CFTR Deletion Confers Mitochondrial Dysfunction and Disrupts Lipid Homeostasis in Intestinal Epithelial Cells

 ,
,
Abstract
1. Introduction
2. Materials and Methods
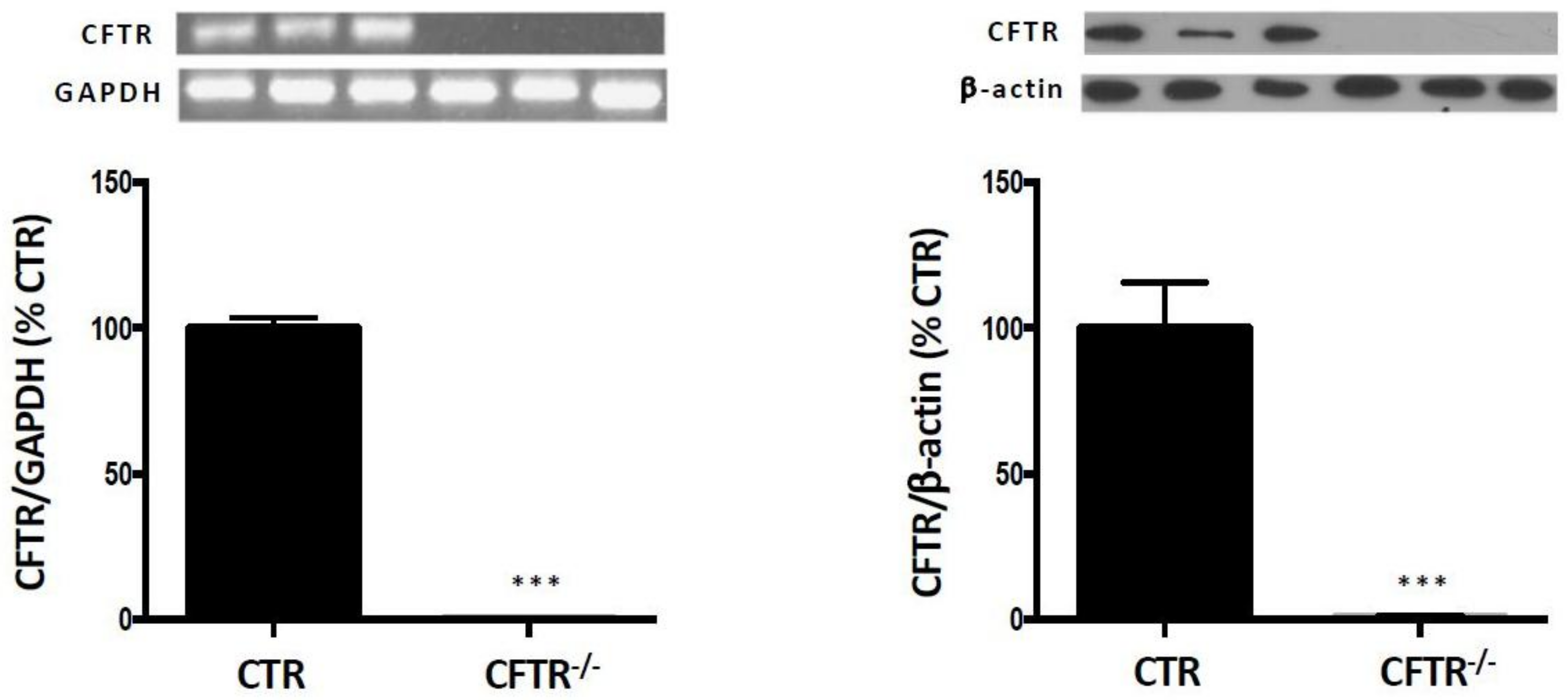
2.1. CFTR-Knockout in Caco-2/15 Cell Line
2.2. Cell Culture
2.3. Isolation of Mitochondria
2.4. Induction of OxS
2.5. Protein Expression Analysis by Immunoblotting
2.6. RNA Isolation and RT-PCR
2.7. Fatty Acid β-Oxidation
2.8. Measurement of Mitochondrial ADP/ATP Ratio
2.9. Lipid and Lipoprotein Assessment
2.10. Lipid Extraction
2.11. Lipid Carrier
2.12. Isolation of Lipoproteins
2.13. De Novo Apolipoproteins Synthesis
2.14. Statistical Analysis
3. Results
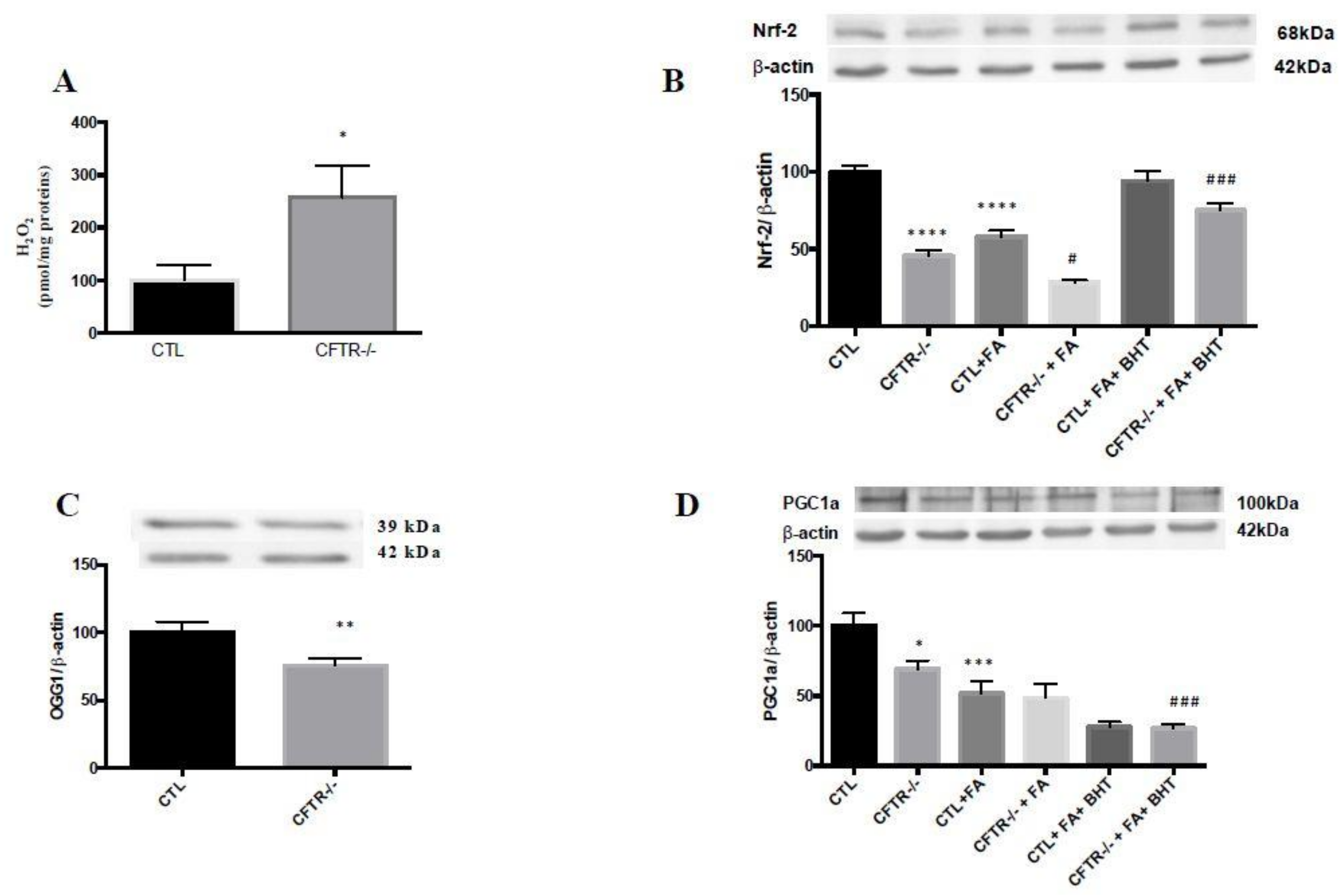
3.1. Mitochondrial Antioxidant Defense
3.2. Fatty Acid β-Oxidation
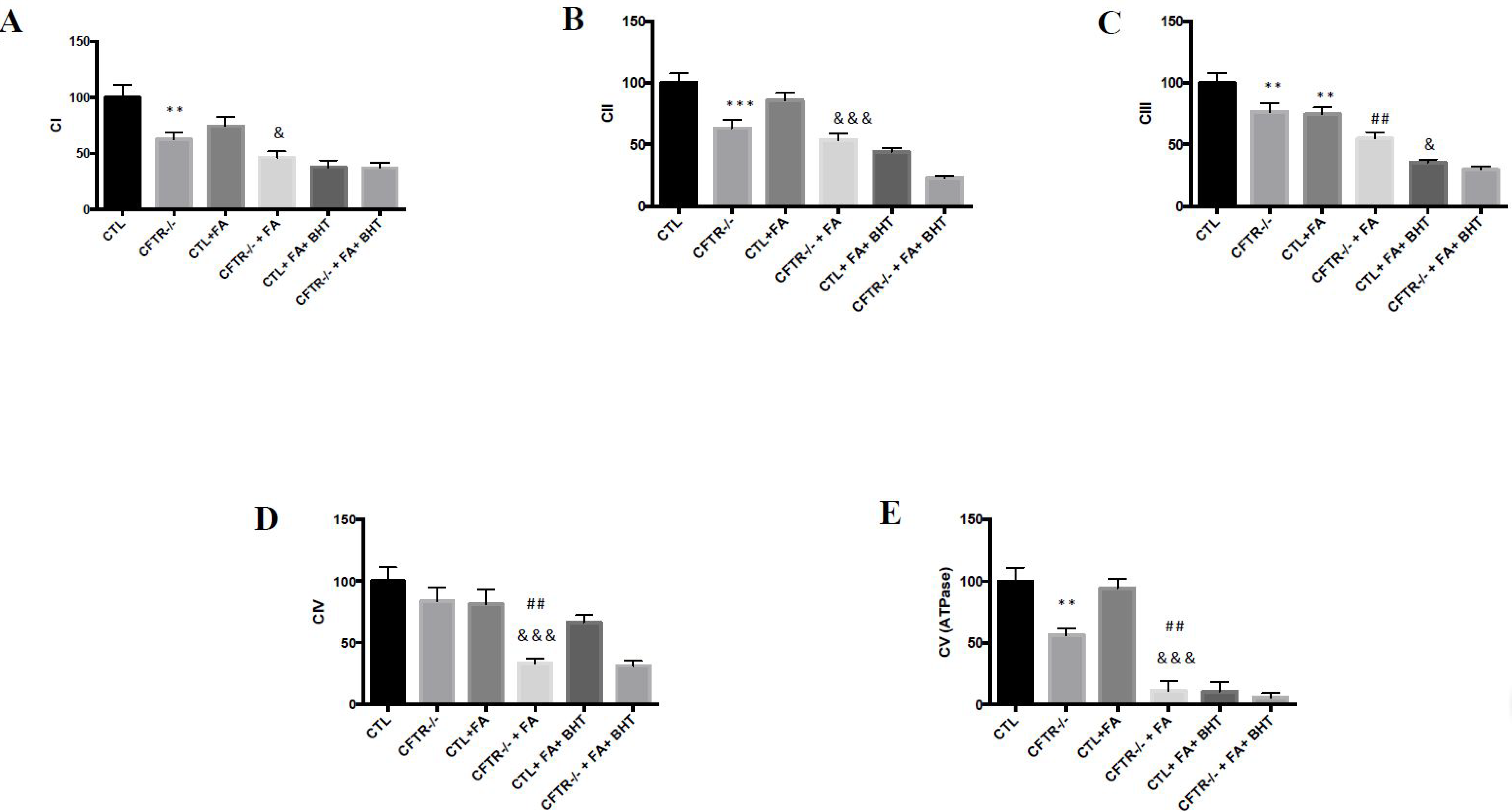
3.3. Oxidative Phosphorylation
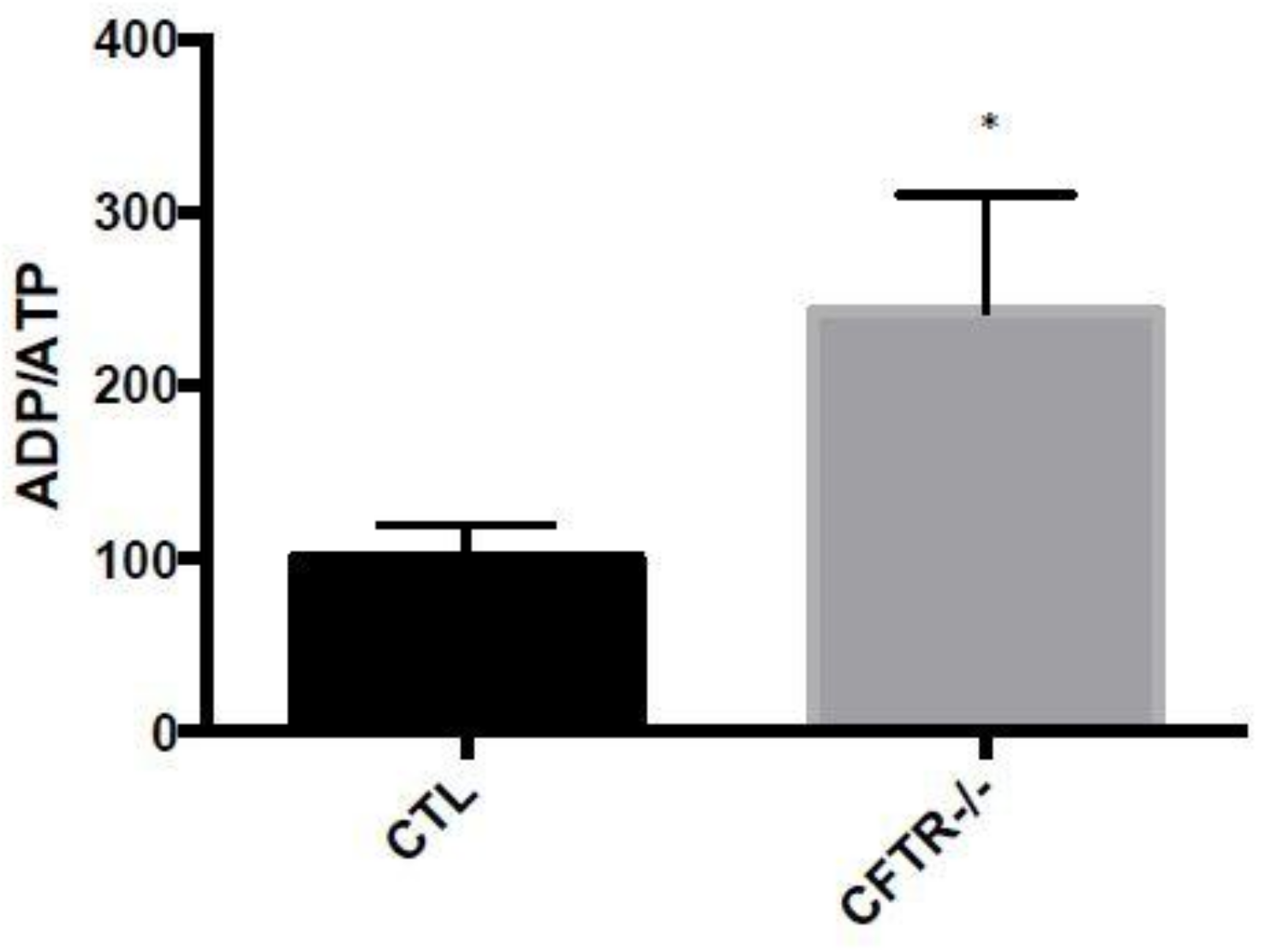
3.4. ADP/ATP Ratio
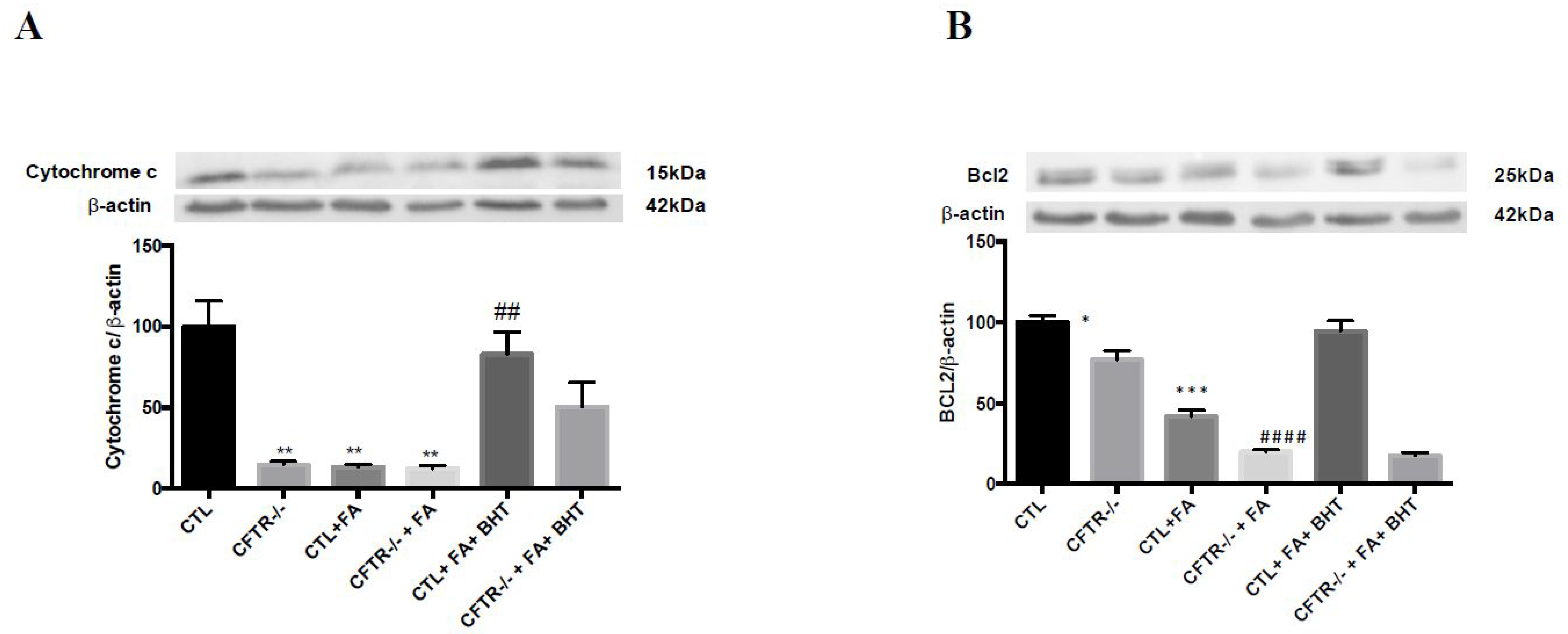
3.5. Apoptosis-Related Proteins
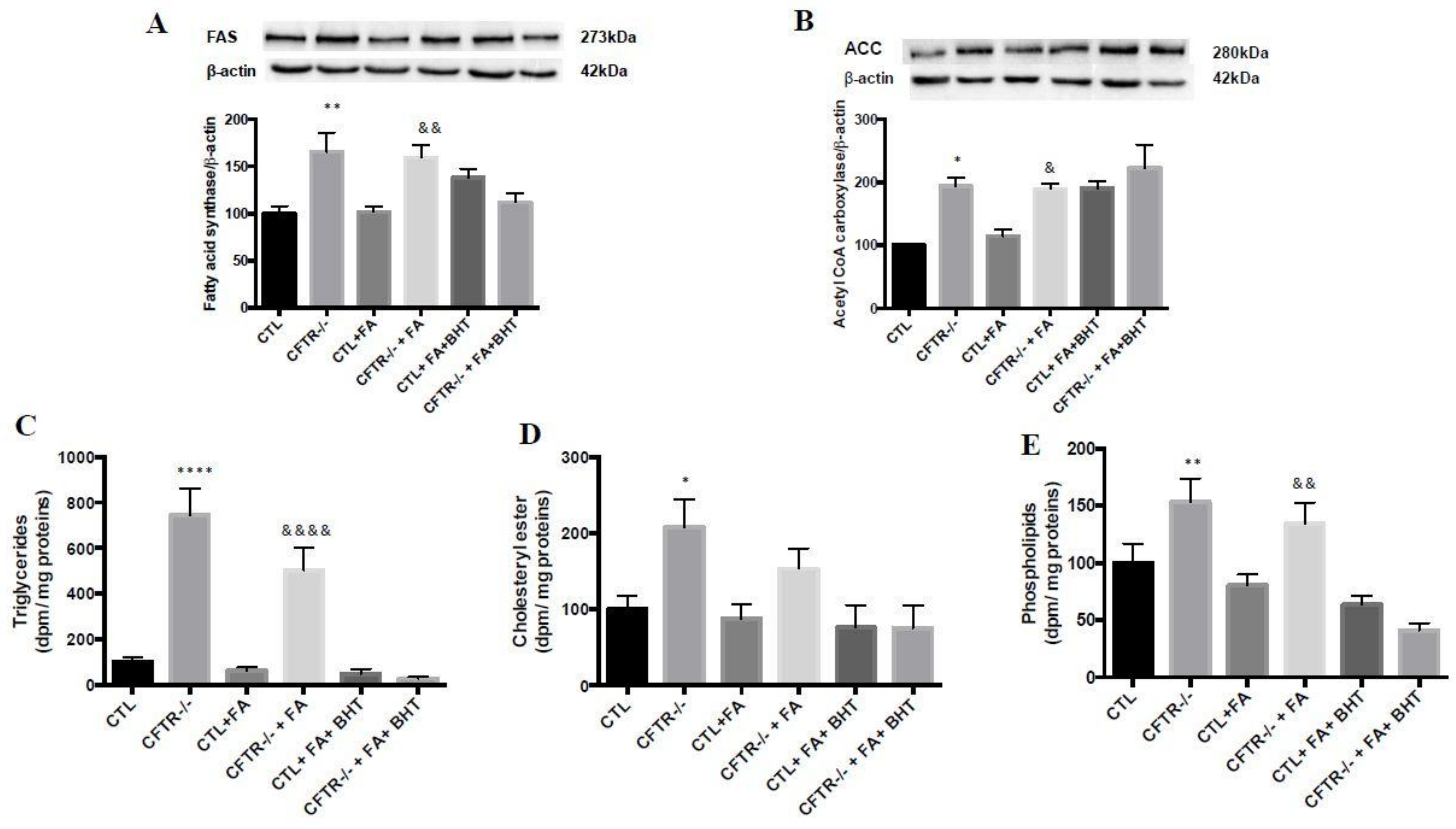
3.6. Lipogenesis
3.7. Lipid Synthesis
3.8. Lipoprotein Production
3.9. Apolipoprotein Biogenesis
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviation
| ACADL | acyl-CoA dehydrogenase long-chain |
| ACC | acetyl CoA carboxylase |
| AMPK | AMP-activated protein kinase |
| ARE | antioxidant response element |
| BHT | butylated hydroxy toluene |
| CF | cystic fibrosis |
| CPT1 | carnitine palmitoyltransferase |
| CTL | controls |
| FAS | fatty acid synthase. |
| Fe/Asc | iron-ascorbate |
| GSH | glutathione |
| Nrf2 | nuclear factor like 2 |
| OGG1 | 8-oxoguanine-DNA glycosylase |
| OxS | oxidative stress |
| PGC-1α | peroxisome proliferator activated receptor gamma coactivator-1-alpha |
| PPARα | peroxisome proliferator-activated receptor alpha |
| ROS | reactive oxygen species |
| ZFN | zinc finger nuclease |
References
- Ravilly, S.; Le Roux, E.; Bellis, G.; Dufour, F. Epidémiologie et physiopathologie de la mucoviscidose. Rev. Francoph. Lab. 2007, 397, 26–36. [Google Scholar] [CrossRef]
- Storni, V.; Claustres, M.; Chinet, T.; Ravilly, S. Diagnostic de la mucoviscidose. Arch. Pédiatr. 2001, 8, 818–832. [Google Scholar] [CrossRef]
- Biswas, S.; Chida, A.S.; Rahman, I. Redox modifications of protein-thiols: Emerging roles in cell signaling. Biochem. Pharmacol. 2006, 71, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Nzengue, Y. Comparaison des Mécanismes de Toxicité Redox du Cadmium, du Cuivre et du Zinc: Place des Métallothionéines et de p53. Ph.D. Thesis, Université Joseph-Fourier-Grenoble I, Joseph Fournier, Grenoble, France, 2008. [Google Scholar]
- Galli, F.; Battistoni, A.; Gambari, R.; Pompella, A.; Bragonzi, A.; Pilolli, F.; Iuliano, L.; Piroddi, M.; Dechecchi, M.C.; Cabrini, G. Oxidative stress and antioxidant therapy in cystic fibrosis. Biochim. Biophys. Acta 2012, 1822, 690–713. [Google Scholar] [CrossRef] [PubMed]
- Kleme, M.L.; Levy, E. Cystic fibrosis-related oxidative stress and intestinal lipid disorders. Antioxid. Redox. Signal. 2015, 22, 614–631. [Google Scholar] [CrossRef] [PubMed]
- Taha, R.; Seidman, E.; Mailhot, G.; Boudreau, F.; Gendron, F.P.; Beaulieu, J.F.; Menard, D.; Delvin, E.; Amre, D.; Levy, E. Oxidative stress and mitochondrial functions in the intestinal Caco-2/15 cell line. PLoS ONE 2010, 5, e11817. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 2012, 23, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Quintans, N.; Prieto, I.; Sanchez-Ramos, C.; Luque, A.; Arza, E.; Olmos, Y.; Monsalve, M. Regulation of endothelial dynamics by PGC-1alpha relies on ROS control of VEGF-A signaling. Free Radic. Biol. Med. 2016, 93, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, N.; Oono, T.; Igarashi, H.; Ito, T.; Nakamura, T.; Uchida, M.; Coy, D.H.; Jensen, R.T.; Takayanagi, R. Vasoactive intestinal peptide reduces oxidative stress in pancreatic acinar cells through the inhibition of NADPH oxidase. Peptides 2011, 32, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Meyerholz, D.K.; Stoltz, D.A.; Pezzulo, A.A.; Welsh, M.J. Pathology of gastrointestinal organs in a porcine model of cystic fibrosis. Am. J. Pathol. 2010, 176, 1377–1389. [Google Scholar] [CrossRef] [PubMed]
- Petrov, M.S. Therapeutic implications of oxidative stress in acute and chronic pancreatitis. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Njoroge, S.W.; Laposata, M.; Katrangi, W.; Seegmiller, A.C. DHA and EPA reverse cystic fibrosis-related FA abnormalities by suppressing FA desaturase expression and activity. J. Lipid. Res. 2012, 53, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Strandvik, B. Fatty acid metabolism in cystic fibrosis. Prostaglandins Leukot. Essent. Fat. Acids 2010, 83, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.S., Jr. Series introduction: the transcription factor NF-kappaB and human disease. J. Clin. Investig. 2001, 107, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Reis, F.J.C.; Damaceno, N. Cystic fibrosis. J. Pediatr. (Rio J.) 1998, 74 (Suppl. 1), S76–S94. [Google Scholar] [CrossRef]
- Kleme, M.L.; Sane, A.T.; Garofalo, C.; Levy, E. Targeted CFTR gene disruption with zinc-finger nucleases in human intestinal epithelial cells induces oxidative stress and inflammation. Int. J. Biochem. Cell Biol. 2016, 74, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Morel, Y.; Barouki, R. Influence du stress oxydant sur la régulation des gènes. Rev. Méd./Sci. 1998, 14, 9. [Google Scholar] [CrossRef]
- Mailhot, G.; Rabasa-Lhoret, R.; Moreau, A.; Berthiaume, Y.; Levy, E. CFTR depletion results in changes in fatty acid composition and promotes lipogenesis in intestinal Caco 2/15 cells. PLoS ONE 2010, 5, e10446. [Google Scholar] [CrossRef]
- Precourt, L.P.; Marcil, V.; Ntimbane, T.; Taha, R.; Lavoie, J.C.; Delvin, E.; Seidman, E.G.; Beaulieu, J.F.; Levy, E. Antioxidative properties of paraoxonase 2 in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G623–G634. [Google Scholar] [CrossRef] [PubMed]
- Escaffit, F.; Pare, F.; Gauthier, R.; Rivard, N.; Boudreau, F.; Beaulieu, J.F. Cdx2 modulates proliferation in normal human intestinal epithelial crypt cells. Biochem. Biophys. Res. Commun. 2006, 342, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Leahy, J.; Spahis, S.; Bonneil, E.; Garofalo, C.; Grimard, G.; Morel, S.; Laverdiere, C.; Krajinovic, M.; Drouin, S.; Delvin, E.; et al. Insight from mitochondrial functions and proteomics to understand cardiometabolic disorders in survivors of acute lymphoblastic leukemia. Metabolism 2018, 85, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Yeganeh, P.R.; Leahy, J.; Spahis, S.; Patey, N.; Desjardins, Y.; Roy, D.; Delvin, E.; Garofalo, C.; Leduc-Gaudet, J.P.; St-Pierre, D.; et al. Apple peel polyphenols reduce mitochondrial dysfunction in mice with DSS-induced ulcerative colitis. J. Nutr. Biochem. 2018, 57, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Denis, M.C.; Roy, D.; Yeganeh, P.R.; Desjardins, Y.; Varin, T.; Haddad, N.; Amre, D.; Sane, A.T.; Garofalo, C.; Furtos, A.; et al. Apple peel polyphenols: a key player in the prevention and treatment of experimental inflammatory bowel disease. Clin. Sci. (London) 2016, 130, 2217–2237. [Google Scholar] [CrossRef] [PubMed]
- Montoudis, A.; Seidman, E.; Boudreau, F.; Beaulieu, J.F.; Menard, D.; Elchebly, M.; Mailhot, G.; Sane, A.T.; Lambert, M.; Delvin, E.; et al. Intestinal fatty acid binding protein regulates mitochondrion beta-oxidation and cholesterol uptake. J. Lipid. Res. 2008, 49, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Mehran, M.; Levy, E.; Bendayan, M.; Seidman, E. Lipid, apolipoprotein, and lipoprotein synthesis and secretion during cellular differentiation in Caco-2 cells. Vitro Cell. Dev. Biol. Anim. 1997, 33, 118–128. [Google Scholar] [CrossRef]
- Levy, E.; Garofalo, C.; Thibault, L.; Dionne, S.; Daoust, L.; Lepage, G.; Roy, C.C. Intraluminal and intracellular phases of fat absorption are impaired in essential fatty acid deficiency. Am. Physi. Soc. 1992, 262, G319–G326. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Sinnett, D.; Thibault, L.; Nguyen, T.D.; Delvin, E.; Menard, D. Insulin modulation of newly synthesized apolipoproteins B-100 and B-48 in human fetal intestine: gene expression and mRNA editing are not involved. FEBS Lett. 1996, 393, 253–258. [Google Scholar] [CrossRef]
- Mailhot, G.; Ravid, Z.; Barchi, S.; Moreau, A.; Rabasa-Lhoret, R.; Levy, E. CFTR knockdown stimulates lipid synthesis and transport in intestinal Caco-2/15 cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G1239–G1249. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Ben Djoudi Ouadda, A.; Spahis, S.; Sane, A.T.; Garofalo, C.; Grenier, E.; Emonnot, L.; Yara, S.; Couture, P.; Beaulieu, J.F.; et al. PCSK9 plays a significant role in cholesterol homeostasis and lipid transport in intestinal epithelial cells. Atherosclerosis 2013, 227, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Morel, S.; Leahy, J.; Fournier, M.; Lamarche, B.; Garofalo, C.; Grimard, G.; Poulain, F.; Delvin, E.; Laverdiere, C.; Krajinovic, M.; et al. Lipid and lipoprotein abnormalities in acute lymphoblastic leukemia survivors. J. Lipid. Res. 2017, 58, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Sane, A.T.; Seidman, E.; Peretti, N.; Kleme, M.L.; Delvin, E.; Deslandres, C.; Garofalo, C.; Spahis, S.; Levy, E. Understanding Chylomicron Retention Disease Through Sar1b Gtpase Gene Disruption: Insight From Cell Culture. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2243–2251. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Garofalo, C.; Rouleau, T.; Gavino, V.; Bendayan, M. Impact of essential fatty acid deficiency on hepatic sterol metabolism in rats. Hepatology 1996, 23, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Bhakat, K.K.; Mokkapati, S.K.; Boldogh, I.; Hazra, T.K.; Mitra, S. Acetylation of human 8-oxoguanine-DNA glycosylase by p300 and its role in 8-oxoguanine repair in vivo. Mol. Cell. Biol. 2006, 26, 1654–1665. [Google Scholar] [CrossRef] [PubMed]
- Kukidome, D.; Nishikawa, T.; Sonoda, K.; Imoto, K.; Fujisawa, K.; Yano, M.; Motoshima, H.; Taguchi, T.; Matsumura, T.; Araki, E. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes 2006, 55, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Miller, S.; Sorscher, E.J. Cystic Fibrosis. N. Engl. J. Med. 2005, 352, 10. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S. Gastrointestinal Manifestations of Cystic Fibrosis. Gastroenterol. Hepatol. (NY) 2016, 12, 43–47. [Google Scholar]
- De Lisle, R.C.; Borowitz, D. The cystic fibrosis intestine. Cold Spring Harb. Perspect Med. 2013, 3, a009753. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Spiegelman, B.M. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 2006, 27, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Courtois, F.; Seidman, G.E.; Delvin, E.; Asselin, C.; Bernotti, S.; Ledoux, M.; Levy, E. Membrane peroxidation by lipopolysaccharide and iron-ascorbate adversely affects Caco-2 cell function: Beneficial role of butyric acid. Am. J. Clin. Nutr. 2003, 77, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Oudshoorn, J.H.; Lecluse, A.L.Y.; Berg, R.v.d.; Wouter, H.J.V.; Laag, J.v.d.; Houwen, R.H.J. Decreased Coenzyme Q10 Concentration in Plasma of Children with Cystic Fibrosis. J. Ped. Gastroenterol. Nutr. 2006, 43, 646–650. [Google Scholar] [CrossRef]
- Day, B.J.; van Heeckeren, A.M.; Min, E.; Velsor, L.W. Role for Cystic Fibrosis Transmembrane Conductance Regulator Protein in a Glutathione Response to Bronchopulmonary Pseudomonas Infection. Infect. Immun. 2004, 72, 2045–2051. [Google Scholar] [CrossRef] [PubMed]
- Naruse, S. cystic fibrosis and related diseases of the pancreas. Best Pract. Res. Clin. Gastroenterol. 2002, 163, 511–526. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Knutti, D.; Kralli, A. PGC-1, a versatile coactivator. Trends Endocrinol. Metab. 2001, 12, 360–365. [Google Scholar] [CrossRef]
- Ichida, M.; Nemoto, S.; Finkel, T. Identification of a specific molecular repressor of the peroxisome proliferator-activated receptor gamma Coactivator-1 alpha (PGC-1alpha). J. Biol. Chem. 2002, 277, 50991–50995. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 1991, 266, 11632–11639. [Google Scholar] [PubMed]
- Zhang, D.D. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug. Metab. Rev. 2006, 38, 769–789. [Google Scholar] [CrossRef] [PubMed]
- Manson, M.E.; Corey, D.A.; White, N.M.; Kelley, T.J. cAMP-mediated regulation of cholesterol accumulation in cystic fibrosis and Niemann-Pick type C cells. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2008, 295, L809–L819. [Google Scholar] [CrossRef] [PubMed]
- Ziady, A.G.; Sokolow, A.; Shank, S.; Corey, D.; Myers, R.; Plafker, S.; Kelley, T.J. Interaction with CREB binding protein modulates the activities of Nrf2 and NF-kappaB in cystic fibrosis airway epithelial cells. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2012, 302, L1221–L1231. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Bravard, A.; Vacher, M.; Gouget, B.; Coutant, A.; de Boisferon, F.H.; Marsin, S.; Chevillard, S.; Radicella, J.P. Redox regulation of human OGG1 activity in response to cellular oxidative stress. Mol. Cell. Biol. 2006, 26, 7430–7436. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Kelly, D.P. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J. Clin. Investig. 2006, 116, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Zechner, C.; Lai, L.; Zechner, J.F.; Geng, T.; Yan, Z.; Rumsey, J.W.; Collia, D.; Chen, Z.; Wozniak, D.F.; Leone, T.C.; et al. Total skeletal muscle PGC-1 deficiency uncouples mitochondrial derangements from fiber type determination and insulin sensitivity. Cell Metab. 2010, 12, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Siow, R.C.; Mann, G.E. Impaired redox signaling and antioxidant gene expression in endothelial cells in diabetes: A role for mitochondria and the nuclear factor-E2-related factor 2-Kelch-like ECH-associated protein 1 defense pathway. Antioxid. Redox. Signal. 2011, 14, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Harmel, E.; Grenier, E.; Bendjoudi Ouadda, A.; El Chebly, M.; Ziv, E.; Beaulieu, J.F.; Sane, A.; Spahis, S.; Laville, M.; Levy, E. AMPK in the small intestine in normal and pathophysiological conditions. Endocrinology 2014, 155, 873–888. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zhang, X.; Zhang, J.T.; Tsang, L.L.; Jiang, X.; Chan, H.C. Defective CFTR-β-catenin interaction promotes NF-κB nuclear translocation and intestinal inflammation in cystic fibrosis. Oncotarget 2016, 7, 64030–64042. [Google Scholar] [PubMed]
- Esposito, S.; Tosco, A.; Villella, V.R.; Raia, V.; Kroemer, G.; Maiuri, L. Manipulating proteostasis to repair the F508del-CFTR defect in cystic fibrosis. Mol. Cell. Pediatr. 2016, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Peretti, N.; Marcil, V.; Drouin, E.; Levy, E. Mechanisms of lipid malabsorption in Cystic Fibrosis: The impact of essential fatty acids deficiency. Nutr. Metab. (London) 2005, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Ishimo, M.C.; Belson, L.; Ziai, S.; Levy, E.; Berthiaume, Y.; Coderre, L.; Rabasa-Lhoret, R. Hypertriglyceridemia is associated with insulin levels in adult cystic fibrosis patients. J. Cyst. Fibros. 2013, 12, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Lepage, G.; Bendayan, M.; Ronco, N.; Thibault, L.; Galeano, N.; Smith, L.; Roy, C.C. Relationship of decreased hepatic lipase activity and lipoprotein abnormalities to essential fatty acid deficiency in cystic fibrosis patients. J. Lipid. Res. 1989, 30, 1197–1209. [Google Scholar] [PubMed]
- Woestenenk, J.W.; Schulkes, D.A.; Schipper, H.S.; van der Ent, C.K.; Houwen, R.H.J. Dietary intake and lipid profile in children and adolescents with cystic fibrosis. J. Cyst. Fibros 2017, 16, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Buehler, T.; Steinmann, M.; Singer, F.; Regamey, N.; Casaulta, C.; Schoeni, M.H.; Simonetti, G.D. Increased arterial stiffness in children with cystic fibrosis. Eur. Respir. J. 2012, 39, 1536–1537. [Google Scholar] [CrossRef] [PubMed]
- Valdivieso, A.G.; Santa-Coloma, T.A. CFTR activity and mitochondrial function. Redox. Biol. 2013, 1, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Favia, M.; Bobba, A.; Guerra, L.; Casavola, V.; Reshkin, S.J. Characterization of mitochondrial function in cells with impaired cystic fibrosis transmembrane conductance regulator (CFTR) function. J. Bioenerg. Biomembr. 2016, 48, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Donepudi, A.C.; More, V.R.; Kulkarni, S.R.; Li, L.; Guo, L.; Yan, B.; Chatterjee, T.; Weintraub, N.; Slitt, A.L. Deficiency in Nrf2 transcription factor decreases adipose tissue mass and hepatic lipid accumulation in leptin-deficient mice. Obesity (Silver Spring) 2015, 23, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Ruotsalainen, A.K.; Lappalainen, J.P.; Heiskanen, E.; Merentie, M.; Sihvola, V.; Napankangas, J.; Lottonen-Raikaslehto, L.; Kansanen, E.; Adinolfi, S.; Kaarniranta, K.; et al. Nrf2 deficiency impairs atherosclerotic lesion development but promotes features of plaque instability in hypercholesterolemic mice. Cardiovasc Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R.; Gupte, A.A.; Ji, R.; Ramirez, M.R.; Minze, L.J.; Liu, J.Z.; Arredondo, M.; Ren, Y.; Deng, T.; Wang, J.; et al. Myeloid deletion of nuclear factor erythroid 2-related factor 2 increases atherosclerosis and liver injury. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2839–2846. [Google Scholar] [CrossRef] [PubMed]
- Ruotsalainen, A.K.; Inkala, M.; Partanen, M.E.; Lappalainen, J.P.; Kansanen, E.; Makinen, P.I.; Heinonen, S.E.; Laitinen, H.M.; Heikkila, J.; Vatanen, T.; et al. The absence of macrophage Nrf2 promotes early atherogenesis. Cardiovasc. Res. 2013, 98, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Ito, K.; Hosoya, T.; Mimura, J.; Maruyama, A.; Noguchi, N.; Yagami, K.; Morito, N.; Takahashi, S.; Maher, J.M.; et al. Nrf2 in bone marrow-derived cells positively contributes to the advanced stage of atherosclerotic plaque formation. Free Radic. Biol. Med. 2012, 53, 2256–2262. [Google Scholar] [CrossRef] [PubMed]
- Sussan, T.E.; Jun, J.; Thimmulappa, R.; Bedja, D.; Antero, M.; Gabrielson, K.L.; Polotsky, V.Y.; Biswal, S. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS ONE 2008, 3, e3791. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VIABILITY CRITERIA | Control Cells | CFTR-/- Cells |
|---|---|---|
| Trypan blue exclusion | >96% | >96% |
| Transepithelial resistance (Ω × cm2) | 100 ± 20 | 96 ± 14 |
| Sucrase activity (UI/g proteins) | 100 ± 21 | 94 ± 15 |
| Villin protein expression (% of controls) | 100 ± 11 | 102 ± 11 |
| Occludin protein expression (% of controls) | 100 ± 6 | 105 ± 5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kleme, M.L.; Sané, A.; Garofalo, C.; Seidman, E.; Brochiero, E.; Berthiaume, Y.; Levy, E. CFTR Deletion Confers Mitochondrial Dysfunction and Disrupts Lipid Homeostasis in Intestinal Epithelial Cells. Nutrients 2018, 10, 836. https://doi.org/10.3390/nu10070836
Kleme ML, Sané A, Garofalo C, Seidman E, Brochiero E, Berthiaume Y, Levy E. CFTR Deletion Confers Mitochondrial Dysfunction and Disrupts Lipid Homeostasis in Intestinal Epithelial Cells. Nutrients. 2018; 10(7):836. https://doi.org/10.3390/nu10070836
Chicago/Turabian StyleKleme, Marie L., Alain Sané, Carole Garofalo, Ernest Seidman, Emmanuelle Brochiero, Yves Berthiaume, and Emile Levy. 2018. "CFTR Deletion Confers Mitochondrial Dysfunction and Disrupts Lipid Homeostasis in Intestinal Epithelial Cells" Nutrients 10, no. 7: 836. https://doi.org/10.3390/nu10070836
APA StyleKleme, M. L., Sané, A., Garofalo, C., Seidman, E., Brochiero, E., Berthiaume, Y., & Levy, E. (2018). CFTR Deletion Confers Mitochondrial Dysfunction and Disrupts Lipid Homeostasis in Intestinal Epithelial Cells. Nutrients, 10(7), 836. https://doi.org/10.3390/nu10070836