

Rhizobiome Signature and Its Alteration Due to Watering in the Wild Plant Moringa oleifera

 , , , , , , , , , , , and
, , , , , , , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Watering Experiment, Soil Collection and Whole Metagenome Sequencing
2.2. DNA Extraction, Library Construction and WGS
2.3. Processing of Sequencing Data and Gene Cataloging
2.4. Gene and Taxonomic Annotation
3. Results
3.1. Statistics of WGS Datasets
3.2. Description of Core/Pan Rarefaction Curves and Venn Diagram
3.3. Correlation Coefficient and Principal Component Analyses
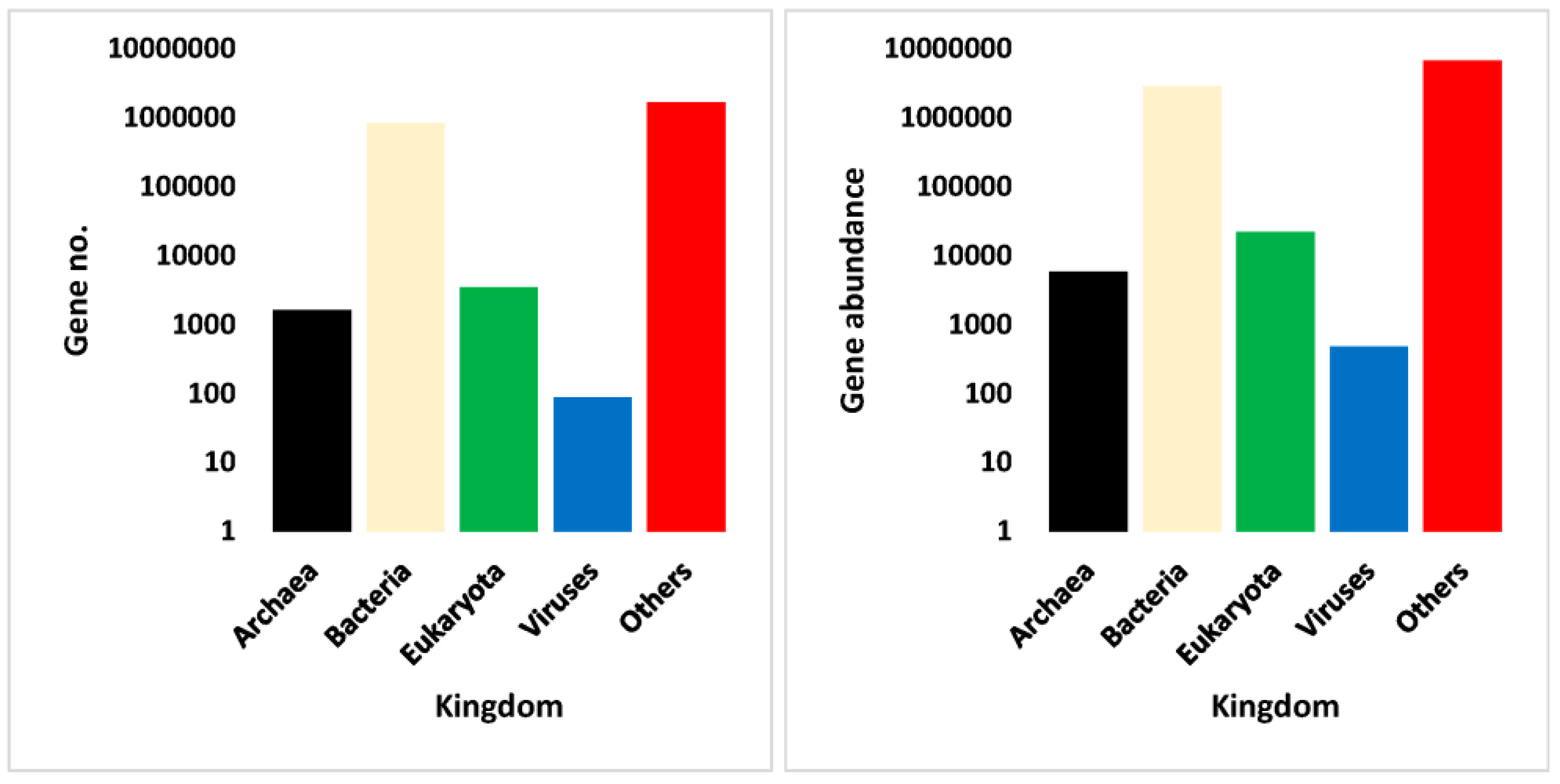
3.4. Annotation Results of Assembled Non-Redundant ORFs
3.5. Differential Microbiome Structure and Abundance
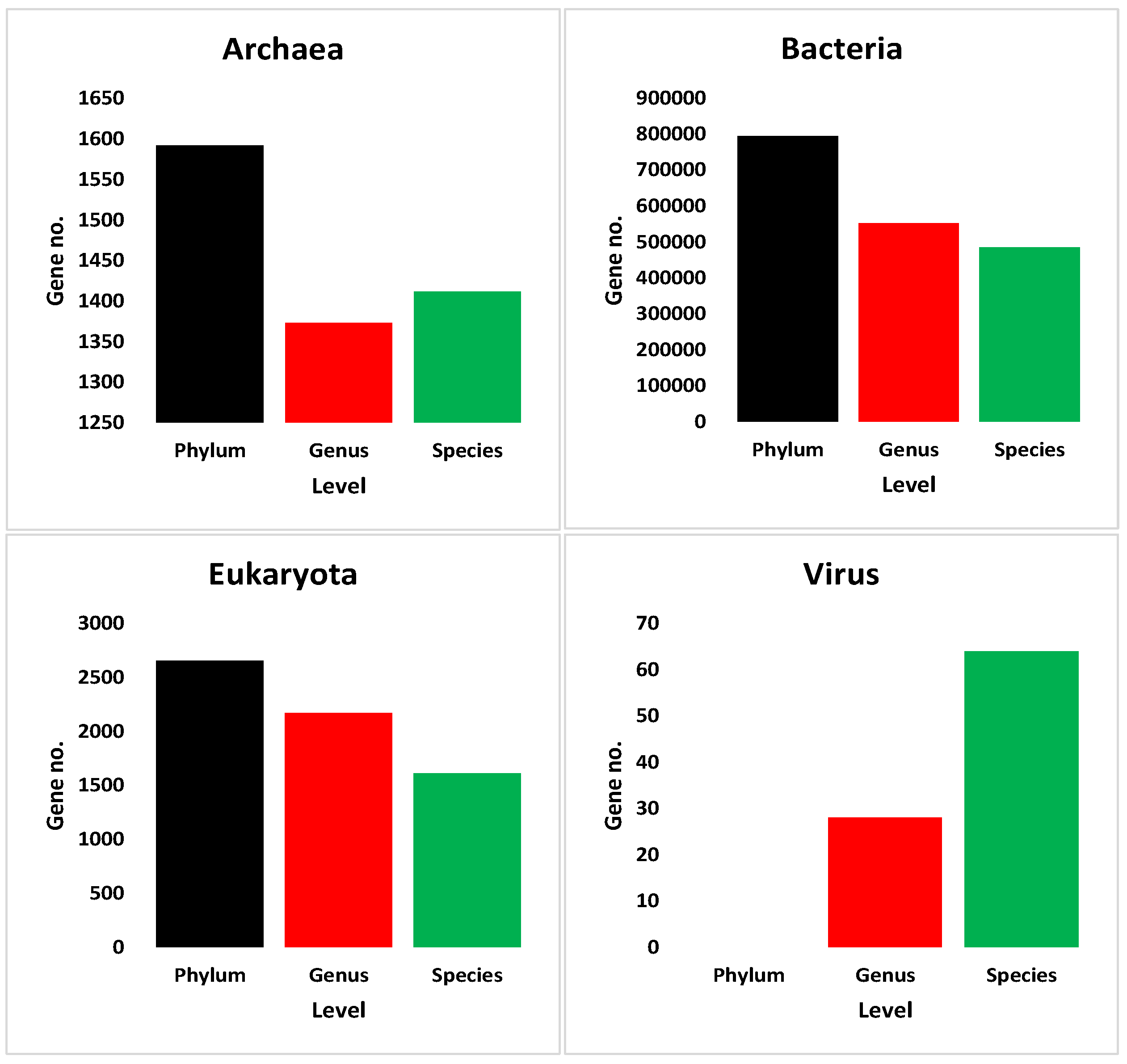
3.5.1. At Phylum Level
3.5.2. At Genus Level
3.5.3. At Species Level
4. Discussion
4.1. Differential Abundance/Relative Abundance of Microbes Based on Soil Type
4.1.1. Higher AB/RAB Taxa in Bulk Soil
4.1.2. Higher AB/RAB in Rhizosphere Soil
4.2. Differential Abundance/Relative Abundance of Microbes Based on Watering Time Points
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gupta, B.; Ahmed, K. Moringa oleifera: A Bibliometric Analysis of International Publications during 1935–2019. Pharmacogn. Rev. 2020, 14, 82–90. [Google Scholar]
- Al-Eisawi, D.M.; Al-Ruzayza, S. The flora of holy Mecca district, Saudi Arabia. Int. J. Biodivers. Conserv. 2015, 7, 173–189. [Google Scholar]
- Gopalakrishnan, L.; Doriya, K.; Kumar, D.S. Moringa oleifera: A review on nutritive importance and its medicinal application. Food Sci. Hum. Wellness 2016, 5, 49–56. [Google Scholar] [CrossRef]
- Rockwood, J.; Anderson, B.; Casamatta, D. Potential uses of Moringa oleifera and an examination of antibiotic efficacy conferred by M. oleifera seed and leaf extracts using crude extraction techniques available to underserved indigenous populations. Int. J. Phytother. Res. 2013, 3, 61–71. [Google Scholar]
- Milla, P.G.; Penalver, R.; Nieto, G. Health Benefits of Uses and Applications of Moringa oleifera in Bakery Products. Plants 2021, 10, 318. [Google Scholar] [CrossRef]
- Koul, B.; Chase, N. Moringa oleifera Lam.: Panacea to several maladies. J. Chem. Pharm. Res. 2015, 7, 687–707. [Google Scholar]
- Kumar, A.; Naaz, F.; Kushwaha, A.; Chaudhary, P.; Srivastav, P. Present review on phytochemistry, neutraceutical, antimicrobial, antidiabetic, biotechnological and pharmacological characteristics of Moringa oleifera Linn. BMR Phytomed. 2016, 2, 1–17. [Google Scholar]
- Saini, R.K.; Sivanesan, I.; Keum, Y.-S. Phytochemicals of Moringa oleifera: A review of their nutritional, therapeutic and industrial significance. 3 Biotech 2016, 6, 1–14. [Google Scholar]
- Fahey, J.W. Moringa oleifera: A review of the medical evidence for its nutritional, therapeutic, and prophylactic properties. Part 1. Trees Life J. 2005, 1, 1–15. [Google Scholar]
- Panda, S.; Kar, A.; Sharma, P.; Sharma, A. Cardioprotective potential of N,alpha-L-rhamnopyranosyl vincosamide, an indole alkaloid, isolated from the leaves of Moringa oleifera in isoproterenol induced cardiotoxic rats: In vivo and in vitro studies. Bioorg. Med. Chem. Lett. 2013, 23, 959–962. [Google Scholar] [CrossRef]
- Ashfaq, M.; Basra, S.M.; Umair, A. Moringa: A miracle plant for agro-forestry. J. Agric. Soc. Sci. 2012, 8, 115–122. [Google Scholar]
- Rivas-Vega, M.E.; López-Pereira, J.L.; Miranda-Baeza, A.; Sandoval-Muy, M.I. Sustitución parcial de harina de sardina con Moringa oleifera en alimentos balanceados para juveniles de tilapia (Oreochromismossambicus x Oreochromisniloticus) cultivada en agua de mar. Biotecnia 2012, 14, 3–10. [Google Scholar]
- Folkard, G.; Sutherland, J. Moringa oleifera un árbol con enormes potencialidades. Agrofor. Today 1996, 8, 5–8. [Google Scholar]
- Gómez, A.V.; Angulo, K.J.O. Revisión de las características y usosde la planta moringa oleífera. Investig. Desarro. 2014, 22, 309–330. [Google Scholar]
- Breitwieser, F.P.; Lu, J.; Salzberg, S.L. A review of methods and databases for metagenomic classification and assembly. Brief Bioinform 2019, 20, 1125–1136. [Google Scholar] [CrossRef]
- Laudadio, I.; Fulci, V.; Palone, F.; Stronati, L.; Cucchiara, S.; Carissimi, C. Quantitative Assessment of Shotgun Metagenomics and 16S rDNA Amplicon Sequencing in the Study of Human Gut Microbiome. Omics 2018, 22, 248–254. [Google Scholar] [CrossRef]
- Campanaro, S.; Treu, L.; Kougias, P.G.; Zhu, X.; Angelidaki, I. Taxonomy of anaerobic digestion microbiome reveals biases associated with the applied high throughput sequencing strategies. Sci. Rep. 2018, 8, 1926. [Google Scholar] [CrossRef]
- Fouhy, F.; Clooney, A.G.; Stanton, C.; Claesson, M.J.; Cotter, P.D. 16S rRNA gene sequencing of mock microbial populations- impact of DNA extraction method, primer choice and sequencing platform. BMC Microbiol. 2016, 16, 123. [Google Scholar] [CrossRef]
- Durazzi, F.; Sala, C.; Castellani, G.; Manfreda, G.; Remondini, D.; De Cesare, A. Comparison between 16S rRNA and shotgun sequencing data for the taxonomic characterization of the gut microbiota. Sci. Rep. 2021, 11, 3030. [Google Scholar]
- Chistoserdova, L. Functional metagenomics: Recent advances and future challenges. Biotechnol. Genet. Eng. Rev. 2009, 26, 335–352. [Google Scholar]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef]
- Pérez-Cobas, A.E.; Gomez-Valero, L.; Buchrieser, C. Metagenomic approaches in microbial ecology: An update on whole-genome and marker gene sequencing analyses. Microb. Genom. 2020, 6, mgen000409. [Google Scholar]
- Dai, L.; Zhang, G.; Yu, Z.; Ding, H.; Xu, Y.; Zhang, Z. Effect of drought stress and developmental stages on microbial community structure and diversity in peanut rhizosphere soil. Int. J. Mol. Sci. 2019, 20, 2265. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Garrido-Oter, R.; Munch, P.C.; Weiman, A.; Droge, J.; Pan, Y.; McHardy, A.C.; Schulze-Lefert, P. Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 2015, 17, 392–403. [Google Scholar] [CrossRef]
- Weese, D.J.; Heath, K.D.; Dentinger, B.T.; Lau, J.A. Long-term nitrogen addition causes the evolution of less-cooperative mutualists. Evolution 2015, 69, 631–642. [Google Scholar] [CrossRef]
- Bouffaud, M.L.; Poirier, M.A.; Muller, D.; Moënne-Loccoz, Y. Root microbiome relates to plant host evolution in maize and other P oaceae. Environ. Microbiol. 2014, 16, 2804–2814. [Google Scholar] [CrossRef]
- Ramirez, K.S.; Craine, J.M.; Fierer, N. Consistent effects of nitrogen amendments on soil microbial communities and processes across biomes. Glob. Chang. Biol. 2012, 18, 1918–1927. [Google Scholar] [CrossRef]
- Jones, D.L.; Nguyen, C.; Finlay, R.D. Carbon flow in the rhizosphere: Carbon trading at the soil–root interface. Plant Soil 2009, 321, 5–33. [Google Scholar] [CrossRef]
- Jones, D.L.; Hodge, A.; Kuzyakov, Y. Plant and mycorrhizal regulation of rhizodeposition. New Phytol. 2004, 163, 459–480. [Google Scholar] [CrossRef]
- Schlaeppi, K.; Dombrowski, N.; Oter, R.G.; Ver Loren van Themaat, E.; Schulze-Lefert, P. Quantitative divergence of the bacterial root microbiota in Arabidopsis thaliana relatives. Proc. Natl. Acad. Sci. USA 2014, 111, 585–592. [Google Scholar] [CrossRef]
- Zachow, C.; Müller, H.; Tilcher, R.; Berg, G. Differences between the rhizosphere microbiome of Beta vulgaris ssp. maritima—Ancestor of all beet crops—And modern sugar beets. Front. Microbiol. 2014, 5, 415. [Google Scholar]
- Geng, L.-L.; Shao, G.-X.; Raymond, B.; Wang, M.-L.; Sun, X.-X.; Shu, C.-L.; Zhang, J. Subterranean infestation by Holotrichia parallela larvae is associated with changes in the peanut (Arachis hypogaea L.) rhizosphere microbiome. Microbiol. Res. 2018, 211, 13–20. [Google Scholar]
- Hurt, R.A.; Qiu, X.; Wu, L.; Roh, Y.; Palumbo, A.V.; Tiedje, J.M.; Zhou, J. Simultaneous recovery of RNA and DNA from soils and sediments. Appl. Environ. Microbiol. 2001, 67, 4495–4503. [Google Scholar] [CrossRef]
- McLean, E. Soil pH and lime requirement. Methods Soil Anal. Part 2 Chem. Microbiol. Prop. 1983, 9, 199–224. [Google Scholar]
- Mende, D.R.; Waller, A.S.; Sunagawa, S.; Järvelin, A.I.; Chan, M.M.; Arumugam, M.; Raes, J.; Bork, P. Assessment of metagenomic assembly using simulated next generation sequencing data. PLoS ONE 2012, 7, e31386. [Google Scholar]
- Karlsson, F.H.; Fåk, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar]
- Oh, J.; Byrd, A.L.; Deming, C.; Conlan, S.; Program, N.C.S.; Kong, H.H.; Segre, J.A. Biogeography and individuality shape function in the human skin metagenome. Nature 2014, 514, 59–64. [Google Scholar] [CrossRef]
- Nielsen, H.B.; Almeida, M.; Juncker, A.S.; Rasmussen, S.; Li, J.; Sunagawa, S.; Plichta, D.R.; Gautier, L.; Pedersen, A.G.; Le Chatelier, E. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 2014, 32, 822–828. [Google Scholar] [CrossRef]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Bohm, J.; Brunetti, F.; Habermann, N.; et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef]
- Sunagawa, S.; Coelho, L.P.; Chaffron, S.; Kultima, J.R.; Labadie, K.; Salazar, G.; Djahanschiri, B.; Zeller, G.; Mende, D.R.; Alberti, A.; et al. Ocean plankton. Structure and function of the global ocean microbiome. Science 2015, 348, 1261359. [Google Scholar] [CrossRef]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN community edition-interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.-J.; Weber, N.; Schuster, S.C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar]
- Wu, L.; Wang, J.; Wu, H.; Chen, J.; Xiao, Z.; Qin, X.; Zhang, Z.; Lin, W. Comparative Metagenomic Analysis of Rhizosphere Microbial Community Composition and Functional Potentials under Rehmannia glutinosa Consecutive Monoculture. Int. J. Mol. Sci. 2018, 19, 2394. [Google Scholar] [CrossRef]
- Luo, C.; Rodriguez-R., L.M.; Johnston, E.R.; Wu, L.; Cheng, L.; Xue, K.; Tu, Q.; Deng, Y.; He, Z.; Shi, J.Z. Soil microbial community responses to a decade of warming as revealed by comparative metagenomics. Appl. Environ. Microbiol. 2014, 80, 1777–1786. [Google Scholar]
- Simon, C.; Daniel, R. Achievements and new knowledge unraveled by metagenomic approaches. Appl. Microbiol. Biotechnol. 2009, 85, 265–276. [Google Scholar] [CrossRef]
- Lucaciu, R.; Pelikan, C.; Gerner, S.M.; Zioutis, C.; Kostlbacher, S.; Marx, H.; Herbold, C.W.; Schmidt, H.; Rattei, T. A Bioinformatics Guide to Plant Microbiome Analysis. Front. Plant Sci. 2019, 10, 1313. [Google Scholar] [CrossRef]
- Xu, W.; Chen, T.; Pei, Y.; Guo, H.; Li, Z.; Yang, Y.; Zhang, F.; Yu, J.; Li, X.; Yang, Y. Characterization of shallow whole-metagenome shotgun sequencing as a high-accuracy and low-cost method by complicated mock microbiomes. Front. Microbiol. 2021, 12, 1723. [Google Scholar] [CrossRef]
- Woese, C.R.; Fox, G.E. Phylogenetic structure of the prokaryotic domain: The primary kingdoms. Proc. Natl. Acad. Sci. USA 1977, 74, 5088–5090. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Consortium, F.B.; List, F.B.C.A.; Bolchacova, E. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Carlton, J.M.; Angiuoli, S.V.; Suh, B.B.; Kooij, T.W.; Pertea, M.; Silva, J.C.; Ermolaeva, M.D.; Allen, J.E.; Selengut, J.D.; Koo, H.L.; et al. Genome sequence and comparative analysis of the model rodent malaria parasite Plasmodium yoelii yoelii. Nature 2002, 419, 512–519. [Google Scholar] [CrossRef]
- Xi, H.; Shen, J.; Qu, Z.; Yang, D.; Liu, S.; Nie, X.; Zhu, L. Effects of Long-term Cotton Continuous Cropping on Soil Microbiome. Sci. Rep. 2019, 9, 18297. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Sun, Y.; Cai, Y.; Liu, L.; Yu, F.; Farrell, M.L.; McKendree, W.; Farmerie, W. ESPRIT: Estimating species richness using large collections of 16S rRNA pyrosequences. Nucleic Acids Res. 2009, 37, e76. [Google Scholar]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef]
- Reinhold-Hurek, B.; Bünger, W.; Burbano, C.S.; Sabale, M.; Hurek, T. Roots shaping their microbiome: Global hotspots for microbial activity. Annu. Rev. Phytopathol. 2015, 53, 403–424. [Google Scholar] [CrossRef]
- Berendsen, R.L.; Pieterse, C.M.; Bakker, P.A. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef]
- Jones, D.; Kemmitt, S.; Wright, D.; Cuttle, S.; Bol, R.; Edwards, A. Rapid intrinsic rates of amino acid biodegradation in soils are unaffected by agricultural management strategy. Soil Biol. Biochem. 2005, 37, 1267–1275. [Google Scholar]
- Saleem, M.; Law, A.D.; Sahib, M.R.; Pervaiz, Z.H.; Zhang, Q. Impact of root system architecture on rhizosphere and root microbiome. Rhizosphere 2018, 6, 47–51. [Google Scholar] [CrossRef]
- Grove, S.; Haubensak, K.A.; Parker, I.M. Direct and indirect effects of allelopathy in the soil legacy of an exotic plant invasion. Plant Ecol. 2012, 213, 1869–1882. [Google Scholar]
- Wu, L.; Chen, J.; Xiao, Z.; Zhu, X.; Wang, J.; Wu, H.; Wu, Y.; Zhang, Z.; Lin, W. Barcoded Pyrosequencing Reveals a Shift in the Bacterial Community in the Rhizosphere and Rhizoplane of Rehmannia glutinosa under Consecutive Monoculture. Int. J. Mol. Sci. 2018, 19, 850. [Google Scholar] [CrossRef]
- Li, X.-G.; Ding, C.-F.; Hua, K.; Zhang, T.-L.; Zhang, Y.-N.; Zhao, L.; Yang, Y.-R.; Liu, J.-G.; Wang, X.-X. Soil sickness of peanuts is attributable to modifications in soil microbes induced by peanut root exudates rather than to direct allelopathy. Soil Biol. Biochem. 2014, 78, 149–159. [Google Scholar] [CrossRef]
- Marquez, N.; Giachero, M.L.; Declerck, S.; Ducasse, D.A. Macrophomina phaseolina: General Characteristics of Pathogenicity and Methods of Control. Front. Plant Sci. 2021, 12, 634397. [Google Scholar] [CrossRef]
- Badri, D.; Vivanco, J. Regulation and function of root exudates Plant. Cell Environ. 2009, 32, 666–681. [Google Scholar] [CrossRef]
- Badri, D.V.; Chaparro, J.M.; Zhang, R.; Shen, Q.; Vivanco, J.M. Application of natural blends of phytochemicals derived from the root exudates of Arabidopsis to the soil reveal that phenolic-related compounds predominantly modulate the soil microbiome. J. Biol. Chem. 2013, 288, 4502–4512. [Google Scholar] [CrossRef]
- de Weert, S.; Vermeiren, H.; Mulders, I.H.; Kuiper, I.; Hendrickx, N.; Bloemberg, G.V.; Vanderleyden, J.; De Mot, R.; Lugtenberg, B.J. Flagella-driven chemotaxis towards exudate components is an important trait for tomato root colonization by Pseudomonas fluorescens. Mol. Plant-Microbe Interact. 2002, 15, 1173–1180. [Google Scholar] [CrossRef]
- Rudrappa, T.; Czymmek, K.J.; Pare, P.W.; Bais, H.P. Root-secreted malic acid recruits beneficial soil bacteria. Plant Physiol. 2008, 148, 1547–1556. [Google Scholar] [CrossRef]
- Argôlo-Filho, R.C.; Loguercio, L.L. Bacillus thuringiensis is an environmental pathogen and host-specificity has developed as an adaptation to human-generated ecological niches. Insects 2013, 5, 62–91. [Google Scholar]
- Fang, Y.; Li, Z.; Liu, J.; Shu, C.; Wang, X.; Zhang, X.; Yu, X.; Zhao, D.; Liu, G.; Hu, S.; et al. A pangenomic study of Bacillus thuringiensis. J. Genet. Genom. 2011, 38, 567–576. [Google Scholar] [CrossRef]
- Raymond, B. The biology, ecology and taxonomy of Bacillus thuringiensis and related bacteria. In Bacillus Thuringiensis and Lysinibacillus sphaericus; Springer: New York, NY, USA, 2017; pp. 19–39. [Google Scholar]
- Polanczyk, R.A.; Zanúncio, J.C.; Alves, S.B. Relationship between chemical properties of the soil and the occurrence of Bacillus thuringiensis. Ciência Rural 2009, 39, 1–5. [Google Scholar]
- Cappello, M.; Bungiro, R.D.; Harrison, L.M.; Bischof, L.J.; Griffitts, J.S.; Barrows, B.D.; Aroian, R.V. A purified Bacillus thuringiensis crystal protein with therapeutic activity against the hookworm parasite Ancylostoma ceylanicum. Proc. Natl. Acad. Sci. USA 2006, 103, 15154–15159. [Google Scholar] [CrossRef]
- Lee, S.C.; Idnurm, A. Fungal Sex: The Mucoromycota. Microbiol. Spectr. 2017, 5, 14. [Google Scholar] [CrossRef]
- Spatafora, J.W.; Aime, M.C.; Grigoriev, I.V.; Martin, F.; Stajich, J.E.; Blackwell, M. The Fungal Tree of Life: From Molecular Systematics to Genome-Scale Phylogenies. Microbiol. Spectr. 2017, 5, 1–34. [Google Scholar] [CrossRef]
- Spatafora, J.W.; Chang, Y.; Benny, G.L.; Lazarus, K.; Smith, M.E.; Berbee, M.L.; Bonito, G.; Corradi, N.; Grigoriev, I.; Gryganskyi, A.; et al. A phylum-level phylogenetic classification of zygomycete fungi based on genome-scale data. Mycologia 2016, 108, 1028–1046. [Google Scholar] [CrossRef]
- Ghosh, T.; Biswas, M.; Guin, C.; Roy, P. A review on characterization, therapeutic approaches and pathogenesis of Macrophomina phaseolina. Plant Cell Biotechnol. Mol. Biol. 2018, 19, 72–84. [Google Scholar]
- Khan, A.N.; Shair, F.; Malik, K.; Hayat, Z.; Khan, M.A.; Hafeez, F.Y.; Hassan, M.N. Molecular Identification and Genetic Characterization of Macrophomina phaseolina Strains Causing Pathogenicity on Sunflower and Chickpea. Front. Microbiol. 2017, 8, 1309. [Google Scholar] [CrossRef]
- Andreo-Jimenez, B.; Vandenkoornhuyse, P.; Le Van, A.; Heutinck, A.; Duhamel, M.; Kadam, N.; Jagadish, K.; Ruyter-Spira, C.; Bouwmeester, H. Plant host and drought shape the root associated fungal microbiota in rice. PeerJ 2019, 7, e7463. [Google Scholar] [CrossRef]
- Rodriguez, R.; White, J.; Redman, R. Fungal endophytes: Diversity and functional roles. New Phytol. 2009, 182, 314–330. [Google Scholar]
- Sinanaj, B.; Hoysted, G.A.; Pressel, S.; Bidartondo, M.I.; Field, K.J. Critical research challenges facing Mucoromycotina ‘fine root endophytes’. New Phytol. 2021, 232, 1528–1534. [Google Scholar]
- Barka, E.A.; Vatsa, P.; Sanchez, L.; Gaveau-Vaillant, N.; Jacquard, C.; Klenk, H.-P.; Clément, C.; Ouhdouch, Y.; van Wezel, G.P. Taxonomy, physiology, and natural products of Actinobacteria. Microbiol. Mol. Biol. Rev. 2016, 80, 1–43. [Google Scholar] [CrossRef]
- Riahi, H.S.; Heidarieh, P.; Fatahi-Bafghi, M. Genus Pseudonocardia: What we know about its biological properties, abilities and current application in biotechnology. J. Appl. Microbiol. 2022, 132, 890–906. [Google Scholar] [CrossRef]
- Ananbeh, H.; Rodrigo, M.A.M.; Jelinkova, P.; Strmiska, V.; Splichal, Z.; Jehmlich, N.; Michalkova, H.; Stojanović, M.; Voberkova, S.; Adam, V. Soil protein as a potential antimicrobial agent against methicillin–resistant Staphylococcus aureus. Environ. Res. 2020, 188, 109320. [Google Scholar]
- Gerken, T.; Wiegner, T.N.; Economy, L.M. A Comparison of Soil Staphylococcus aureus and Fecal Indicator Bacteria Concentrations across Land Uses in a Hawaiian Watershed; 0047-2425; Wiley Online Library: Hoboken, NJ, USA, 2022. [Google Scholar]
- Lazcano, C.; Boyd, E.; Holmes, G.; Hewavitharana, S.; Pasulka, A.; Ivors, K. The rhizosphere microbiome plays a role in the resistance to soil-borne pathogens and nutrient uptake of strawberry cultivars under field conditions. Sci. Rep. 2021, 11, 1–17. [Google Scholar] [CrossRef]
- Chaiharn, M.; Pathom-Aree, W.; Sujada, N.; Lumyong, S. Characterization of phosphate solubilizing Streptomyces as a biofertilizer. Chiang Mai J. Sci 2018, 45, 701–716. [Google Scholar]
- Jog, R.; Pandya, M.; Nareshkumar, G.; Rajkumar, S. Mechanism of phosphate solubilization and antifungal activity of Streptomyces spp. isolated from wheat roots and rhizosphere and their application in improving plant growth. Microbiology 2014, 160, 778–788. [Google Scholar]
- Devi, R.; Kaur, T.; Kour, D.; Yadav, A.; Yadav, A.N.; Suman, A.; Ahluwalia, A.S.; Saxena, A.K. Minerals solubilizing and mobilizing microbiomes: A sustainable approaches for managing minerals deficiency in agricultural soil. J. Appl. Microbiol. 2022, 133, 1245–1272. [Google Scholar]
- Sathya, A.; Vijayabharathi, R.; Gopalakrishnan, S. Plant growth-promoting actinobacteria: A new strategy for enhancing sustainable production and protection of grain legumes. 3 Biotech 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Tian, J.; Ge, F.; Zhang, D.; Deng, S.; Liu, X. Roles of Phosphate Solubilizing Microorganisms from Managing Soil Phosphorus Deficiency to Mediating Biogeochemical P Cycle. Biology 2021, 10, 158. [Google Scholar] [CrossRef]
- Divjot, K.; Rana, K.L.; Tanvir, K.; Yadav, N.; Yadav, A.N.; Kumar, M.; Kumar, V.; Dhaliwal, H.S.; Saxena, A.K. Biodiversity, current developments and potential biotechnological applications of phosphorus-solubilizing and-mobilizing microbes: A review. Pedosphere 2021, 31, 43–75. [Google Scholar]
- Hamdali, H.; Smirnov, A.; Esnault, C.; Ouhdouch, Y.; Virolle, M.J. Physiological studies and comparative analysis of rock phosphate solubilization abilities of Actinomycetales originating from Moroccan phosphate mines and of Streptomyces lividans. Appl. Soil Ecol. 2010, 44, 24–31. [Google Scholar]
- Hamdali, H.; Bouizgarne, B.; Hafidi, M.; Lebrihi, A.; Virolle, M.J.; Ouhdouch, Y. Screening for rock phosphate solubilizing Actinomycetes from Moroccan phosphate mines. Appl. Soil Ecol. 2008, 38, 12–19. [Google Scholar]
- Niu, B.; Paulson, J.N.; Zheng, X.; Kolter, R. Simplified and representative bacterial community of maize roots. Proc. Natl. Acad. Sci. USA 2017, 114, E2450–E2459. [Google Scholar] [CrossRef]
- Lidbury, I.; Borsetto, C.; Murphy, A.R.J.; Bottrill, A.; Jones, A.M.E.; Bending, G.D.; Hammond, J.P.; Chen, Y.; Wellington, E.M.H.; Scanlan, D.J. Niche-adaptation in plant-associated Bacteroidetes favours specialisation in organic phosphorus mineralisation. ISME J. 2021, 15, 1040–1055. [Google Scholar] [CrossRef]
- Cordell, D.; Drangert, J.-O.; White, S. The story of phosphorus: Global food security and food for thought. Glob. Environ. Chang. 2009, 19, 292–305. [Google Scholar]
- Tyrrell, T. The relative influences of nitrogen and phosphorus on oceanic primary production. Nature 1999, 400, 525–531. [Google Scholar] [CrossRef]
- Goll, D.S.; Brovkin, V.; Parida, B.; Reick, C.H.; Kattge, J.; Reich, P.B.; Van Bodegom, P.; Niinemets, Ü. Nutrient limitation reduces land carbon uptake in simulations with a model of combined carbon, nitrogen and phosphorus cycling. Biogeosciences 2012, 9, 3547–3569. [Google Scholar]
- White, P.J.; Hammond, J.P. Phosphorus nutrition of terrestrial plants. In The Ecophysiology of Plant-Phosphorus Interactions; Springer: New York, NY, USA, 2008; pp. 51–81. [Google Scholar]
- Schachtman, D.P.; Reid, R.J.; Ayling, S.M. Phosphorus Uptake by Plants: From Soil to Cell. Plant Physiol. 1998, 116, 447–453. [Google Scholar] [CrossRef]
- Thomas, F.; Hehemann, J.H.; Rebuffet, E.; Czjzek, M.; Michel, G. Environmental and gut bacteroidetes: The food connection. Front. Microbiol. 2011, 2, 93. [Google Scholar] [CrossRef]
- Bauer, M.; Kube, M.; Teeling, H.; Richter, M.; Lombardot, T.; Allers, E.; Würdemann, C.A.; Quast, C.; Kuhl, H.; Knaust, F. Whole genome analysis of the marine Bacteroidetes ‘Gramella forsetii’reveals adaptations to degradation of polymeric organic matter. Environ. Microbiol. 2006, 8, 2201–2213. [Google Scholar]
- Martens, E.C.; Koropatkin, N.M.; Smith, T.J.; Gordon, J.I. Complex glycan catabolism by the human gut microbiota: The Bacteroidetes Sus-like paradigm. J. Biol. Chem. 2009, 284, 24673–24677. [Google Scholar] [CrossRef]
- Castelle, C.J.; Banfield, J.F. Major new microbial groups expand diversity and alter our understanding of the tree of life. Cell 2018, 172, 1181–1197. [Google Scholar] [CrossRef]
- Starr, E.P.; Shi, S.; Blazewicz, S.J.; Probst, A.J.; Herman, D.J.; Firestone, M.K.; Banfield, J.F. Stable isotope informed genome-resolved metagenomics reveals that Saccharibacteria utilize microbially-processed plant-derived carbon. Microbiome 2018, 6, 1–12. [Google Scholar] [CrossRef]
- Xie, B.; Wang, J.; Nie, Y.; Chen, D.; Hu, B.; Wu, X.; Du, W. EpicPCR-directed cultivation of a Candidatus Saccharibacteria symbiont reveals a type IV pili-dependent epibiotic lifestyle. Proc. Natl. Acad. Sci. USA 2021, 119, e2215990119. [Google Scholar] [CrossRef]
- Tsuda, K.; Sato, M.; Glazebrook, J.; Cohen, J.D.; Katagiri, F. Interplay between MAMP-triggered and SA-mediated defense responses. Plant J. 2008, 53, 763–775. [Google Scholar] [CrossRef]
- Lu, H. Dissection of salicylic acid-mediated defense signaling networks. Plant Signal Behav. 2009, 4, 713–717. [Google Scholar] [CrossRef]
- Takahashi, Y.; Fujitani, H.; Hirono, Y.; Tago, K.; Wang, Y.; Hayatsu, M.; Tsuneda, S. Enrichment of Comammox and Nitrite-Oxidizing Nitrospira from Acidic Soils. Front. Microbiol. 2020, 11, 1737. [Google Scholar] [CrossRef]
- Tarre, S.; Green, M. High-rate nitrification at low pH in suspended-and attached-biomass reactors. Appl. Environ. Microbiol. 2004, 70, 6481–6487. [Google Scholar] [CrossRef]
- Cytryn, E.; Levkovitch, I.; Negreanu, Y.; Dowd, S.; Frenk, S.; Silber, A. Impact of short-term acidification on nitrification and nitrifying bacterial community dynamics in soilless cultivation media. Appl. Environ. Microbiol. 2012, 78, 6576–6582. [Google Scholar] [CrossRef]
- Hu, J.; Zhao, Y.; Yao, X.; Wang, J.; Zheng, P.; Xi, C.; Hu, B. Dominance of comammox Nitrospira in soil nitrification. Sci. Total Environ. 2021, 780, 146558. [Google Scholar] [CrossRef]
- Wrage, N.; Velthof, G.L.; Van Beusichem, M.L.; Oenema, O. Role of nitrifier denitrification in the production of nitrous oxide. Soil Biol. Biochem. 2001, 33, 1723–1732. [Google Scholar]
- Vitousek, P.M.; Aber, J.D.; Howarth, R.W.; Likens, G.E.; Matson, P.A.; Schindler, D.W.; Schlesinger, W.H.; Tilman, D.G. Human alteration of the global nitrogen cycle: Sources and consequences. Ecol. Appl. 1997, 7, 737–750. [Google Scholar]
- Koch, H.; van Kessel, M.A.; Lücker, S. Complete nitrification: Insights into the ecophysiology of comammox Nitrospira. Appl. Microbiol. Biotechnol. 2019, 103, 177–189. [Google Scholar] [CrossRef]
- Graham, E.B.; Knelman, J.E.; Schindlbacher, A.; Siciliano, S.; Breulmann, M.; Yannarell, A.; Beman, J.; Abell, G.; Philippot, L.; Prosser, J. Microbes as engines of ecosystem function: When does community structure enhance predictions of ecosystem processes? Front. Microbiol. 2016, 7, 214. [Google Scholar]
- Séneca, J.; Pjevac, P.; Canarini, A.; Herbold, C.W.; Zioutis, C.; Dietrich, M.; Simon, E.; Prommer, J.; Bahn, M.; Pötsch, E.M. Composition and activity of nitrifier communities in soil are unresponsive to elevated temperature and CO2, but strongly affected by drought. ISME J. 2020, 14, 3038–3053. [Google Scholar]
- Dieleman, W.I.; Vicca, S.; Dijkstra, F.A.; Hagedorn, F.; Hovenden, M.J.; Larsen, K.S.; Morgan, J.A.; Volder, A.; Beier, C.; Dukes, J.S.; et al. Simple additive effects are rare: A quantitative review of plant biomass and soil process responses to combined manipulations of CO2 and temperature. Glob. Chang. Biol. 2012, 18, 2681–2693. [Google Scholar] [CrossRef]
- Song, J.; Wan, S.; Piao, S.; Knapp, A.K.; Classen, A.T.; Vicca, S.; Ciais, P.; Hovenden, M.J.; Leuzinger, S.; Beier, C.; et al. A meta-analysis of 1119 manipulative experiments on terrestrial carbon-cycling responses to global change. Nat. Ecol. Evol. 2019, 3, 1309–1320. [Google Scholar] [CrossRef]
- Sghaier, H.; Hezbri, K.; Ghodhbane-Gtari, F.; Pujic, P.; Sen, A.; Daffonchio, D.; Boudabous, A.; Tisa, L.S.; Klenk, H.P.; Armengaud, J.; et al. Stone-dwelling actinobacteria Blastococcus saxobsidens, Modestobacter marinus and Geodermatophilus obscurus proteogenomes. ISME J. 2016, 10, 21–29. [Google Scholar] [CrossRef]
- Normand, P.; Daffonchio, D.; Gtari, M. The family geodermatophilaceae. In The Prokaryotes: Actinobacteria; Springer: Berlin/Heidelberg, Germay, 2014; pp. 361–379. [Google Scholar]
- Chukwuneme, C.F.; Babalola, O.O.; Kutu, F.R.; Ojuederie, O.B. Characterization of actinomycetes isolates for plant growth promoting traits and their effects on drought tolerance in maize. J. Plant Interact. 2020, 15, 93–105. [Google Scholar]
- Denner, E.B.M.; Kolari, M.; Hoornstra, D.; Tsitko, I.; Kampfer, P.; Busse, H.J.; Salkinoja-Salonen, M. Rubellimicrobium thermophilum gen. nov., sp. nov., a red-pigmented, moderately thermophilic bacterium isolated from coloured slime deposits in paper machines. Int. J. Syst. Evol. Microbiol. 2006, 56, 1355–1362. [Google Scholar] [CrossRef]
- Dastager, S.G.; Lee, J.C.; Ju, Y.J.; Park, D.J.; Kim, C.J. Rubellimicrobium mesophilum sp. nov., a mesophilic, pigmented bacterium isolated from soil. Int. J. Syst. Evol. Microbiol. 2008, 58, 1797–1800. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, H.; Sun, L.; Qi, G.; Chen, S.; Zhao, X. Microbial community composition is related to soil biological and chemical properties and bacterial wilt outbreak. Sci. Rep. 2017, 7, 343. [Google Scholar] [CrossRef]
- Msaddak, A.; Rejili, M.; Duran, D.; Rey, L.; Imperial, J.; Palacios, J.M.; Ruiz-Argueso, T.; Mars, M. Members of Microvirga and Bradyrhizobium genera are native endosymbiotic bacteria nodulating Lupinus luteus in Northern Tunisian soils. FEMS Microbiol. Ecol. 2017, 93, fix068. [Google Scholar] [CrossRef]
- Zhang, Y.Q.; Chen, J.; Liu, H.Y.; Zhang, Y.Q.; Li, W.J.; Yu, L.Y. Geodermatophilus ruber sp. nov., isolated from rhizosphere soil of a medicinal plant. Int. J. Syst. Evol. Microbiol. 2011, 61, 190–193. [Google Scholar] [CrossRef]
- Camarena-Pozos, D.A.; Flores-Núñez, V.M.; López, M.G.; López-Bucio, J.; Partida-Martínez, L.P. Smells from the desert: M icrobial volatiles that affect plant growth and development of native and non-native plant species. Plant Cell Environ. 2019, 42, 1368–1380. [Google Scholar]
- Flores-Núñez, V.M.; Fonseca-García, C.; Desgarennes, D.; Eloe-Fadrosh, E.; Woyke, T.; Partida-Martínez, L.P. Functional signatures of the epiphytic prokaryotic microbiome of agaves and cacti. Front. Microbiol. 2020, 10, 3044. [Google Scholar]
- Zhao, Y.; Fu, W.; Hu, C.; Chen, G.; Xiao, Z.; Chen, Y.; Wang, Z.; Cheng, H. Variation of rhizosphere microbial community in continuous mono-maize seed production. Sci. Rep. 2021, 11, 1544. [Google Scholar] [CrossRef]
- Yoon, J.-H.; Park, Y. The genus Nocardioides. Prokaryotes 2006, 3, 1099–1113. [Google Scholar]
- Kim, H.; Nishiyama, M.; Kunito, T.; Senoo, K.; Kawahara, K.; Murakami, K.; Oyaizu, H. High population of Sphingomonas species on plant surface. J. Appl. Microbiol. 1998, 85, 731–736. [Google Scholar] [CrossRef]
- Monciardini, P.; Cavaletti, L.; Schumann, P.; Rohde, M.; Donadio, S. Conexibacter woesei gen. nov., sp. nov., a novel representative of a deep evolutionary line of descent within the class Actinobacteria. Int. J. Syst. Evol. Microbiol. 2003, 53, 569–576. [Google Scholar] [CrossRef]
- Pold, G.; Billings, A.F.; Blanchard, J.L.; Burkhardt, D.B.; Frey, S.D.; Melillo, J.M.; Schnabel, J.; van Diepen, L.T.; DeAngelis, K.M. Long-Term Warming Alters Carbohydrate Degradation Potential in Temperate Forest Soils. Appl. Environ. Microbiol. 2016, 82, 6518–6530. [Google Scholar] [CrossRef]
- Naylor, D.; DeGraaf, S.; Purdom, E.; Coleman-Derr, D. Drought and host selection influence bacterial community dynamics in the grass root microbiome. ISME J. 2017, 11, 2691–2704. [Google Scholar] [CrossRef]
- Berg, G.; Grube, M.; Schloter, M.; Smalla, K. Unraveling the plant microbiome: Looking back and future perspectives. Front. Microbiol. 2014, 5, 148. [Google Scholar] [CrossRef]
- Berg, G. Plant–microbe interactions promoting plant growth and health: Perspectives for controlled use of microorganisms in agriculture. Appl. Microbiol. Biotechnol. 2009, 84, 11–18. [Google Scholar] [CrossRef]
- Buscardo, E.; Souza, R.C.; Meir, P.; Geml, J.; Schmidt, S.K.; Da Costa, A.C.; Nagy, L. Effects of natural and experimental drought on soil fungi and biogeochemistry in an Amazon rain forest. Commun. Earth Environ. 2021, 2, 55. [Google Scholar]
- Yang, N.; Nesme, J.; Roder, H.L.; Li, X.; Zuo, Z.; Petersen, M.; Burmolle, M.; Sorensen, S.J. Emergent bacterial community properties induce enhanced drought tolerance in Arabidopsis. NPJ Biofilms Microbiomes 2021, 7, 82. [Google Scholar] [CrossRef]
- Yandigeri, M.S.; Meena, K.K.; Singh, D.; Malviya, N.; Singh, D.P.; Solanki, M.K.; Yadav, A.K.; Arora, D.K. Drought-tolerant endophytic actinobacteria promote growth of wheat (Triticum aestivum) under water stress conditions. Plant Growth Regul. 2012, 68, 411–420. [Google Scholar]
- Lasudee, K.; Tokuyama, S.; Lumyong, S.; Pathom-Aree, W. Actinobacteria associated with arbuscular mycorrhizal Funneliformis mosseae spores, taxonomic characterization and their beneficial traits to plants: Evidence obtained from mung bean (Vigna radiata) and Thai jasmine rice (Oryza sativa). Front. Microbiol. 2018, 9, 1247. [Google Scholar]
- Cha-Um, S.; Yooyongwech, S.; Supaibulwatana, K. Water deficit stress in the reproductive stage of four indica rice (Oryza sativa L.) genotypes. Pak. J. Bot. 2010, 42, 3387–3398. [Google Scholar]
- Pandey, V.; Shukla, A. Acclimation and tolerance strategies of rice under drought stress. Rice Sci. 2015, 22, 147–161. [Google Scholar]
- Kaushal, M.; Wani, S.P. Plant-growth-promoting rhizobacteria: Drought stress alleviators to ameliorate crop production in drylands. Ann. Microbiol. 2016, 66, 35–42. [Google Scholar]
- Forni, C.; Duca, D.; Glick, B.R. Mechanisms of plant response to salt and drought stress and their alteration by rhizobacteria. Plant Soil 2017, 410, 335–356. [Google Scholar] [CrossRef]
- Vurukonda, S.S.; Vardharajula, S.; Shrivastava, M.; Sk, Z.A. Enhancement of drought stress tolerance in crops by plant growth promoting rhizobacteria. Microbiol. Res. 2016, 184, 13–24. [Google Scholar] [CrossRef]
- Dimkpa, C.; Weinand, T.; Asch, F. Plant–rhizobacteria interactions alleviate abiotic stress conditions. Plant Cell Environ. 2009, 32, 1682–1694. [Google Scholar] [CrossRef]
- Yang, J.; Kloepper, J.W.; Ryu, C.M. Rhizosphere bacteria help plants tolerate abiotic stress. Trends Plant Sci. 2009, 14, 1–4. [Google Scholar] [CrossRef]
- Lugtenberg, B.; Kamilova, F. Plant-growth-promoting rhizobacteria. Annu. Rev. Microbiol. 2009, 63, 541–556. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Group | Raw Data (Gb) 1 | Raw Reads (no.) 2 | Ns (%) 3 | Clean Data (Gb) 4 | Effective (%) 5 |
|---|---|---|---|---|---|---|
| S1 | A | 6.02 | 40,123,352 | 0.34 | 6.00 | 99.73 |
| S2 | B | 8.03 | 53,530,450 | 0.34 | 8.00 | 99.69 |
| S3 | C | 8.47 | 56,437,554 | 0.2 | 8.45 | 99.77 |
| R1 | A | 7.03 | 46,870,664 | 0.11 | 7.01 | 99.75 |
| R2 | B | 8.14 | 54,268,014 | 0.21 | 8.12 | 99.71 |
| R3 | C | 7.48 | 49,871,224 | 0.45 | 7.46 | 99.71 |
| Sample ID | Group | Total Len. (bp) 1 | No. Scaftigs 2 | Average Len. (bp) 3 | Max Len. (bp) 4 |
|---|---|---|---|---|---|
| S1 | A | 387,720,527 | 413,461 | 937.74 | 140,598 |
| S2 | B | 298,717,745 | 346,445 | 862.24 | 118,379 |
| S3 | C | 399,538,190 | 487,285 | 819.93 | 117,898 |
| R1 | A | 421,596,529 | 450,710 | 935.41 | 67,510 |
| R2 | B | 280,594,281 | 334,491 | 838.87 | 39,284 |
| R3 | C | 407,398,380 | 429,698 | 948.1 | 58,099 |
| NOVO_MIX 7 | 2,169,967,683 | 2,553,156 | 849.92 | 20,354 |
| Sample ID | Group | ORFs No. 1 | Integrity:None 2 | Integrity:Start 3 | Integrity:End 4 | Integrity:All 5 | Total Len. (Mb) 6 | Average Len. (bp) 7 |
|---|---|---|---|---|---|---|---|---|
| S1 | A | 367,793 | 21,809 | 106,909 | 73,846 | 165,229 | 123.87 | 336.81 |
| S2 | B | 338,997 | 25,804 | 100,893 | 77,249 | 135,051 | 123.67 | 364.8 |
| S3 | C | 534,234 | 55,782 | 162,132 | 144,520 | 171,800 | 207.75 | 388.88 |
| R1 | A | 709,892 | 83,195 | 212,224 | 216,176 | 198,297 | 345.42 | 486.57 |
| R2 | B | 499,735 | 65,343 | 149,218 | 162,133 | 123,041 | 227.86 | 455.97 |
| R3 | C | 688,207 | 81,551 | 201,411 | 208,372 | 196,873 | 336.92 | 489.57 |
| NOVO_MIX 8 | 3,486,643 | 399,752 | 1,101,816 | 1,073,746 | 911,329 | 1508.40 | 432.62 | |
| Taxon | Soil Type | Time after Watering | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Bulk | Rhizosphere | A (0 h) | B (24 h) | C (48 h) | ||||||
| AB | RAB | AB | RAB | AB | RAB | AB | RAB | AB | RAB | |
| Phylum | ||||||||||
| Firmicutes | ||||||||||
| Verrucomicrobia | ||||||||||
| Eu:Ascomycota | ||||||||||
| Eu:Mucoromycota | ||||||||||
| Actinobacteria | ||||||||||
| Bacteroidetes | ||||||||||
| Candidatus Saccharibacteria | ||||||||||
| Nitrospirae | ||||||||||
| Acidobacteria | ||||||||||
| Verrucomicrobia | ||||||||||
| Gemmatimonadetes | ||||||||||
| Genus | ||||||||||
| Pseudonocardia | ||||||||||
| Staphylococcus | ||||||||||
| Blastococcus | ||||||||||
| Microvirga | ||||||||||
| Geodermatophilus | ||||||||||
| Arthrobacter | ||||||||||
| Solirubrobacter | ||||||||||
| Mycobacterium | ||||||||||
| Belnapia | ||||||||||
| Marmoricola | ||||||||||
| Streptomyces | ||||||||||
| Bacillus | ||||||||||
| Ramlibacter | ||||||||||
| Gemmatirosa | ||||||||||
| Microbacterium | ||||||||||
| Rubellimicrobium | ||||||||||
| Sphingomonas | ||||||||||
| Species | ||||||||||
| Staphylococcus aureus | ||||||||||
| Pseudonocardia sp. CNS-004 | ||||||||||
| Pseudonocardia sp. MH-G8 | ||||||||||
| Bacillus thuringiensis | ||||||||||
| Sphingomonas sp. URHD0057 | ||||||||||
| Blastococcus sp. DSM 46786 | ||||||||||
| Solirubrobacter sp. URHD0082 | ||||||||||
| Geodermatophilus sabuli | ||||||||||
| Microvirga sp. BSC39 | ||||||||||
| Microvirga massiliensis | ||||||||||
| Arthrobacter crystallopoietes | ||||||||||
| Rubellimicrobium mesophilum | ||||||||||
| Conexibacter woesei | ||||||||||
| Sphingomonas jaspsi | ||||||||||
| Gemmatirosa kalamazoonesis | ||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Refai, M.Y.; Abulfaraj, A.A.; Hakeem, I.J.; Shaer, N.A.; Alqahtani, M.D.; Alomran, M.M.; Alotaibi, N.M.; Sonbol, H.S.; Alhashimi, A.M.; Al-Abbas, N.S.; et al. Rhizobiome Signature and Its Alteration Due to Watering in the Wild Plant Moringa oleifera. Sustainability 2023, 15, 2745. https://doi.org/10.3390/su15032745
Refai MY, Abulfaraj AA, Hakeem IJ, Shaer NA, Alqahtani MD, Alomran MM, Alotaibi NM, Sonbol HS, Alhashimi AM, Al-Abbas NS, et al. Rhizobiome Signature and Its Alteration Due to Watering in the Wild Plant Moringa oleifera. Sustainability. 2023; 15(3):2745. https://doi.org/10.3390/su15032745
Chicago/Turabian StyleRefai, Mohammed Y., Aala A. Abulfaraj, Israa J. Hakeem, Nehad A. Shaer, Mashael D. Alqahtani, Maryam M. Alomran, Nahaa M. Alotaibi, Hana S. Sonbol, Abdulrahman M. Alhashimi, Nouf S. Al-Abbas, and et al. 2023. "Rhizobiome Signature and Its Alteration Due to Watering in the Wild Plant Moringa oleifera" Sustainability 15, no. 3: 2745. https://doi.org/10.3390/su15032745
APA StyleRefai, M. Y., Abulfaraj, A. A., Hakeem, I. J., Shaer, N. A., Alqahtani, M. D., Alomran, M. M., Alotaibi, N. M., Sonbol, H. S., Alhashimi, A. M., Al-Abbas, N. S., Ashy, R. A., Alshareef, S. A., & Jalal, R. S. (2023). Rhizobiome Signature and Its Alteration Due to Watering in the Wild Plant Moringa oleifera. Sustainability, 15(3), 2745. https://doi.org/10.3390/su15032745