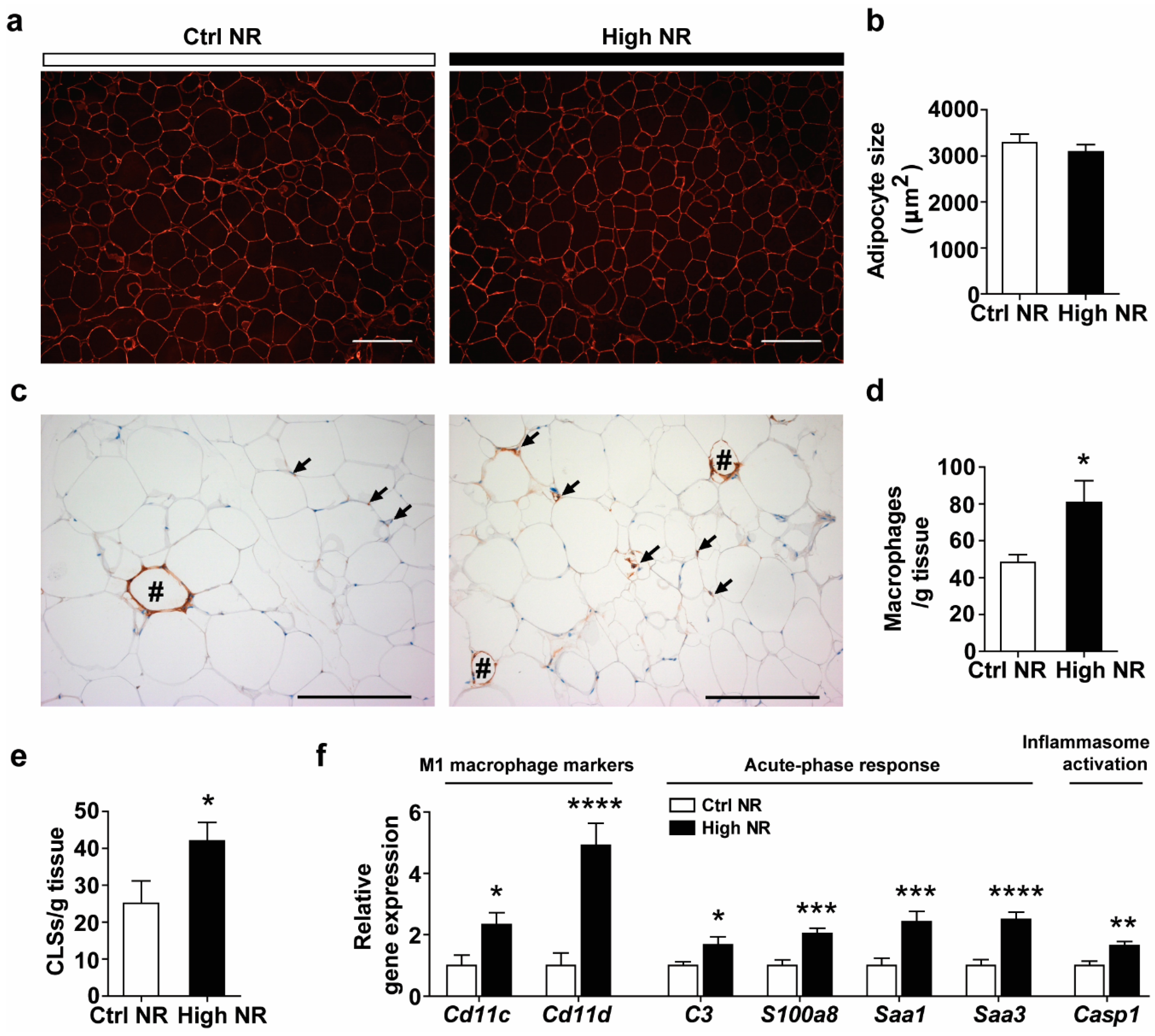
3.4. High-Dose NR Leads to More Severe WAT Inflammation
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Wan, P.; Moat, S.; Anstey, A. Pellagra: A review with emphasis on photosensitivity. Br. J. Dermatol. 2011, 164, 1188–1200. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Gariani, K.; Menzies, K.J.; Ryu, D.; Wegner, C.J.; Wang, X.; Ropelle, E.R.; Moullan, N.; Zhang, H.; Perino, A.; Lemos, V.; et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 2016, 63, 1190–1204. [Google Scholar] [CrossRef] [PubMed]
- Trammell, S.A.; Weidemann, B.J.; Chadda, A.; Yorek, M.S.; Holmes, A.; Coppey, L.J.; Obrosov, A.; Kardon, R.H.; Yorek, M.A.; Brenner, C. Nicotinamide riboside opposes type 2 diabetes and neuropathy in mice. Sci. Rep. 2016, 6, 26933. [Google Scholar] [CrossRef] [PubMed]
- Jukarainen, S.; Heinonen, S.; Ramo, J.T.; Rinnankoski-Tuikka, R.; Rappou, E.; Tummers, M.; Muniandy, M.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; et al. Obesity is associated with low NAD+/SIRT pathway expression in adipose tissue of BMI-discordant monozygotic twins. J. Clin. Endocrinol. Metab. 2016, 101, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Elhassan, Y.S.; Philp, A.A.; Lavery, G.G. Targeting NAD+ in metabolic disease: New insights into an old molecule. J. Endocr. Soc. 2017, 1, 816–835. [Google Scholar] [CrossRef] [PubMed]
- Horwitt, M.K.; Harper, A.E.; Henderson, L.M. Niacin-tryptophan relationships for evaluating niacin equivalents. Am. J. Clin. Nutr. 1981, 34, 423–427. [Google Scholar] [CrossRef]
- Fukuwatari, T.; Shibata, K. Nutritional aspect of tryptophan metabolism. Int. J. Tryptophan Res. 2013, 6, 3–8. [Google Scholar] [CrossRef]
- Bogan, K.L.; Brenner, C. Nicotinic acid, nicotinamide, and nicotinamide riboside: A molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef]
- Khan, N.A.; Auranen, M.; Paetau, I.; Pirinen, E.; Euro, L.; Forsstrom, S.; Pasila, L.; Velagapudi, V.; Carroll, C.J.; Auwerx, J.; et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol. Med. 2014, 6, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Ryu, D.; Zhang, H.; Ropelle, E.R.; Sorrentino, V.; Mazala, D.A.; Mouchiroud, L.; Marshall, P.L.; Campbell, M.D.; Ali, A.S.; Knowels, G.M.; et al. NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci. Transl. Med. 2016, 8, 361ra139. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Su, X.; Quinn, W.J., 3rd; Hui, S.; Krukenberg, K.; Frederick, D.W.; Redpath, P.; Zhan, L.; Chellappa, K.; White, E.; et al. Quantitative analysis of NAD synthesis-breakdown fluxes. Cell Metab. 2018, 27, 1067–1080. [Google Scholar] [CrossRef] [PubMed]
- Trammell, S.A.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 12948. [Google Scholar] [CrossRef] [PubMed]
- Dollerup, O.L.; Christensen, B.; Svart, M.; Schmidt, M.S.; Sulek, K.; Ringgaard, S.; Stodkilde-Jorgensen, H.; Moller, N.; Brenner, C.; Treebak, J.T.; et al. A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: Safety, insulin-sensitivity, and lipid-mobilizing effects. Am. J. Clin. Nutr. 2018, 108, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Martens, C.R.; Denman, B.A.; Mazzo, M.R.; Armstrong, M.L.; Reisdorph, N.; McQueen, M.B.; Chonchol, M.; Seals, D.R. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD+ in healthy middle-aged and older adults. Nat. Commun. 2018, 9, 1286. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Mariman, E.; Renes, J.; Keijer, J. The secretory function of adipocytes in the physiology of white adipose tissue. J. Cell. Physiol. 2008, 216, 3–13. [Google Scholar] [CrossRef]
- Stromsdorfer, K.L.; Yamaguchi, S.; Yoon, M.J.; Moseley, A.C.; Franczyk, M.P.; Kelly, S.C.; Qi, N.; Imai, S.; Yoshino, J. NAMPT-mediated NAD+ biosynthesis in adipocytes regulates adipose tissue function and multi-organ insulin sensitivity in mice. Cell Rep. 2016, 16, 1851–1860. [Google Scholar] [CrossRef]
- Shi, W.; Hegeman, M.A.; van Dartel, D.A.M.; Tang, J.; Suarez, M.; Swarts, H.; van der Hee, B.; Arola, L.; Keijer, J. Effects of a wide range of dietary nicotinamide riboside (NR) concentrations on metabolic flexibility and white adipose tissue (WAT) of mice fed a mildly obesogenic diet. Mol. Nutr. Food Res. 2017, 61, 1600878. [Google Scholar] [CrossRef]
- Kourtzidis, I.A.; Stoupas, A.T.; Gioris, I.S.; Veskoukis, A.S.; Margaritelis, N.V.; Tsantarliotou, M.; Taitzoglou, I.; Vrabas, I.S.; Paschalis, V.; Kyparos, A.; et al. The NAD+ precursor nicotinamide riboside decreases exercise performance in rats. J. Int. Soc. Sports Nutr. 2016, 13, 32. [Google Scholar] [CrossRef]
- Kourtzidis, I.A.; Dolopikou, C.F.; Tsiftsis, A.N.; Margaritelis, N.V.; Theodorou, A.A.; Zervos, I.A.; Tsantarliotou, M.P.; Veskoukis, A.S.; Vrabas, I.S.; Paschalis, V.; et al. Nicotinamide riboside supplementation dysregulates redox and energy metabolism in rats: Implications for exercise performance. Exp. Physiol. 2018, 103, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Van Rossum, C.; Buurma-Rethans, E.; Vennemann, F.; Beukers, M.; Brants, H.A.; De Boer, E.; Ocké, M.C. The Diet of the Dutch: Results of the First Two Years of the Dutch National Food Consumption Survey 2012–2016; RIVM: Bilthoven, The Netherlands, 2016. [Google Scholar]
- Duivenvoorde, L.P.; van Schothorst, E.M.; Swarts, H.J.; Keijer, J. Assessment of metabolic flexibility of old and adult mice using three noninvasive, indirect calorimetry-based treatments. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, T.; Laskewitz, A.; Grefhorst, A.; Boer, T.S.; Bloks, V.W.; Kuipers, F.; Groen, A.K.; Reijngoud, D.-J. A novel approach to monitor glucose metabolism using stable isotopically labelled glucose in longitudinal studies in mice. Lab. Anim. 2013, 47, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Trammell, S.A.; Brenner, C. Targeted, LCMS-based Metabolomics for Quantitative Measurement of NAD+ Metabolites. Comput. Struct. Biotechnol. J. 2013, 4, e201301012. [Google Scholar] [CrossRef] [PubMed]
- Purhonen, J.; Rajendran, J.; Tegelberg, S.; Smolander, O.P.; Pirinen, E.; Kallijarvi, J.; Fellman, V. NAD+ repletion produces no therapeutic effect in mice with respiratory chain complex III deficiency and chronic energy deprivation. FASEB J. 2018, 32, 5913–5926. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557. [Google Scholar] [CrossRef] [PubMed]
- Beg, M.; Abdullah, N.; Thowfeik, F.S.; Altorki, N.K.; McGraw, T.E. Distinct Akt phosphorylation states are required for insulin regulated Glut4 and Glut1-mediated glucose uptake. Elife 2017, 6, e26896. [Google Scholar] [CrossRef]
- Wu, Z.; Xie, Y.; Morrison, R.F.; Bucher, N.L.; Farmer, S.R. PPARgamma induces the insulin-dependent glucose transporter GLUT4 in the absence of C/EBPalpha during the conversion of 3T3 fibroblasts into adipocytes. J. Clin. Investig. 1998, 101, 22–32. [Google Scholar] [CrossRef]
- Fang, J.; Ianni, A.; Smolka, C.; Vakhrusheva, O.; Nolte, H.; Kruger, M.; Wietelmann, A.; Simonet, N.G.; Adrian-Segarra, J.M.; Vaquero, A.; et al. Sirt7 promotes adipogenesis in the mouse by inhibiting autocatalytic activation of Sirt1. Proc. Natl. Acad. Sci. USA 2017, 114, E8352–E8361. [Google Scholar] [CrossRef]
- Mueckler, M. Facilitative glucose transporters. Eur. J. Biochem. 1994, 219, 713–725. [Google Scholar] [CrossRef]
- Galgani, J.E.; Moro, C.; Ravussin, E. Metabolic flexibility and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1009–E1017. [Google Scholar] [CrossRef] [PubMed]
- van Ommen, B.; Keijer, J.; Heil, S.G.; Kaput, J. Challenging homeostasis to define biomarkers for nutrition related health. Mol. Nutr. Food Res. 2009, 53, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Czech, M.P. The GLUT4 glucose transporter. Cell Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.D.; Peroni, O.; Kim, J.K.; Kim, Y.B.; Boss, O.; Hadro, E.; Minnemann, T.; Shulman, G.I.; Kahn, B.B. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 2001, 409, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Banks, A.S.; Estall, J.L.; Kajimura, S.; Boström, P.; Laznik, D.; Ruas, J.L.; Chalmers, M.J.; Kamenecka, T.M.; Blüher, M. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARγ by Cdk5. Nature 2010, 466, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.C.; Ljubicic, S.; Leibiger, B.; Kern, M.; Leibiger, I.B.; Moede, T.; Kelly, M.E.; Bhowmick, D.C.; Murano, I.; Cohen, P.; et al. Adipsin is an adipokine that improves beta cell function in diabetes. Cell 2014, 158, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Flier, J.S.; Cook, K.S.; Usher, P.; Spiegelman, B.M. Severely impaired adipsin expression in genetic and acquired obesity. Science 1987, 237, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Kasus-Jacobi, A.; Perdereau, D.; Auzan, C.; Clauser, E.; Van Obberghen, E.; Mauvais-Jarvis, F.; Girard, J.; Burnol, A.F. Identification of the rat adapter Grb14 as an inhibitor of insulin actions. J. Biol. Chem. 1998, 273, 26026–26035. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Capitaine, N.; Le Marcis, V.; Vega, N.; Bereziat, V.; Kergoat, M.; Laville, M.; Girard, J.; Vidal, H.; Burnol, A.F. Increased adipose tissue expression of Grb14 in several models of insulin resistance. FASEB J. 2004, 18, 965–967. [Google Scholar] [CrossRef]
- Beale, E.G.; Hammer, R.E.; Antoine, B.; Forest, C. Disregulated glyceroneogenesis: PCK1 as a candidate diabetes and obesity gene. Trends Endocrinol. Metab. 2004, 15, 129–135. [Google Scholar] [CrossRef]
- Dragos, S.M.; Bergeron, K.F.; Desmarais, F.; Suitor, K.; Wright, D.C.; Mounier, C.; Mutch, D.M. Reduced SCD1 activity alters markers of fatty acid reesterification, glyceroneogenesis, and lipolysis in murine white adipose tissue and 3T3-L1 adipocytes. Am. J. Physiol. Cell Physiol. 2017, 313, C295–C304. [Google Scholar] [CrossRef] [PubMed]
- Millward, C.A.; Desantis, D.; Hsieh, C.W.; Heaney, J.D.; Pisano, S.; Olswang, Y.; Reshef, L.; Beidelschies, M.; Puchowicz, M.; Croniger, C.M. Phosphoenolpyruvate carboxykinase (Pck1) helps regulate the triglyceride/fatty acid cycle and development of insulin resistance in mice. J. Lipid Res. 2010, 51, 1452–1463. [Google Scholar] [CrossRef] [PubMed]
- Sekimoto, R.; Kishida, K.; Nakatsuji, H.; Nakagawa, T.; Funahashi, T.; Shimomura, I. High circulating levels of S100A8/A9 complex (calprotectin) in male Japanese with abdominal adiposity and dysregulated expression of S100A8 and S100A9 in adipose tissues of obese mice. Biochem. Biophys. Res. Commun. 2012, 419, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.Z.; Lee, M.J.; Hu, H.; Pollin, T.I.; Ryan, A.S.; Nicklas, B.J.; Snitker, S.; Horenstein, R.B.; Hull, K.; Goldberg, N.H.; et al. Acute-phase serum amyloid A: An inflammatory adipokine and potential link between obesity and its metabolic complications. PLoS Med. 2006, 3, e287. [Google Scholar] [CrossRef] [PubMed]
- Han, C.Y.; Subramanian, S.; Chan, C.K.; Omer, M.; Chiba, T.; Wight, T.N.; Chait, A. Adipocyte-derived serum amyloid A3 and hyaluronan play a role in monocyte recruitment and adhesion. Diabetes 2007, 56, 2260–2273. [Google Scholar] [CrossRef] [PubMed]
- Munkonda, M.N.; Lapointe, M.; Miegueu, P.; Roy, C.; Gauvreau, D.; Richard, D.; Cianflone, K. Recombinant acylation stimulating protein administration to C3−/− mice increases insulin resistance via adipocyte inflammatory mechanisms. PLoS ONE 2012, 7, e46883. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Renes, J.; Bouwman, F.; Bunschoten, A.; Mariman, E.; Keijer, J. Absence of an adipogenic effect of rosiglitazone on mature 3T3-L1 adipocytes: Increase of lipid catabolism and reduction of adipokine expression. Diabetologia 2007, 50, 654–665. [Google Scholar] [CrossRef]
- Hiuge-Shimizu, A.; Maeda, N.; Hirata, A.; Nakatsuji, H.; Nakamura, K.; Okuno, A.; Kihara, S.; Funahashi, T.; Shimomura, I. Dynamic changes of adiponectin and S100A8 levels by the selective peroxisome proliferator–activated receptor-γ agonist rivoglitazone. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 792–799. [Google Scholar] [CrossRef]
- Stienstra, R.; Joosten, L.A.; Koenen, T.; van Tits, B.; van Diepen, J.A.; van den Berg, S.A.; Rensen, P.C.; Voshol, P.J.; Fantuzzi, G.; Hijmans, A.; et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010, 12, 593–605. [Google Scholar] [CrossRef]
- Parlee, S.D.; Lentz, S.I.; Mori, H.; MacDougald, O.A. Quantifying size and number of adipocytes in adipose tissue. Methods Enzymol. 2014, 537, 93–122. [Google Scholar] [CrossRef]
- Toye, A.A.; Lippiat, J.D.; Proks, P.; Shimomura, K.; Bentley, L.; Hugill, A.; Mijat, V.; Goldsworthy, M.; Moir, L.; Haynes, A.; et al. A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia 2005, 48, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Freeman, H.C.; Hugill, A.; Dear, N.T.; Ashcroft, F.M.; Cox, R.D. Deletion of nicotinamide nucleotide transhydrogenase: A new quantitive trait locus accounting for glucose intolerance in C57BL/6J mice. Diabetes 2006, 55, 2153–2156. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.Y.; Lin, Y.T.; Lin, G.; Wu, P.R.; Cheng, M.L. Nicotinamide nucleotide transhydrogenase (NNT) deficiency dysregulates mitochondrial retrograde signaling and impedes proliferation. Redox Biol. 2017, 12, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Meimaridou, E.; Kowalczyk, J.; Guasti, L.; Hughes, C.R.; Wagner, F.; Frommolt, P.; Nurnberg, P.; Mann, N.P.; Banerjee, R.; Saka, H.N.; et al. Mutations in NNT encoding nicotinamide nucleotide transhydrogenase cause familial glucocorticoid deficiency. Nat. Genet. 2012, 44, 740–742. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, W.; Hegeman, M.A.; Doncheva, A.; Bekkenkamp-Grovenstein, M.; de Boer, V.C.J.; Keijer, J. High Dose of Dietary Nicotinamide Riboside Induces Glucose Intolerance and White Adipose Tissue Dysfunction in Mice Fed a Mildly Obesogenic Diet. Nutrients 2019, 11, 2439. https://doi.org/10.3390/nu11102439
Shi W, Hegeman MA, Doncheva A, Bekkenkamp-Grovenstein M, de Boer VCJ, Keijer J. High Dose of Dietary Nicotinamide Riboside Induces Glucose Intolerance and White Adipose Tissue Dysfunction in Mice Fed a Mildly Obesogenic Diet. Nutrients. 2019; 11(10):2439. https://doi.org/10.3390/nu11102439
Chicago/Turabian StyleShi, Wenbiao, Maria A. Hegeman, Atanaska Doncheva, Melissa Bekkenkamp-Grovenstein, Vincent C. J. de Boer, and Jaap Keijer. 2019. "High Dose of Dietary Nicotinamide Riboside Induces Glucose Intolerance and White Adipose Tissue Dysfunction in Mice Fed a Mildly Obesogenic Diet" Nutrients 11, no. 10: 2439. https://doi.org/10.3390/nu11102439
APA StyleShi, W., Hegeman, M. A., Doncheva, A., Bekkenkamp-Grovenstein, M., de Boer, V. C. J., & Keijer, J. (2019). High Dose of Dietary Nicotinamide Riboside Induces Glucose Intolerance and White Adipose Tissue Dysfunction in Mice Fed a Mildly Obesogenic Diet. Nutrients, 11(10), 2439. https://doi.org/10.3390/nu11102439