


RNA-Pol II Transcription Elongation Factor FpRtfA Is Required for Virulence and Secondary Metabolism in Fusarium pseudograminearum

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
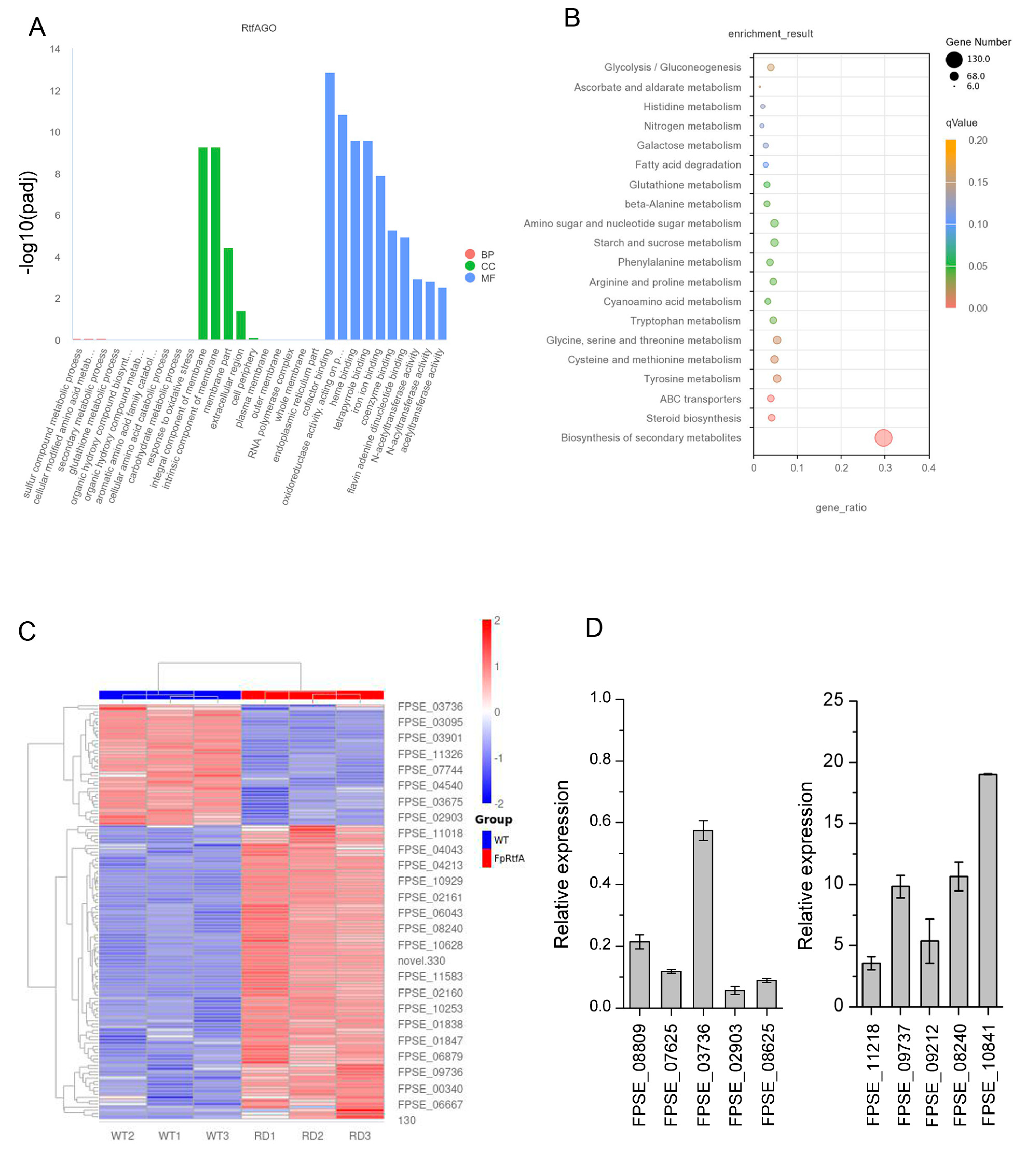
Abstract
1. Introduction
2. Materials and Methods
2.1. Strains and Growth Conditions
2.2. Transformants of Deletion and Complementarity
2.3. Assays for Virulence and Infection
2.4. RNA-Seq and Bioinformatics Analysis
2.5. Determination of DON Production
2.6. Data Statistics
3. Results
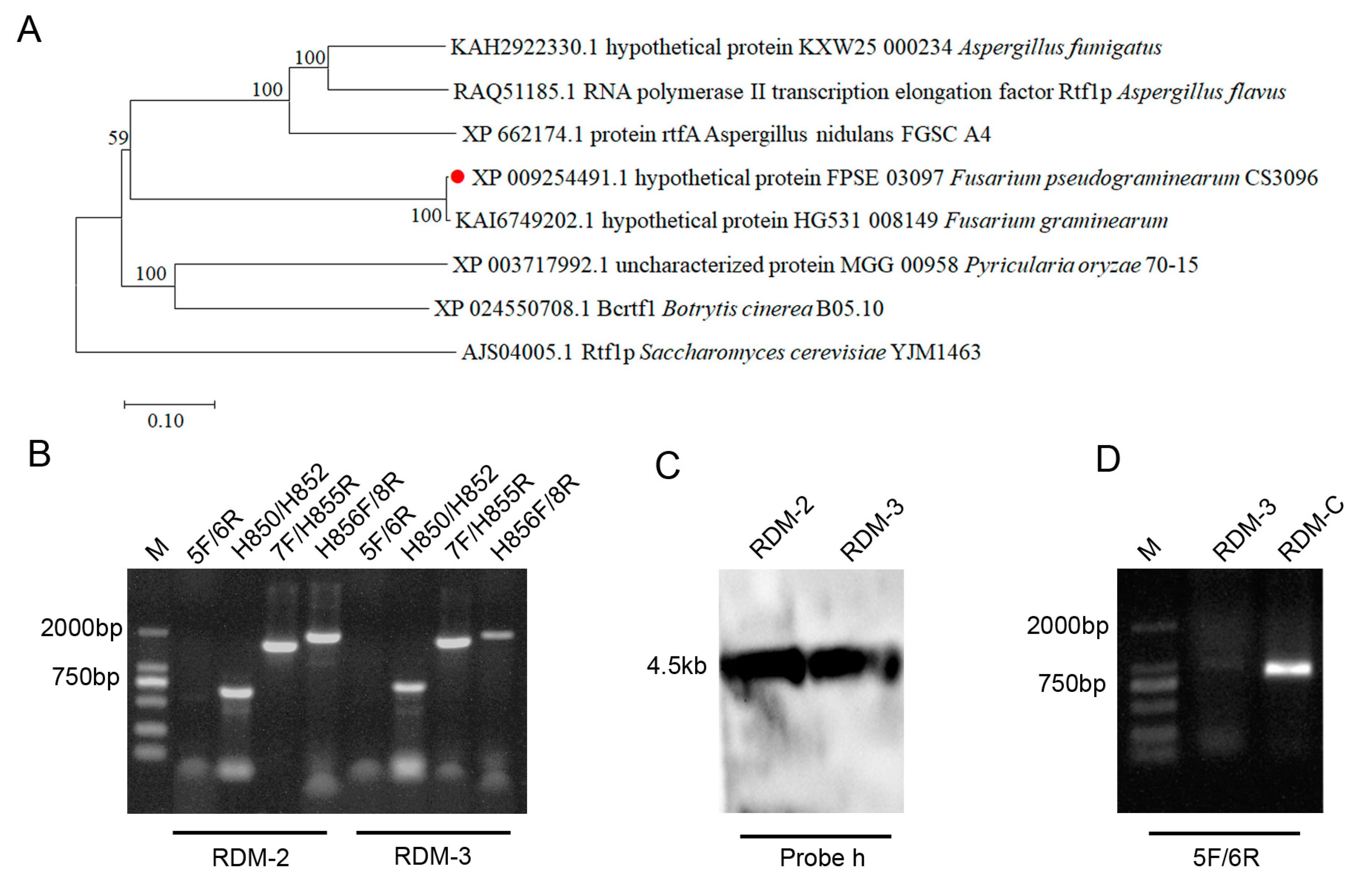
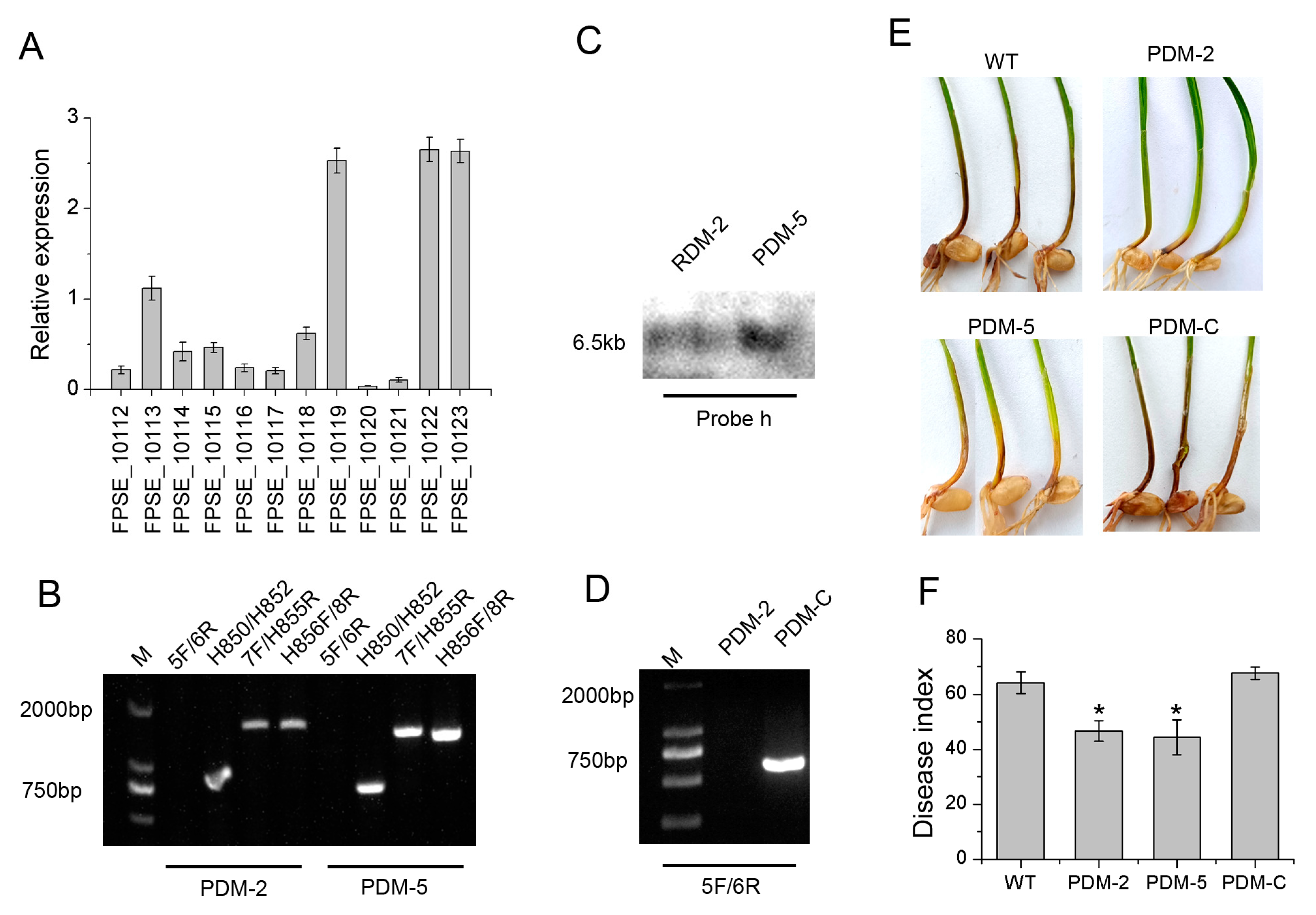
3.1. Deletion and Complementarity of FpRtfA
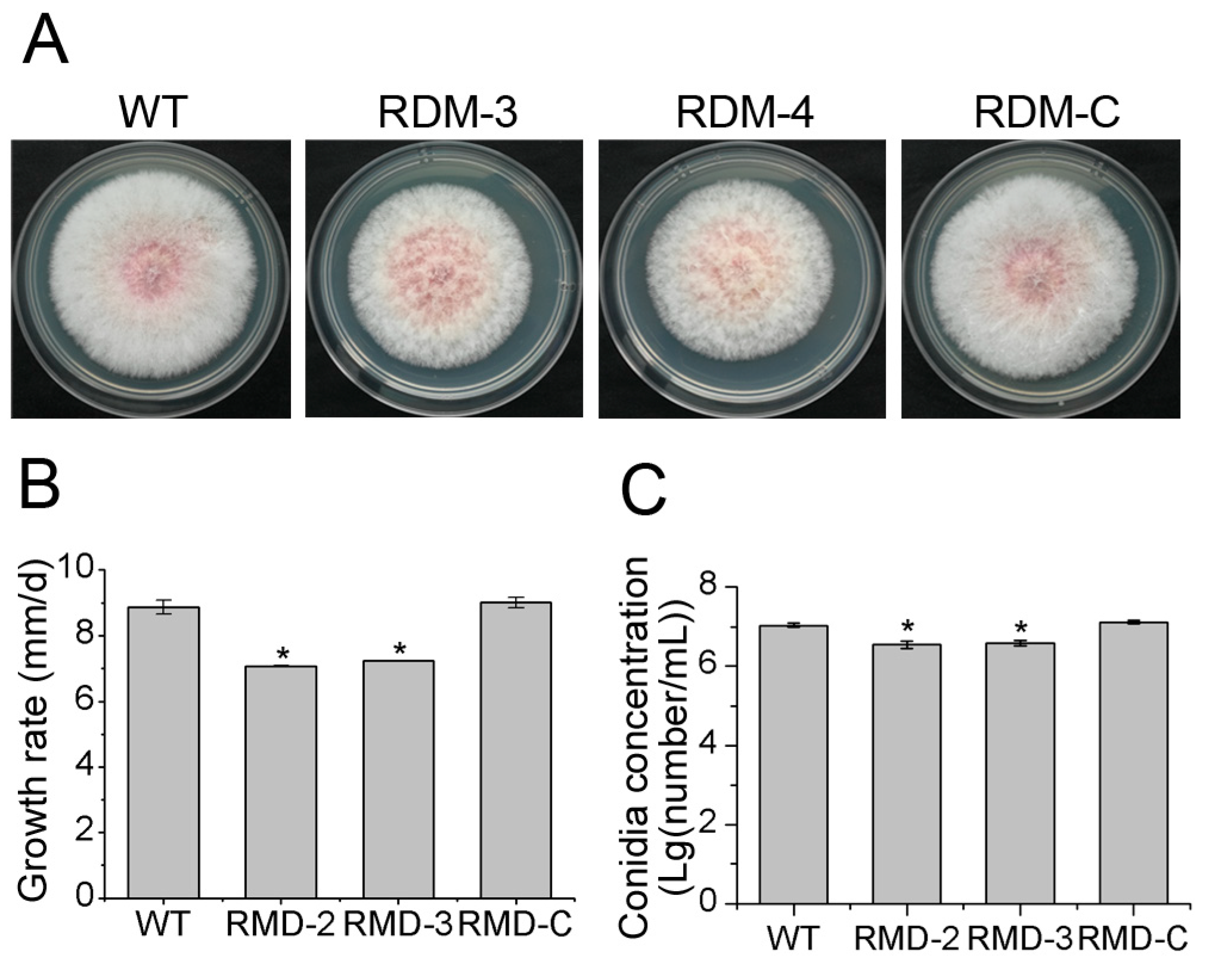
3.2. FpRtfA Is Necessary for Normal Growth and Conidiation
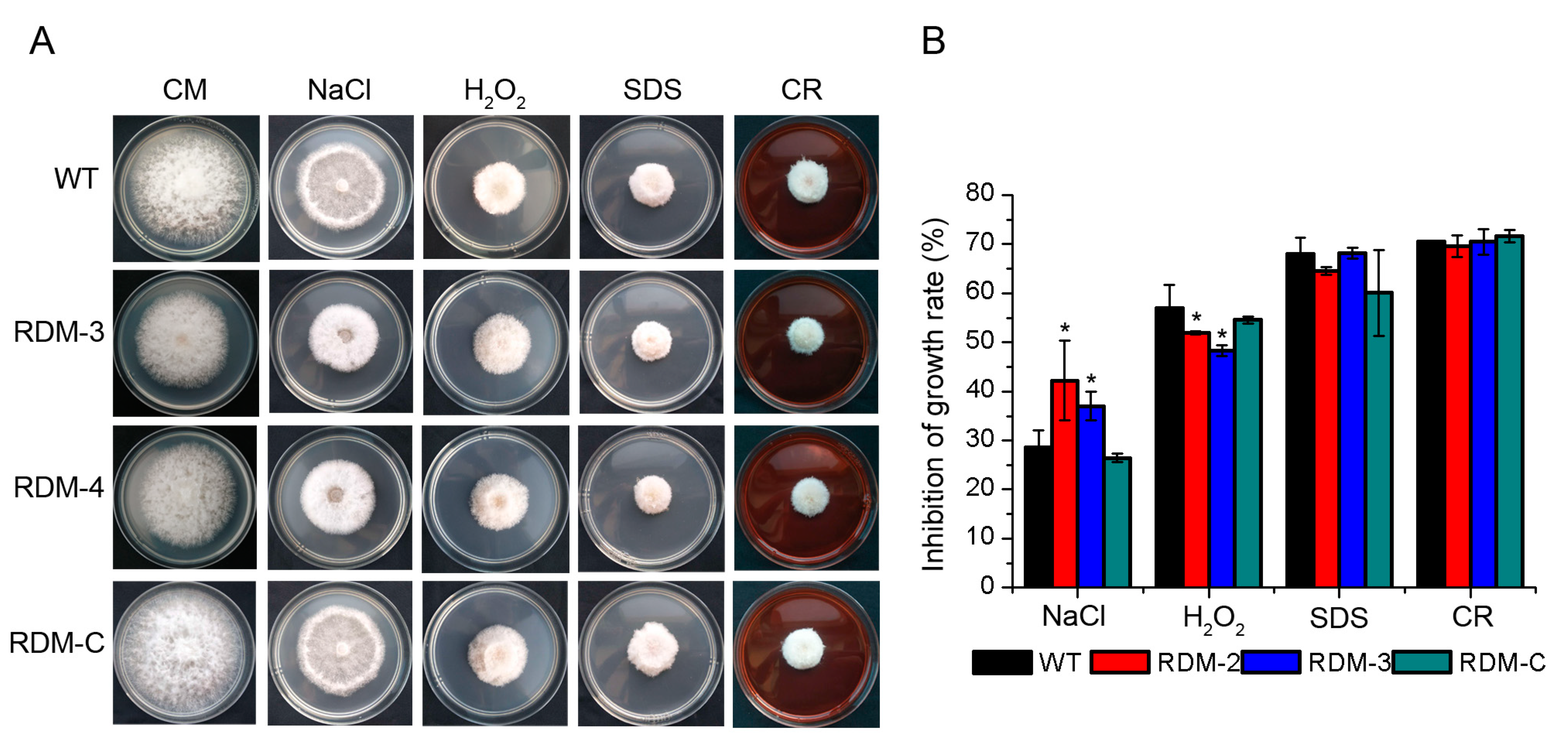
3.3. FpRtfA Affects Responses of F. pseudograminearum to Different Stress Conditions
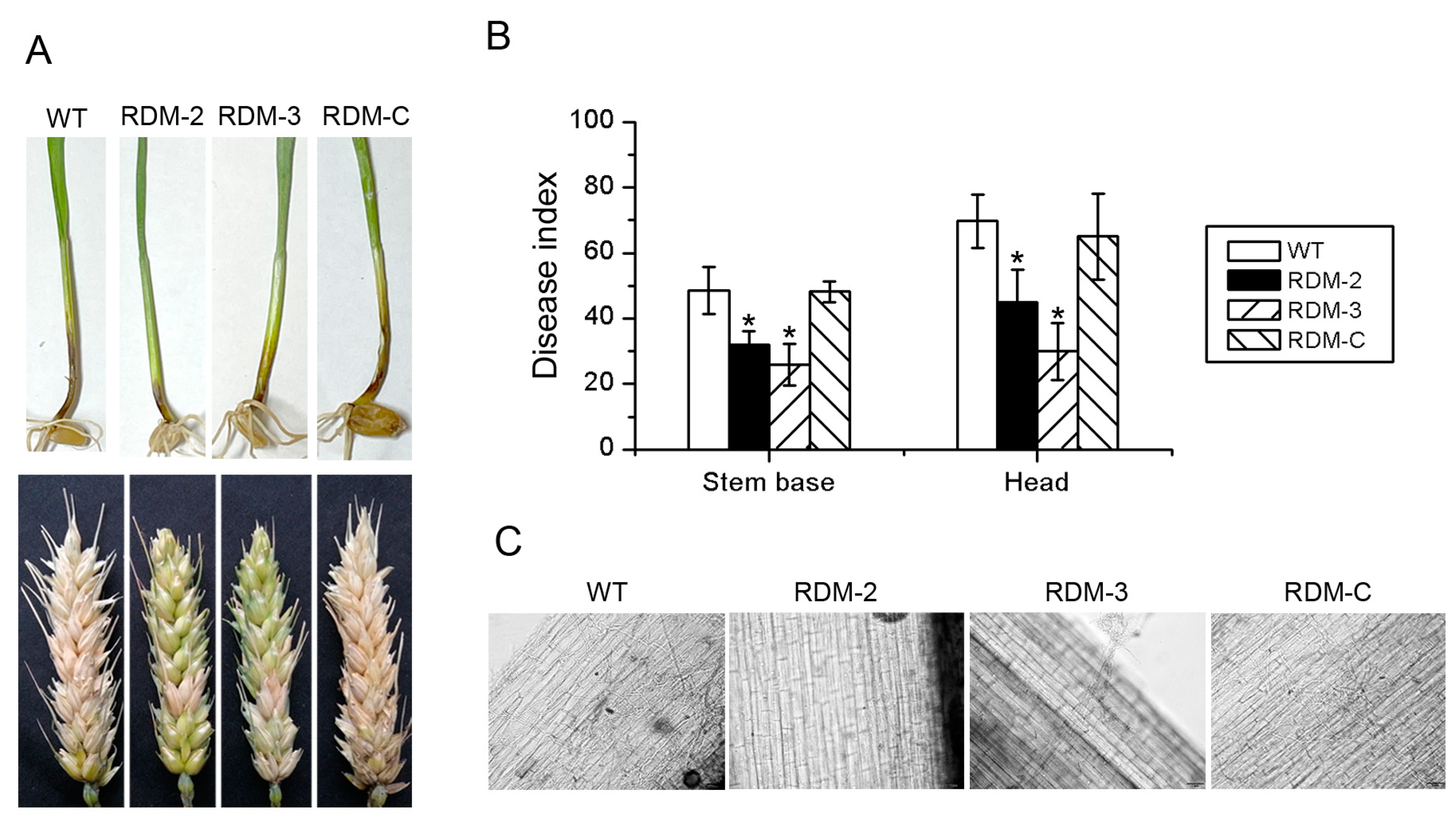
3.4. FpRtfA Is Required for Full Virulence in F. pseudograminearum
3.5. FpRtfA Regulates Genes Expression of Secondary Metabolites but Not DON
3.6. A Cluster of PKS Regulated by FpRtfA Is Associated with Virulence in F. pseudograminearum
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smiley, R.W.; Gourlie, J.A.; Easley, S.A.; Patterson, L.-M.; Whittaker, R.G. Crop damage estimates for crown rot of wheat and barley in the Pacific Northwest. Plant Dis. 2005, 89, 595–604. [Google Scholar] [CrossRef]
- Murray, G.M.; Brennan, J.P. Estimating disease losses to the Australian wheat industry. Australas. Plant Pathol. 2009, 38, 558–570. [Google Scholar] [CrossRef]
- Li, H.; Yuan, H.; Fu, B.; Xing, X.; Sun, B.; Tang, W. First report of Fusarium pseudograminearum causing crown rot of wheat in Henan, China. Plant Dis. 2012, 96, 1065. [Google Scholar] [CrossRef]
- Ji, L.; Kong, L.; Li, Q.; Wang, L.; Chen, D.; Ma, P. First report of Fusarium pseudograminearum causing Fusarium head blight of wheat in Hebei Province, China. Plant Dis. 2016, 100, 220. [Google Scholar] [CrossRef]
- Kazan, K.; Gardiner, D.M. Fusarium crown rot caused by Fusarium pseudograminearum in cereal crops: Recent progress and future prospects. Mol. Plant Pathol. 2018, 19, 1547–1562. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Liu, C.J.; Mitter, V.; Scott, J.B.; Akinsanmi, O.A.; Ali, S.; Dill-Macky, R.; Nicol, J.; Backhouse, D.; Simpfendorfer, S. Pathogen population structure and epidemiology are keys to wheat crown rot and Fusarium head blight management. Australas. Plant Pathol. 2006, 35, 643–655. [Google Scholar] [CrossRef]
- Zhou, H.; He, X.; Wang, S.; Ma, Q.; Sun, B.; Ding, S.; Chen, L.; Zhang, M.; Li, H. Diversity of the Fusarium pathogens associated with crown rot in the Huanghuai wheat-growing region of China. Environ. Microbiol. 2019, 21, 2740–2754. [Google Scholar] [CrossRef] [PubMed]
- Doehlemann, G.; Okmen, B.; Zhu, W.; Sharon, A. Plant Pathogenic Fungi. Microbiol. Spectr. 2017, 5, 1–23. [Google Scholar] [CrossRef]
- Pestka, J.J.; Smolinski, A.T. Deoxynivalenol: Toxicology and potential effects on humans. J. Toxicol. Environ. Health Part B 2005, 8, 39–69. [Google Scholar] [CrossRef]
- Desjardins, A.E. Gibberella from A (venaceae) to Z (eae). Annu. Rev. Phytopathol. 2003, 41, 177–198. [Google Scholar] [CrossRef]
- Chen, Y.; Kistler, H.C.; Ma, Z. Fusarium graminearum Trichothecene Mycotoxins: Biosynthesis, Regulation, and Management. Annu. Rev. Phytopathol. 2019, 57, 15–39. [Google Scholar] [CrossRef]
- Powell, J.J.; Carere, J.; Fitzgerald, T.L.; Stiller, J.; Covarelli, L.; Xu, Q.; Gubler, F.; Colgrave, M.L.; Gardiner, D.M.; Manners, J.M.; et al. The Fusarium crown rot pathogen Fusarium pseudograminearum triggers a suite of transcriptional and metabolic changes in bread wheat (Triticum aestivum L.). Ann. Bot. 2017, 119, 853–867. [Google Scholar] [PubMed]
- Tunali, B.; Obanor, F.; Erginbas, G.; Westecott, R.A.; Nicol, J.; Chakraborty, S. Fitness of three Fusarium pathogens of wheat. FEMS Microbiol. Ecol. 2012, 81, 596–609. [Google Scholar] [CrossRef]
- Monds, R.D.; Cromey, M.G.; Lauren, D.R.; Di, M.M.; Marshall, J. Fusarium graminearum, F. cortaderiae and F. pseudograminearum in New Zealand: Molecular phylogenetic analysis, mycotoxin chemotypes and co-existence of species. Mycol. Res. 2005, 109, 410. [Google Scholar] [CrossRef] [PubMed]
- Hansen, F.T.; Gardiner, D.M.; Lysøe, E.; Fuertes, P.R.; Tudzynski, B.; Wiemann, P.; Sondergaard, T.E.; Giese, H.; Brodersen, D.E.; Sørensen, J.L. An update to polyketide synthase and non-ribosomal synthetase genes and nomenclature in Fusarium. Fungal Genet. Biol. 2015, 75, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Brakhage, A.A. Regulation of fungal secondary metabolism. Nat. Rev. Microbiol. 2013, 11, 21–32. [Google Scholar] [CrossRef]
- Stolinski, L.A.; Eisenmann, D.M.; Arndt, K.M. Identification of RTF1, a novel gene important for TATA site selection by TATA box-binding protein in Saccharomyces cerevisiae. Mol. Cell. Biol. 1997, 17, 4490–4500. [Google Scholar] [CrossRef]
- Ng, H.H.; Dole, S.; Struhl, K. The Rtf1 Component of the Paf1 Transcriptional Elongation Complex Is Required for Ubiquitination of Histone H2B. J. Biol. Chem. 2003, 278, 33625–33628. [Google Scholar] [CrossRef]
- Ramamoorthy, V.; Shantappa, S.; Dhingra, S.; Calvo, A.M. veA-dependent RNA-pol II transcription elongation factor-like protein, RtfA, is associated with secondary metabolism and morphological development in Aspergillus nidulans. Mol. Microbiol. 2012, 85, 795–814. [Google Scholar] [CrossRef]
- Myers, R.R.; Smith, T.D.; Elsawa, S.F.; Puel, O.; Tadrist, S.; Calvo, A.M. rtfA controls development, secondary metabolism, and virulence in Aspergillus fumigatus. PLoS ONE 2017, 12, e0176702. [Google Scholar] [CrossRef]
- Lohmar, J.M.; Harris-Coward, P.Y.; Cary, J.W.; Dhingra, S.; Calvo, A.M. rtfA, a putative RNA-Pol II transcription elongation factor gene, is necessary for normal morphological and chemical development in Aspergillus flavus. Appl. Microbiol. Biotechnol. 2016, 100, 5029–5041. [Google Scholar] [CrossRef]
- Lohmar, J.M.; Puel, O.; Cary, J.W.; Calvo, A.M. The rtfA gene regulates plant and animal pathogenesis and secondary metabolism in Aspergillus flavus. Appl. Environ. Microbiol. 2019, 85, e02446-18. [Google Scholar] [CrossRef]
- Chen, L.; Ma, Y.; Peng, M.; Chen, W.; Xia, H.; Zhao, J.; Zhang, Y.; Fan, Z.; Xing, X.; Li, H. Analysis of Apoptosis-Related Genes Reveals that Apoptosis Functions in Conidiation and Pathogenesis of Fusarium pseudograminearum. mSphere 2021, 6, e01140-20. [Google Scholar] [CrossRef]
- Mohamed Zubi, W.S.; Mohd, M.H.; Mohamed Nor, N.M.I.; Zakaria, L. Fusarium Species in Mangrove Soil in Northern Peninsular Malaysia and the Soil Physico-Chemical Properties. Microorganisms 2021, 9, 497. [Google Scholar] [CrossRef]
- Yu, J.-H.; Hamari, Z.; Han, K.-H.; Seo, J.-A.; Reyes-Domínguez, Y.; Scazzocchio, C. Double-joint PCR: A PCR-based molecular tool for gene manipulations in filamentous fungi. Fungal Genet. Biol. 2004, 41, 973–981. [Google Scholar] [CrossRef]
- Liu, Z.; Friesen, T.L. Polyethylene Glycol (PEG)-Mediated Transformation in Filamentous Fungal Pathogens. In Plant Fungal Pathogens: Methods and Protocols; Bolton, M.D., Thomma, B.P.H.J., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 365–375. [Google Scholar]
- Tang, G.; Zhang, C.; Ju, Z.; Zheng, S.; Wen, Z.; Xu, S.; Chen, Y.; Ma, Z. The mitochondrial membrane protein FgLetm1 regulates mitochondrial integrity, production of endogenous reactive oxygen species and mycotoxin biosynthesis in Fusarium graminearum. Mol. Plant Pathol. 2018, 19, 1595–1611. [Google Scholar] [CrossRef] [PubMed]
- Bruno Kenneth, S.; Tenjo, F.; Li, L.; Hamer John, E.; Xu, J.-R. Cellular Localization and Role of Kinase Activity of PMK1 in Magnaporthe grisea. Eukaryot. Cell 2004, 3, 1525–1532. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Heyer, C.; Choi, Y.-E.; Mehrabi, R.; Xu, J.-R. The CID1 cyclin C-like gene is important for plant infection in Fusarium graminearum. Fungal Genet. Biol. 2010, 47, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Li, H.B.; Zhou, M.X.; Liu, C.J. A major QTL conferring crown rot resistance in barley and its association with plant height. Theor. Appl. Genet. 2009, 118, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-Y.; Xie, Y.-S.; Cui, Y.-Y.; Xu, J.; He, W.; Chen, H.-G.; Guo, J.-H. Conjunctively screening of biocontrol agents (BCAs) against fusarium root rot and fusarium head blight caused by Fusarium graminearum. Microbiol. Res. 2015, 177, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, D.M.; McDonald, M.C.; Covarelli, L.; Solomon, P.S.; Rusu, A.G.; Marshall, M.; Kazan, K.; Chakraborty, S.; McDonald, B.A.; Manners, J.M. Comparative pathogenomics reveals horizontally acquired novel virulence genes in fungi infecting cereal hosts. PLoS Pathog. 2012, 8, e1002952. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xie, S.; Zhang, Y.; Kang, R.; Zhang, M.; Wang, M.; Li, H.; Chen, L.; Yuan, H.; Ding, S.; et al. The FpPPR1 Gene Encodes a Pentatricopeptide Repeat Protein That Is Essential for Asexual Development, Sporulation, and Pathogenesis in Fusarium pseudograminearum. Front. Genet. 2020, 11, 535622. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L.; Liang, S.; Zhang, P.; Kang, R.; Zhang, M.; Wang, M.; Chen, L.; Yuan, H.; Ding, S.; et al. FpDep1, a component of Rpd3L histone deacetylase complex, is important for vegetative development, ROS accumulation, and pathogenesis in Fusarium pseudograminearum. Fungal Genet. Biol. 2020, 135, 103299. [Google Scholar] [CrossRef]
- Gardiner, D.M.; Kazan, K.; Manners, J.M. Nutrient profiling reveals potent inducers of trichothecene biosynthesis in Fusarium graminearum. Fungal Genet. Biol. 2009, 46, 604–613. [Google Scholar] [CrossRef]
- Soleimany, F.; Jinap, S.; Faridah, A.; Khatib, A. A UPLC–MS/MS for simultaneous determination of aflatoxins, ochratoxin A, zearalenone, DON, fumonisins, T-2 toxin and HT-2 toxin, in cereals. Food Control 2012, 25, 647–653. [Google Scholar] [CrossRef]
- Duan, S.; Jin, J.; Gao, Y.; Jin, C.; Mu, J.; Zhen, W.; Sun, Q.; Xie, C.; Ma, J. Integrated transcriptome and metabolite profiling highlights the role of benzoxazinoids in wheat resistance against Fusarium crown rot. Crop J. 2022, 10, 407–417. [Google Scholar] [CrossRef]
- Xia, H.; Chen, L.; Fan, Z.; Peng, M.; Zhao, J.; Chen, W.; Li, H.; Shi, Y.; Ding, S.; Li, H. Heat Stress Tolerance Gene FpHsp104 Affects Conidiation and Pathogenicity of Fusarium pseudograminearum. Front. Microbiol. 2021, 12, 695535. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, L.; Liang, Y.; Yu, J. FgPEX4 is involved in development, pathogenicity, and cell wall integrity in Fusarium graminearum. Curr. Genet. 2019, 65, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.; Sun, Y.; Liu, Y.; Li, R.; Chen, Y.; Zhou, T. Cinnamon Oil Inhibits Penicillium expansum Growth by Disturbing the Carbohydrate Metabolic Process. J. Fungi 2021, 7, 123. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Wang, Y.; Han, S.; Li, Q.; Kong, L. RNA-Pol II Transcription Elongation Factor FpRtfA Is Required for Virulence and Secondary Metabolism in Fusarium pseudograminearum. Sustainability 2023, 15, 11401. https://doi.org/10.3390/su151411401
Wu Y, Wang Y, Han S, Li Q, Kong L. RNA-Pol II Transcription Elongation Factor FpRtfA Is Required for Virulence and Secondary Metabolism in Fusarium pseudograminearum. Sustainability. 2023; 15(14):11401. https://doi.org/10.3390/su151411401
Chicago/Turabian StyleWu, Yuxing, Yajiao Wang, Sen Han, Qiusheng Li, and Lingxiao Kong. 2023. "RNA-Pol II Transcription Elongation Factor FpRtfA Is Required for Virulence and Secondary Metabolism in Fusarium pseudograminearum" Sustainability 15, no. 14: 11401. https://doi.org/10.3390/su151411401
APA StyleWu, Y., Wang, Y., Han, S., Li, Q., & Kong, L. (2023). RNA-Pol II Transcription Elongation Factor FpRtfA Is Required for Virulence and Secondary Metabolism in Fusarium pseudograminearum. Sustainability, 15(14), 11401. https://doi.org/10.3390/su151411401