Oxidative Stress Levels in the Brain Are Determined by Post-Mortem Interval and Ante-Mortem Vitamin C State but Not Alzheimer’s Disease Status

Abstract
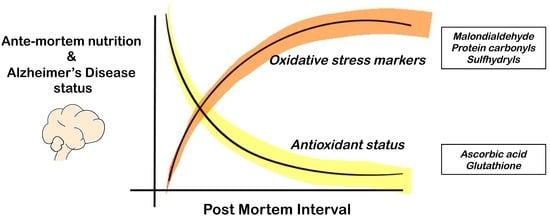
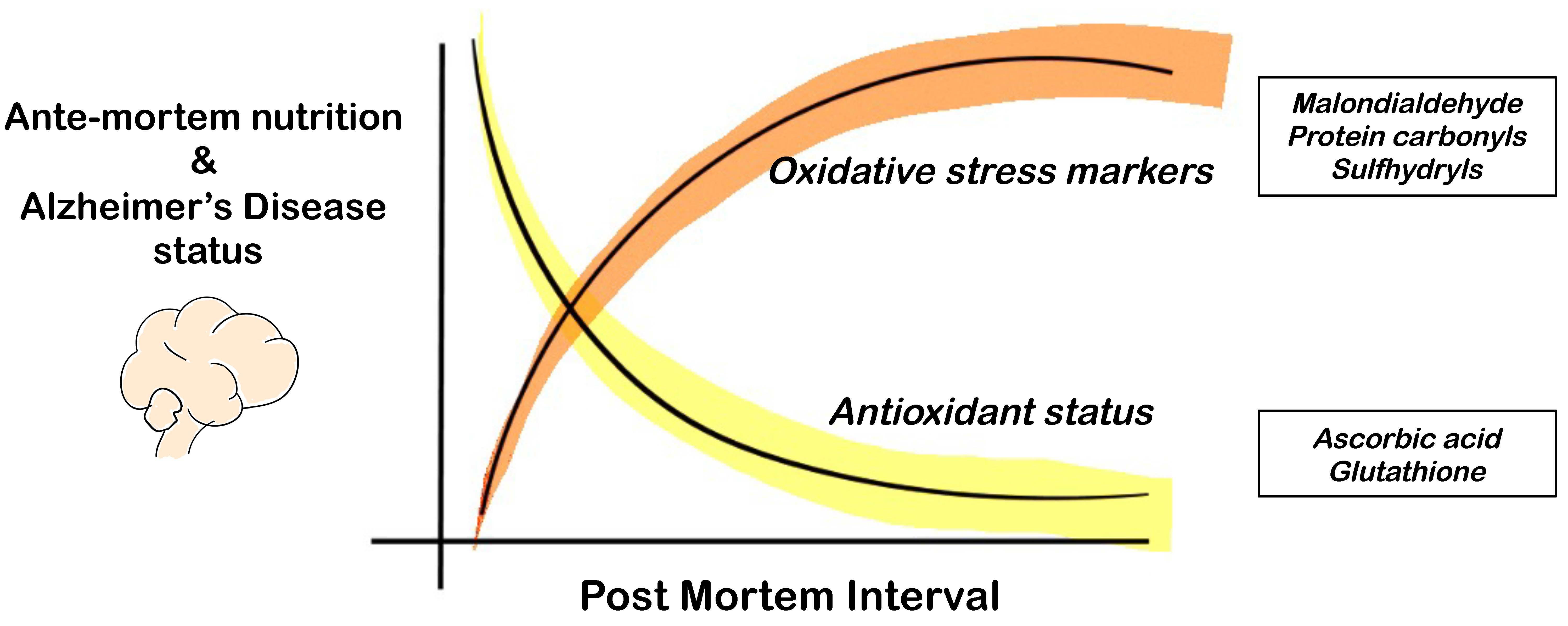
1. Introduction
2. Methods
2.1. Animals
2.2. Human Tissue Acquisition
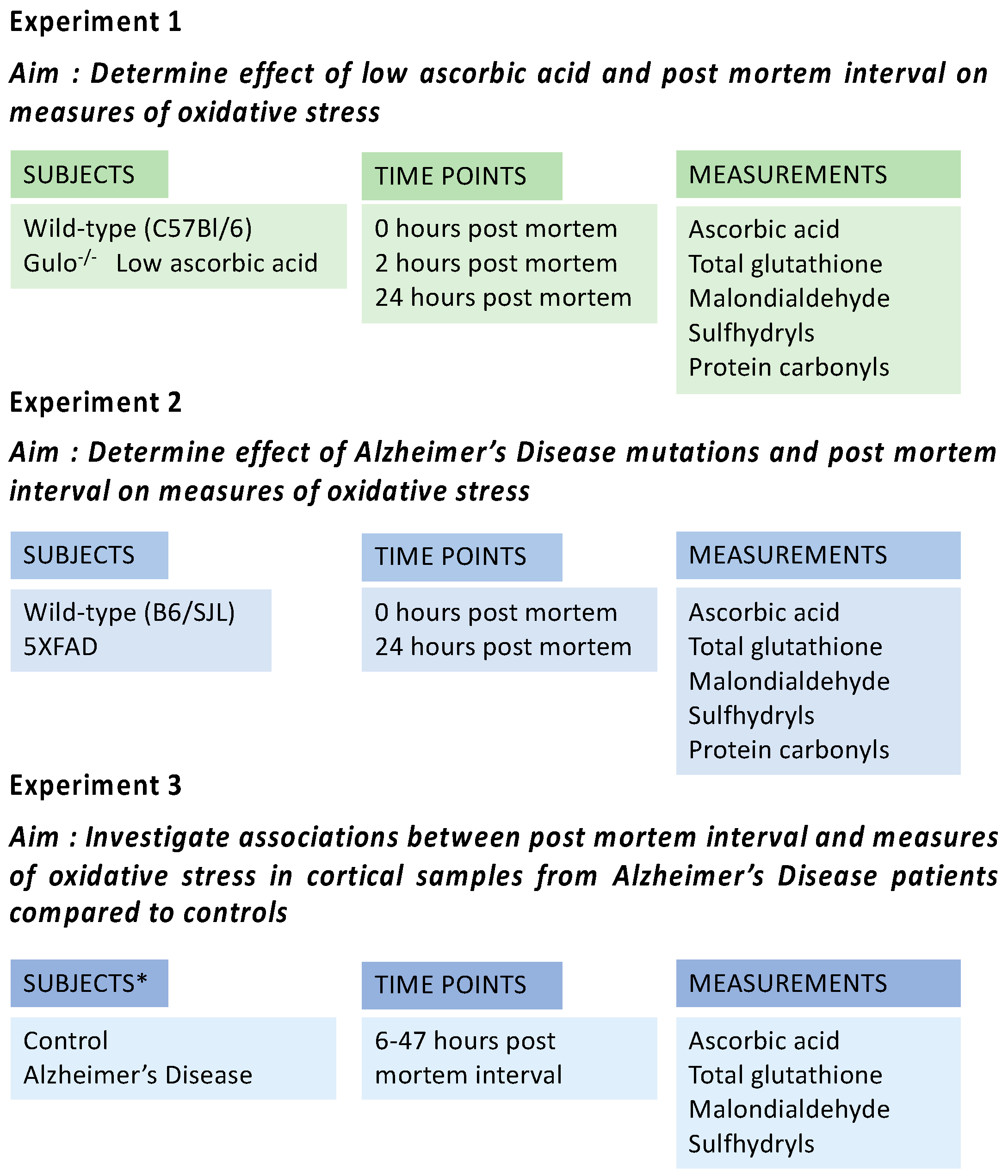
2.3. Experimental Design
2.4. Biochemical Analyses
2.5. Statistical Analyses
3. Results
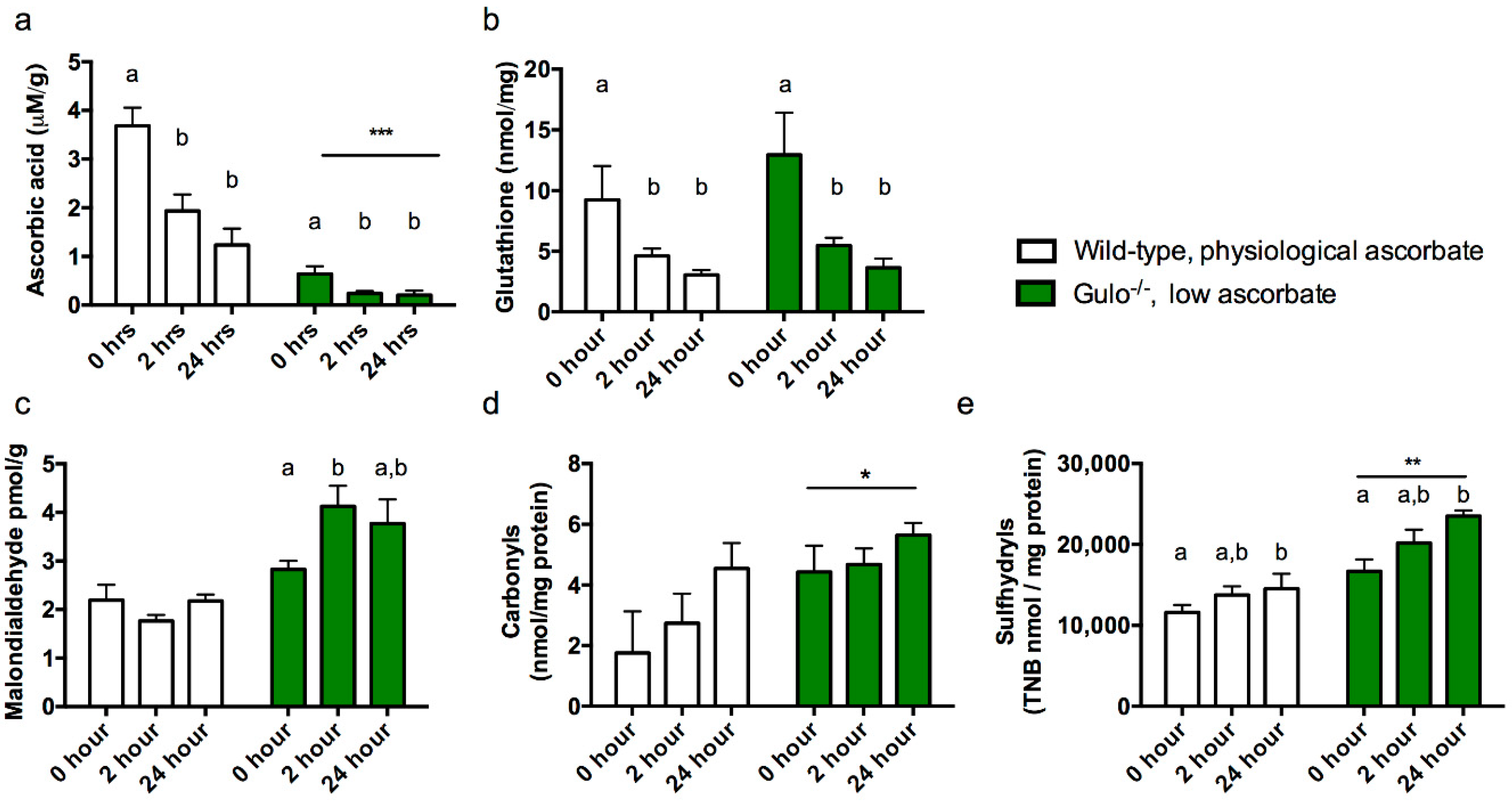
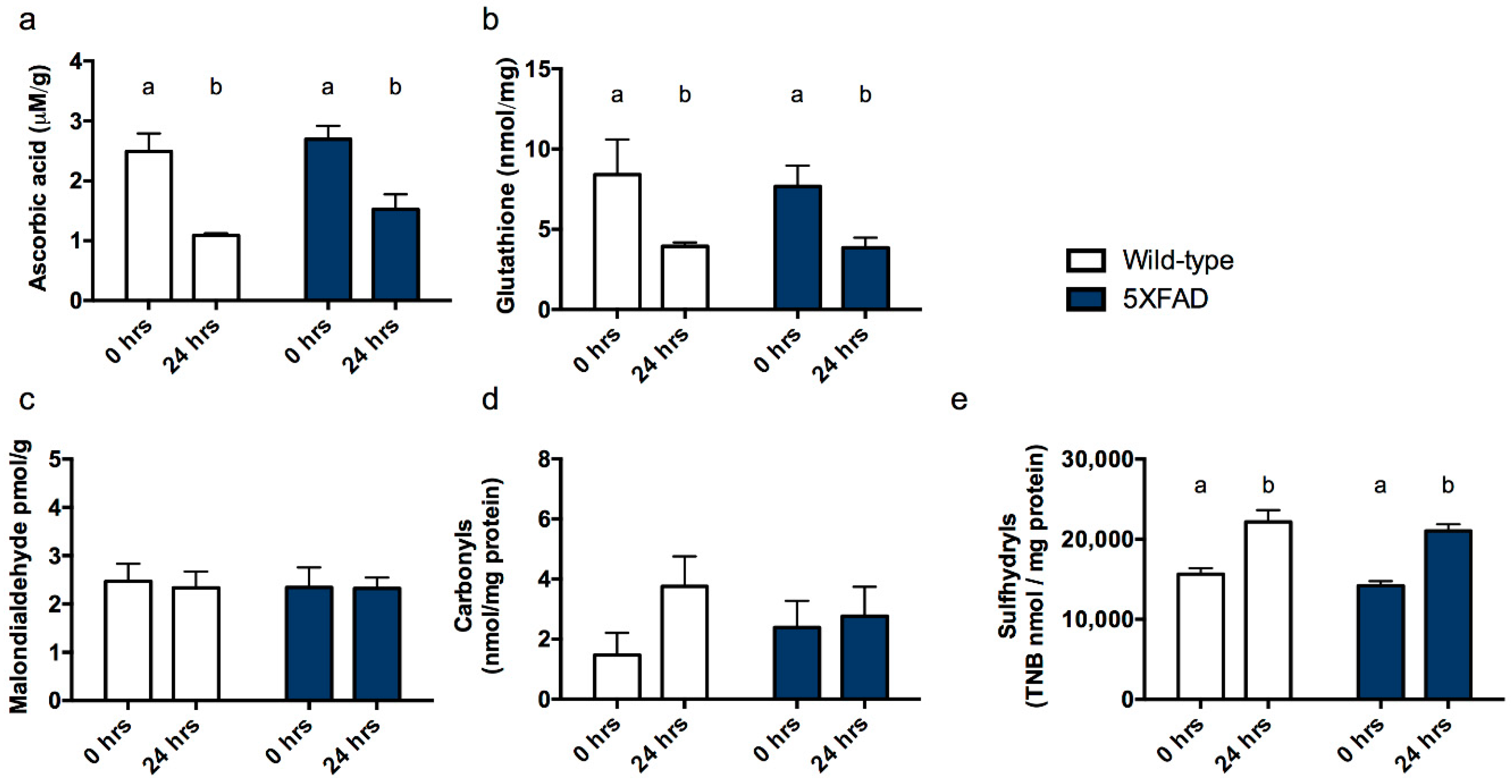
3.1. Experiment 1
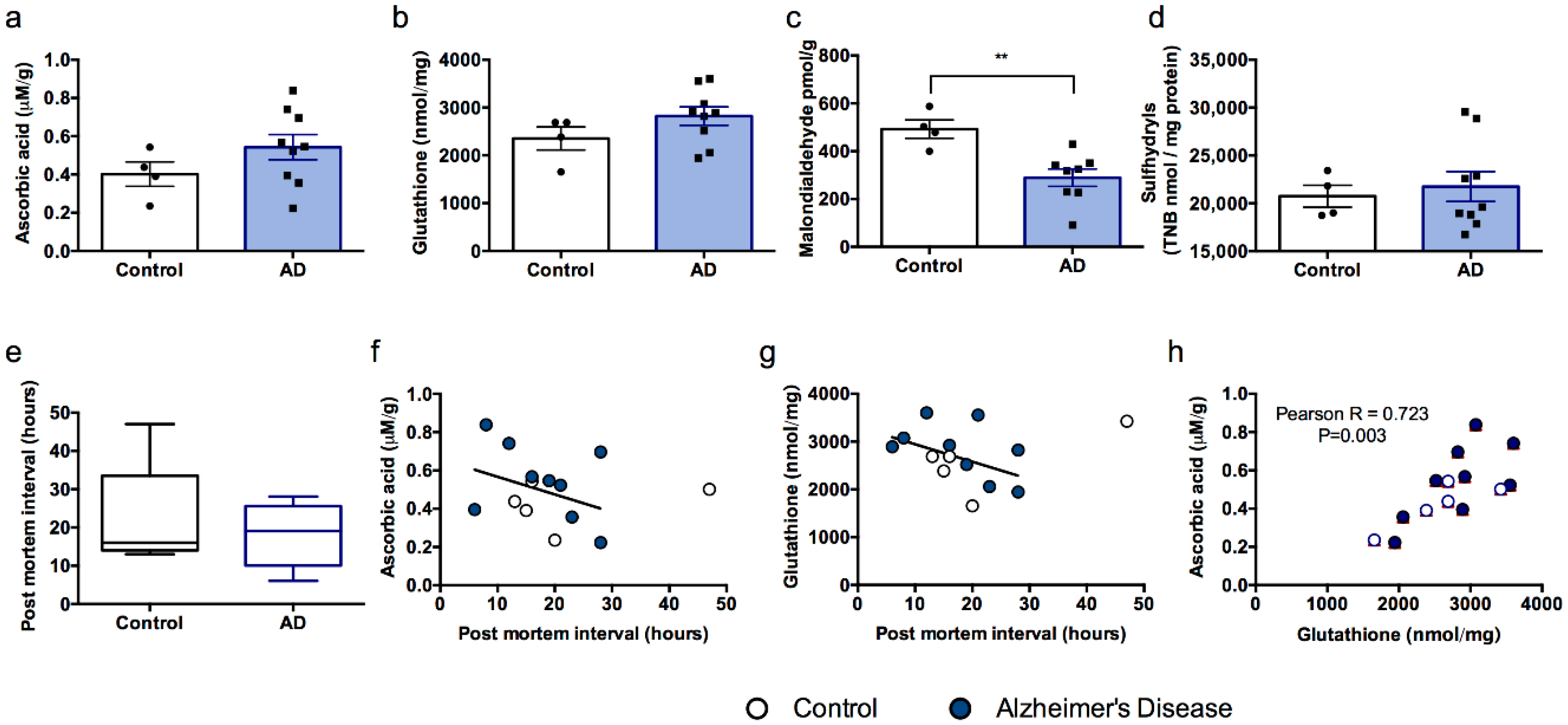
3.2. Experiment 2
3.3. Experiment 3
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.L.; Weissman, L.; Bohr, V.A.; Mattson, M.P. Mitochondrial DNA damage and repair in neurodegenerative disorders. DNA Repair 2008, 7, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Texel, S.J.; Mattson, M.P. Impaired adaptive cellular responses to oxidative stress and the pathogenesis of Alzheimer’s disease. Antioxid. Redox Signal. 2011, 14, 1519–1534. [Google Scholar] [CrossRef] [PubMed]
- Mecocci, P. Oxidative stress in mild cognitive impairment and Alzheimer disease: A continuum. J. Alzheimer's Dis. 2004, 6, 159–163. [Google Scholar] [CrossRef]
- Schrag, M.; Mueller, C.; Zabel, M.; Crofton, A.; Kirsch, W.M.; Ghribi, O.; Squitti, R.; Perry, G. Oxidative stress in blood in Alzheimer’s disease and mild cognitive impairment: A meta-analysis. Neurobiol. Dis. 2013, 59, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Weindruch, R.; Prolla, T.A. Gene-expression profile of the ageing brain in mice. Nature Genet. 2000, 25, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Melov, S.; Adlard, P.A.; Morten, K.; Johnson, F.; Golden, T.R.; Hinerfeld, D.; Schilling, B.; Mavros, C.; Masters, C.L.; Volitakis, I.; et al. Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS ONE 2007, 2, e536. [Google Scholar] [CrossRef] [PubMed]
- Zabel, M.; Nackenoff, A.; Kirsch, W.M.; Harrison, F.E.; Perry, G.; Schrag, M. Markers of oxidative damage to lipids, nucleic acids and proteins and antioxidant enzymes activities in Alzheimer’s disease brain: A meta-analysis in human pathological specimens. Free Radic. Biol. Med. 2018, 115, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging-Alzheimer’s A. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Clement, C.; Hill, J.M.; Dua, P.; Culicchia, F.; Lukiw, W.J. Analysis of RNA from Alzheimer’s Disease Post-mortem Brain Tissues. Mol. Neurobiol. 2016, 53, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Skoumalova, A.; Hort, J. Blood markers of oxidative stress in Alzheimer’s disease. J. Cell. Mol. Med. 2012, 16, 2291–2300. [Google Scholar] [CrossRef] [PubMed]
- Montine, T.J.; Quinn, J.F.; Milatovic, D.; Silbert, L.C.; Dang, T.; Sanchez, S.; Terry, E.; Roberts, L.J.; Kaye, J.A.; Morrow, J.D. Peripheral F2-isoprostanes and F4-neuroprostanes are not increased in Alzheimer’s disease. Ann. Neurol. 2002, 52, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; Green, R.J.; Dawes, S.M.; May, J.M. Vitamin C distribution and retention in the mouse brain. Brain Res. 2010, 1348, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; Yu, S.S.; Van Den Bossche, K.L.; Li, L.; May, J.M.; McDonald, M.P. Elevated oxidative stress and sensorimotor deficits but normal cognition in mice that cannot synthesize ascorbic acid. J. Neurochem. 2008, 106, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.S.; Lamb, J.; May, J.M.; Harrison, F.E. Behavioral and monoamine changes following severe vitamin C deficiency. J. Neurochem. 2013, 124, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Mirra, S.S.; Heyman, A.; McKeel, D.; Sumi, S.M.; Crain, B.J.; Brownlee, L.M.; Vogel, F.S.; Hughes, J.P.; van Belle, G.; Berg, L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991, 41, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; Hosseini, A.H.; McDonald, M.P.; May, J.M. Vitamin C reduces spatial learning deficits in middle-aged and very old APP/PSEN1 transgenic and wild-type mice. Pharmacol. Biochem.Behav. 2009, 93, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; Kode, A.; Biswas, S.K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 2006, 1, 3159–3165. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; Hosseini, A.H.; Dawes, S.M.; Weaver, S.; May, J.M. Ascorbic acid attenuates scopolamine-induced spatial learning deficits in the water maze. Behav. Brain Res. 2009, 205, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.L.; Morgan, P.E.; Davies, M.J. Quantification of protein modification by oxidants. Free Radic. Biol. Med. 2009, 46, 965–988. [Google Scholar] [CrossRef] [PubMed]
- Sgaravatti, A.M.; Magnusson, A.S.; Oliveira, A.S.; Mescka, C.P.; Zanin, F.; Sgarbi, M.B.; Pederzolli, C.D.; Wyse, A.T.; Wannmacher, C.M.; Wajner, M.; et al. Effects of 1,4-butanediol administration on oxidative stress in rat brain: Study of the neurotoxicity of gamma-hydroxybutyric acid in vivo. Metab. Brain Dis. 2009, 24, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, M.Y.; Markesbery, W.R. Changes in thiol content and expression of glutathione redox system genes in the hippocampus and cerebellum in Alzheimer’s disease. Neurosci. Lett. 2001, 302, 141–145. [Google Scholar] [CrossRef]
- Harrison, F.E.; Meredith, M.E.; Dawes, S.M.; Saskowski, J.L.; May, J.M. Low ascorbic acid and increased oxidative stress in gulo(-/-) mice during development. Brain Res. 2010, 1349, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; May, J.M.; McDonald, M.P. Vitamin C deficiency increases basal exploratory activity but decreases scopolamine-induced activity in APP/PSEN1 transgenic mice. Pharmacol. Biochem. Behav. 2010, 94, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Sofic, E.; Rausch, W.D.; Schmidt, B.; Reynolds, G.P.; Jellinger, K.; Youdim, M.B. Transition metals, ferritin, glutathione and ascorbic acid in parkinsonian brains. J. Neurochem. 1989, 52, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Schaus, R. The ascorbic acid content of human pituitary, cerebral cortex, heart and skeletal muscle and its relation to age. Am. J. Clin. Nutr. 1957, 5, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Gan, R.; Eintracht, S.; Hoffer, L.J. Vitamin C deficiency in a university teaching hospital. J. Am. Coll. Nutr. 2008, 27, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Gale, C.R.; Martyn, C.N.; Cooper, C. Cognitive impairment and mortality in a cohort of elderly people. BMJ 1996, 312, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Lindeman, R.D.; Romero, L.J.; Koehler, K.M.; Liang, H.C.; LaRue, A.; Baumgartner, R.N.; Garry, P.J. Serum vitamin B12, C and folate concentrations in the New Mexico elder health survey: Correlations with cognitive and affective functions. J. Am. Coll. Nutr. 2000, 19, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Hynd, M.R.; Lewohl, J.M.; Scott, H.L.; Dodd, P.R. Biochemical and molecular studies using human autopsy brain tissue. J. Neurochem. 2003, 85, 543–562. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Nicola, D.; Boche, D. Post-mortem analysis of neuroinflammatory changes in human Alzheimer’s disease. Alzheimer's Res. Ther. 2015, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Eck, P.; Erichsen, H.C.; Taylor, J.G.; Corpe, C.; Chanock, S.J.; Levine, M. Genomic and functional analysis of the sodium-dependent vitamin C transporter SLC23A1-SVCT1. Genes Nutr. 2007, 2, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Cahill, L.E.; El-Sohemy, A. Vitamin C Transporter Gene Polymorphisms, Dietary Vitamin C and Serum Ascorbic Acid. J. Nutrigenet. Nutrigenom. 2009, 2, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Michels, A.J.; Hagen, T.M.; Frei, B. Human genetic variation influences vitamin C homeostasis by altering vitamin C transport and antioxidant enzyme function. Ann. Rev. Nutr. 2013, 33, 45–70. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Age | Sex | Post Mortem Interval (Hours) |
|---|---|---|---|
| Alzheimer’s Disease 1 | 92 | Female | 16 |
| Alzheimer’s Disease 2 | 81 | Female | 21 |
| Alzheimer’s Disease 3 | 63 | Male | 28 |
| Alzheimer’s Disease 4 | 78 | Female | 6 |
| Alzheimer’s Disease 5 | 54 | Male | 8 |
| Alzheimer’s Disease 6 | 65 | Female | 19 |
| Alzheimer’s Disease 7 | 64 | Female | 28 |
| Alzheimer’s Disease 8 | 86 | Male | 23 |
| Alzheimer’s Disease 9 | 70 | Male | 12 |
| Control 1 | 102 | Female | 47 |
| Control 2 | 78 | Male | 20 |
| Control 3 | 62 | Male | 15 |
| Control 4 | 59 | Female | 16 |
| Control 5 | 27 | Male | 13 |
| Ascorbate | Malondialdehyde | Glutathione | Sulfhydryls | Post Mortem Interval | ||
|---|---|---|---|---|---|---|
| Ascorbate | Pearson Correlation | −0.123 | 0.768 ** | 0.16 | −0.337 | |
| P (2-tailed) | 0.704 | 0.002 | 0.601 | 0.26 | ||
| N | 12 | 13 | 13 | 13 | ||
| Malondialdehyde | Pearson Correlation | -0.024 | −0.303 | −0.437 | ||
| P (2-tailed) | 0.942 | 0.339 | 0.156 | |||
| N | 12 | 12 | 12 | |||
| Glutathione | Pearson Correlation | 0.015 | −0.432 | |||
| P (2-tailed) | 0.962 | 0.141 | ||||
| N | 13 | 13 | ||||
| Sulfhydryls | Pearson Correlation | 0.044 | ||||
| P (2-tailed) | 0.885 | |||||
| N | 13 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eckman, J.; Dixit, S.; Nackenoff, A.; Schrag, M.; Harrison, F.E. Oxidative Stress Levels in the Brain Are Determined by Post-Mortem Interval and Ante-Mortem Vitamin C State but Not Alzheimer’s Disease Status. Nutrients 2018, 10, 883. https://doi.org/10.3390/nu10070883
Eckman J, Dixit S, Nackenoff A, Schrag M, Harrison FE. Oxidative Stress Levels in the Brain Are Determined by Post-Mortem Interval and Ante-Mortem Vitamin C State but Not Alzheimer’s Disease Status. Nutrients. 2018; 10(7):883. https://doi.org/10.3390/nu10070883
Chicago/Turabian StyleEckman, Jared, Shilpy Dixit, Alex Nackenoff, Matthew Schrag, and Fiona E. Harrison. 2018. "Oxidative Stress Levels in the Brain Are Determined by Post-Mortem Interval and Ante-Mortem Vitamin C State but Not Alzheimer’s Disease Status" Nutrients 10, no. 7: 883. https://doi.org/10.3390/nu10070883
APA StyleEckman, J., Dixit, S., Nackenoff, A., Schrag, M., & Harrison, F. E. (2018). Oxidative Stress Levels in the Brain Are Determined by Post-Mortem Interval and Ante-Mortem Vitamin C State but Not Alzheimer’s Disease Status. Nutrients, 10(7), 883. https://doi.org/10.3390/nu10070883