
Electrochemistry of Sulfides: Process and Environmental Aspects




{kind=link}
Abstract
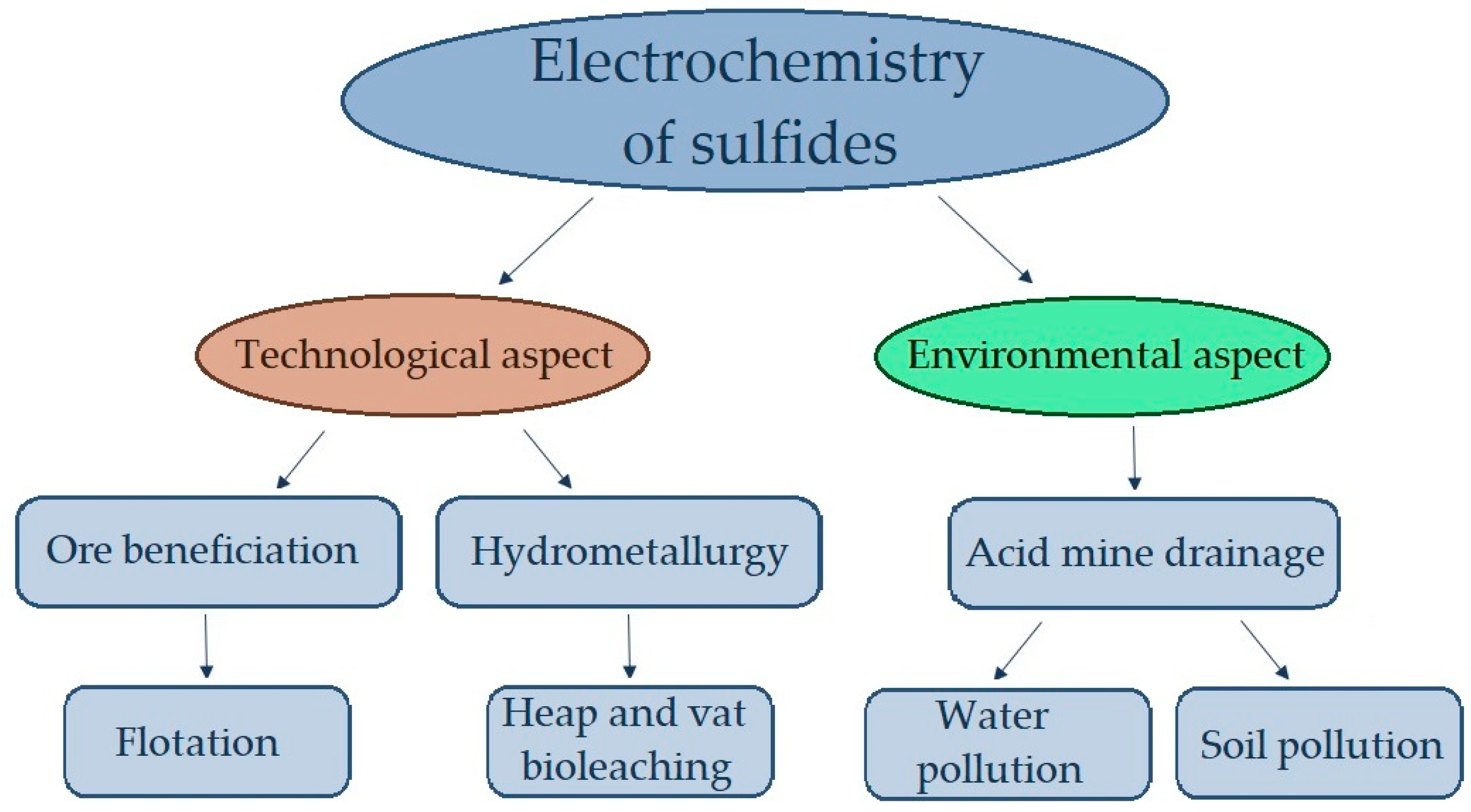
:1. Introduction
2. Electrochemistry of Sulfides in Aqueous Solutions
2.1. Iron Sulfides
2.2. Copper Sulfides
2.3. Nickel Sulfides
2.4. Lead Sulfide (Galena)
2.5. Zinc Sulfide (Sphalerite)
3. Reduction of Oxygen on the Surface of Sulfides
H2O2 + 2e− = 2OH−.
4. Electrochemical Properties of Sulfides in Technological Processes
4.1. Electrochemical Properties of Sulfides in Flotation Processes
4.2. Galvanic Interactions of Sulfides in Hydrometallurgical Processes
5. Oxidation of Sulfide Minerals during Mining Waste Storage
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Plaksin, I.N.; Shafeev, R.S.; Chanturiya, V.A. The relationship between the energy structure of mineral crystals and their flotation properties. In Proceedings of the VIII International Mineral Processing Congress; Mekhanobr: Leningrad, Russia, 1969; Volume 2, pp. 235–245. (In Russian). [Google Scholar]
- Plaksin, I.N.; Shafeev, R.S. Effects of some semiconductor properties of the surface on the interaction of xanthate with galena. Dokl. Akad. Nauk. USSR 1960, 132, 399–401. (In Russian) [Google Scholar]
- Plaksin, I.N.; Shafeev, R.S. On the effects of the electrochemical potential on the distribution of xanthate across the surface of sulfides. Dokl. Akad. Nauk. USSR 1958, 118, 546–548. (In Russian) [Google Scholar]
- Abramov, A.A.; Avdohin, V.M. Oxidation of Sulfide Minerals in Benefication Processes; Gordon and Breach Science Publishers: Philadelphia, PA, USA, 1997; 321p. [Google Scholar]
- Chanturiya, V.A.; Vigdergauz, V.E. Electrochemistry of Sulfides. Theory and Practice of Flotation; Publishing House Ore and Metals: Moscow, Russia, 2009; 280p. [Google Scholar]
- Buckley, A.N.; Hamilton, I.C.; Woods, R. Flotation of sulphide minerals. In Developments in Mineral Processing; Forssberg, K.S.E., Ed.; Elsevier: Amsterdam, The Netherlands, 1985; pp. 41–115. [Google Scholar]
- Woods, R. Flotation of sulfide minerals. In Reagents in Mineral Technology; Somasundaran, P., Moudgil, B.M., Eds.; Marcel Dekker: New York, NY, USA, 1987; pp. 39–78. [Google Scholar]
- Bozkurt, V.; Xu, Z.; Finch, J.A. Pentlandite/pyrrhotite interaction and xanthate adsorbtion. Int. J. Miner. Process. 1998, 52, 203–214. [Google Scholar] [CrossRef]
- Chanturiya, V.A. Scientific foundations of the electrochemical technology of mineral processing processes. Vestn. Akad. Nauk. USSR 1985, 9, 39–47. (In Russian) [Google Scholar]
- Chanturiya, V.A.; Nazarova, G.N. Electrochemical Technology for Beneficiation and Hydrometallurgical Processes; Nauka: Moscow, Russia, 1977; 160p. (In Russian) [Google Scholar]
- Watling, H.R. Review of biohydrometallurgical metals extraction from polymetallic mineral resources. Minerals 2015, 5, 1–60. [Google Scholar] [CrossRef]
- Muñoz, A.; Bevilaqua, D.; Garcia, O., Jr. Leaching of Ni and Cu from mine wastes (tailings and slags) using acid solutions and A. Ferrooxidans. Adv. Mater. Res. 2009, 71–73, 425–428. [Google Scholar] [CrossRef]
- Riekkola-Vanhanen, M.; Palmu, L. Talvivaara Nickel Mine–from a project to a mine and beyond. In Proceedings of the Symposium Ni-Co 2013, San Antonio, TA, USA, 3–7 March 2013; pp. 269–278. [Google Scholar]
- Tanne, C.K.; Schippers, A. Electrochemical applications in metal bioleaching. Adv. Biochem. Eng. Biotechnol. 2017, 167, 327–359. [Google Scholar]
- Tanne, C.K.; Schippers, A. Electrochemical process engineering in biohydrometallurgical metal recovery from mineral sulfides. Solid State Phenom. 2017, 262, 118–121. [Google Scholar] [CrossRef]
- Holmes, P.R.; Crundwell, F.K. Kinetic aspects of galvanic interactions between minerals during dissolution. Hydrometallurgy 1995, 39, 353–375. [Google Scholar] [CrossRef]
- Kalinnikov, V.T.; Makarov, D.V.; Makarov, V.N. Oxidation sequence of sulfide minerals in operating and out-of-service mine waste storage. Theor. Found. Chem. Eng. 2001, 35, 63–68. [Google Scholar] [CrossRef]
- Parbhakar-Fox, A.; Lottermoser, B.; Bradshaw, D. Evaluating waste rock mineralogy and microtexture during kinetic testing for improved acid rock drainage prediction. Miner. Eng. 2013, 52, 111–124. [Google Scholar] [CrossRef]
- Chopard, A.; Plante, B.; Benzaazoua, M.; Bouzahzah, H.; Marion, P. Geochemical investigation of the galvanic effects during oxidation of pyrite and base-metals sulfides. Chemosphere 2017, 166, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Kwong, Y.T.J.; Swerhone, G.W.; Lawrence, J.R. Galvanic sulphide oxidation as a metal-leaching mechanism and its environmental implications. Geochem. Explor. Environ. Anal. 2003, 3, 337–343. [Google Scholar] [CrossRef]
- Radyushkina, K.A.; Wigdergauz, V.E.; Tarasevich, M.R.; Chanturiya, V.A. Electrochemistry of sulfide minerals. Electrochemical processes on the surface of pyrite and pyrrhotite in aqueous solutions of electrolytes. Elektrokhimjya 1986, 22, 1394–1398. (In Russian) [Google Scholar]
- Chanturiya, V.A.; Makarov, V.N.; Makarov, D.V.; Vasil’eva, T.N.; Belyaevskii, A.T. Electrochemical oxidation of pyrrhotine in alkaline media. Russ. J. Electrochem. 1999, 35, 759–763. [Google Scholar]
- Andriamanana, A.; Lamache, M. Etude electrochimique de la pyrite en milieu acide. Electrochim. Acta 1983, 28, 177–183. [Google Scholar] [CrossRef]
- Hamilton, I.C.; Woods, R. An investigation of surface oxidation of pyrite and pyrrhotite by linear potential sweep voltammetry. J. Electroanal. Chem. 1979, 101, 327–343. [Google Scholar] [CrossRef]
- Mischra, K.K.; Osseo-Asare, K. Aspects of the interfacial electrochemistry of semiconductor pyrite (FeS2). J. Electrochem. Soc. 1988, 135, 2502–2509. [Google Scholar] [CrossRef]
- Moses, C.O.; Nordström, D.K.; Herman, J.S.; Mills, A.L. Aqueous pyrite oxidation by dissolved oxygen and by ferric ion. Geochim. Cosmochim. Acta 1987, 51, 1561–1571. [Google Scholar] [CrossRef]
- Yin, Q.; Kelsall, G.H.; Vaughan, D.J.; Welham, N.J. Rotating ring(Pt)-disc(FeS2) electrode behavior in hydrocloric solutions. J. Colloid Interface Sci. 1999, 210, 375–383. [Google Scholar] [CrossRef]
- Kelsall, G.H.; Yin, Q.; Vaughan, D.J.; England, K.E.R.; Brandon, N.P. Electrochemical oxidation of pyrite (FeS2) in aqueous electrolytes. J. Electroanal. Chem. 1999, 47, 116–125. [Google Scholar] [CrossRef]
- Wang, X.-H. Interfacial electrochemistry of pyrite oxidation and flotation. J. Colloid Interface Sci. 1996, 178, 628–637. [Google Scholar] [CrossRef]
- Chernyshova, I.V. Pyrite oxidation mechanism in aqueous solutions: An in situ FTIR study. Russ. J. Electrochem. 2004, 40, 69–77. [Google Scholar] [CrossRef]
- Holmes, P.R.; Crundwell, F.K. Polysulfides do not cause passivation: Results from the dissolution of pyrite and implications for other sulfide minerals. Hydrometallurgy 2013, 139, 101–110. [Google Scholar] [CrossRef]
- Mikhlin, Y. Reactivity of pyrrhotite surfaces: An electrochemical study. Phys. Chem. Chem. Phys. 2000, 2, 5672–5677. [Google Scholar] [CrossRef]
- Kuklinskii, A.V.; Mikhlin, Y.L.; Pashkov, G.L.; Kargin, V.F.; Asanov, I.P. Conditions for the formation of a nonequilibrium nonstoichiometric layer on pyrrhotite in acid solutions. Russ. J. Electrochem. 2001, 37, 1269–1276. [Google Scholar] [CrossRef]
- Mikhlin, Y.L.; Kuklinskii, A.V.; Pashkov, G.L.; Asanov, I.P. Pyrrhotite electrooxidation in acid solutions. Russ. J. Electrochem. 2001, 37, 1277–1282. [Google Scholar] [CrossRef]
- Habashl, F.; Torres-Âcuna, N. The anodic dissolution of copper(l) sulfide and the direct recovery of copper and elemental sulfur from white metal. Trans. Metall. Eng. AIME 1968, 242, 780–787. [Google Scholar]
- Nechvoglod, O.V.; Pikalov, S.M. Mechanism of passive layer formation during electrochemical oxidation of copper(I) sulfide. Russ. J. Electrochem. 2021, 57, 580–586. [Google Scholar] [CrossRef]
- O’Dell, C.S.; Walker, G.W.; Richardson, P.E. Electrochemistry of the chalcocite-xanthate system. J. Appl. Electrochem. 1996, 16, 544–554. [Google Scholar] [CrossRef]
- Ghali, E.; Lewenstam, A. Electrodissolution of synthetic covellite in hydrochloric acid. J. Appl. Electrochem. 1982, 12, 369–376. [Google Scholar] [CrossRef]
- Wigdergauz, V.E.; Chanturiya, V.A.; Teplyakova, M.V. Potentiometric study of chalcopyrite electroleaching. In Combined Ore Processing Methods; IPKON, Academy of Sciences of the USSR: Moscow, Russia, 1988; pp. 13–22. (In Russian) [Google Scholar]
- Biegler, T.; Swift, D.A. Anodic electrochemistry of chalcopyrite. J. Appl. Electrochem. 1979, 9, 545–554. [Google Scholar] [CrossRef]
- Biegler, T.; Swift, D.A. The electrolytic reduction of chalcopyrite in acid solution. J. Appl. Electrochem. 1977, 6, 229–235. [Google Scholar] [CrossRef]
- Radyushkina, K.A.; Wigdergauz, V.E.; Tarasevich, M.R.; Chanturiya, V.A. Electrochemistry of sulfide minerals. Surface redox transformations of chalcopyrite and chalcocite in aqueous solutions of electrolytes. Electrochimiya 1986, 22, 1491–1496. (In Russian) [Google Scholar]
- Chander, S. Electrochemistry of sulfide flotation: Growth characteristics of surface coating and their properties, with special reference to chalcopyrite and pyrite. Int. J. Miner. Process. 1991, 33, 121–134. [Google Scholar] [CrossRef]
- Velásquez, P.; Gómez, H.; Ramos-Barrado, J.R.; Leinen, D. Voltammetry and XPS analysis of a chalcopyrite CuFeS2 electrode. Colloids Surf. A 1998, 140, 369–375. [Google Scholar] [CrossRef]
- Velásquez, P.; Gómez, H.; Leinen, D.; Ramos-Barrado, J.R. Electrochemical impedance spectroscopy of chalcopyrite CuFeS2 electrode. Colloids Surf. A 1998, 140, 177–182. [Google Scholar] [CrossRef]
- Moyo, T.; Petersen, J.; Nicol, M.J. The electrochemistry and kinetics of the oxidative dissolution of chalcopyrite in ammoniacal solutions. Part I–Anodic reactions. Hydrometallurgy 2018, 182, 97–103. [Google Scholar] [CrossRef]
- Moyo, T.; Petersen, J.; Nicol, M.J. The electrochemistry and kinetics of the oxidative dissolution of chalcopyrite in ammoniacal solutions. Part II–Cathodic reactions. Hydrometallurgy 2019, 184, 67–74. [Google Scholar] [CrossRef]
- Nicol, M.J. The electrochemistry of chalcopyrite in alkaline solutions. Hydrometallurgy 2019, 187, 134–140. [Google Scholar] [CrossRef]
- Price, D.C.; Chilton, J.P. The anodic reactions of bornite in sulfuric acid solution. Hydrometallurgy 1981, 7, 117–133. [Google Scholar] [CrossRef]
- Zhao, H.; Huang, X.; Hu, M.; Zhang, C.; Zhang, Y.; Wang, J.; Qin, W.; Qiu, G. Insights into the surface transformation and electrochemical dissolution process of bornite in bioleaching. Minerals 2018, 8, 173. [Google Scholar] [CrossRef]
- Ásbjornsson, J.; Kelsall, G.H.; Pattrick, R.A.D.; Vaughan, D.J.; Wincott, P.L.; Hope, G.A. Electrochemical and surface analytical studies of enargite in acid solution. J. Electrochem. Soc. 2004, 151, E250–E256. [Google Scholar] [CrossRef]
- Ma, Y.; Yang, Y.; Skinner, W.; Chen, M. Electrochemical and spectroscopic analysis of enargite (Cu3AsS4) dissolution mechanism in sulfuric acid solution. Hydrometallurgy 2020, 194, 105346. [Google Scholar] [CrossRef]
- Nicol, M.J.; Ruiz-Sanchez, A.; Senanayake, G.; Tjandrawan, V.; Lapidus, G.T. The electrochemical behaviour of enargite in ammoniacal solutions. I. Anodic reactions. Hydrometallurgy 2019, 188, 92–100. [Google Scholar] [CrossRef]
- Nicol, M.J.; Ruiz-Sanchez, A.; Senanayake, G.; Tjandrawan, V.; Lapidus, G.T. The electrochemical behaviour of enargite in ammoniacal solutions. II. Cathodic reactions. Hydrometallurgy 2019, 189, 105115. [Google Scholar] [CrossRef]
- Buckley, A.N.; Woods, R. Electrochemical and XPS studies of the surface oxidation of synthetic heazlewoodite (Ni3S2). J. Appl. Electrochem. 1991, 21, 575–582. [Google Scholar] [CrossRef]
- Power, G.P. The electrochemistry of the nickel sulfides. 1. NiS. Aust. J. Chem. 1981, 34, 2287–2291. [Google Scholar] [CrossRef]
- Power, G.P. The electrochemistry of the nickel sulfides–2. Ni3S2. Electrochim. Acta 1982, 27, 359–364. [Google Scholar] [CrossRef]
- Aromaa, J. Electrochemical dissolution of synthetic heazlewoodite (Ni3S2). Physicochem. Probl. Miner. Processing 2011, 46, 51–64. [Google Scholar]
- Thornber, M.R. Mineralogical and electrochemical stability of the nickel –iron sulphides–pentlandite and violarite. J. Appl. Electrochem. 1983, 13, 253–267. [Google Scholar] [CrossRef]
- Warner, T.E.; Rice, N.M.; Taylor, N. An electrochemical study of the oxidative dissolution of synthetic pentlandite in aqueous media. Hydrometallurgy 1992, 31, 55–90. [Google Scholar] [CrossRef]
- Warner, T.E.; Rice, N.M.; Taylor, N. Electrochemical study of oxidative dissolution of synthetic violarite in aqueous media. In Hydrometallurgy’94; Springer: Berlin/Heidelberg, Germany, 1994; pp. 273–287. [Google Scholar]
- Makarov, D.V.; Forsling, W.; Makarov, V.N. Electrooxidation of pentlandite in a carbonate solution. Russ. J. Electrochem. 2004, 40, 420–423. (In Russian) [Google Scholar] [CrossRef]
- Mikhlin, Y.L.; Galkin, P.S.; Kopteva, S.A. Electrochemical study of galena surfaces in acid solutions. Izv. Sib. Otd. Akad. Nauk. SSSR. Ser. Him. 1988, 2, 11–17. (In Russian) [Google Scholar]
- Chernyshova, I.V. In situ study of the oxidation of galenite (natural PbS) in alkaline media by FTIR spectroscopy: Anode processes in the absence of oxygen. Russ. J. Electrochem. 2001, 37, 579–584. [Google Scholar] [CrossRef]
- Ahlberg, E.; Elfstrom, B.A. Anodic polarization of galena in relation to flotation. Int. J. Miner. Process. 1997, 33, 135–142. [Google Scholar] [CrossRef]
- Cisneros-Gonzalez, I.; Oropeza-Guzman, M.T.; Gonzalez, I. Cyclic voltammetry applied to the characterisation of galena. Hydrometallurgy 1999, 53, 133–144. [Google Scholar] [CrossRef]
- Gardner, J.R.; Woods, R. Study of surface oxidation of galena using cyclic voltammetry. J. Electroanal. Chem. 1979, 100, 447–459. [Google Scholar] [CrossRef]
- Nicol, M.J.; Paul, R.L.; Diggle, J.W.; Saunders, A.P. The electrochemical behaviour of galena (lead sulphide): II. Catodic reduction. Electrochim. Acta 1978, 23, 635–639. [Google Scholar] [CrossRef]
- Paul, R.L.; Nicol, M.J.; Diggle, J.W.; Saunders, A.P. The electrochemical behaviour of galena (lead sulphide): I. Anodic dissolution. Electrochim. Acta 1978, 23, 625–633. [Google Scholar] [CrossRef]
- Pritzker, M.D.; Yoon, R.H. Thermodynamic calculation on sulfide flotation system. 2. Comparison with electrochemical experiments on the galena-ethylxanthate system. Int. J. Miner. Process. 1987, 20, 267–290. [Google Scholar] [CrossRef]
- Mikhlin, Y.L. State of the Real Surface and Features of the Kinetics of Dissolution and Oxidation of Metal Sulfides in Interaction with Acid Solutions. Extended abstract. Ph.D. Thesis, Institute of Chemistry and Chemical Technologies of the SB RAS, Krasnoyarsk, Russia, 2003; 40p. (In Russian). [Google Scholar]
- Ahlberg, E.; Ásbjörnsson, J. Carbon paste electrodes in mineral processing: An electrochemical study of sphalerite. Hydrometallurgy 1994, 36, 19–37. [Google Scholar] [CrossRef]
- Choi, W.K.; Torma, A.E.; Ohline, R.W.; Ghali, E. Electrochemical aspects of zinc sulphide leaching by Thiobacillus ferrooxidans. Hydrometallurgy 1993, 33, 137–152. [Google Scholar] [CrossRef]
- Lotens, J.P.; Wesker, E. The behaviour of sulphur in the oxidative leaching of sulphidic minerals. Hydrometallurgy 1987, 18, 39–54. [Google Scholar] [CrossRef]
- Nava, J.L.; Oropeza, M.T.; Gonzalez, I. Oxidation of Mineral Species as a Function of the Anodic Potential of Zinc Concentrate in Sulfuric Acid. J. Electrochem. Soc. 2004, 151, B387–B393. [Google Scholar] [CrossRef]
- Narasagoudar, R.A.; Johnson, J.W.; O’Keefe, T.J. The anodic dissolution of ZnS electrodes in sulfuric acid solutions. Hydrometallurgy 1982, 9, 37–55. [Google Scholar] [CrossRef]
- Srinivasan, G.N.; Iyer, S.V. Cyclic voltammetric studies on sphalerite electrodes. Bull. Electrochem. 2000, 16, 5–9. [Google Scholar]
- Urbano, G.; Meléndez, A.M.; Reyes, V.E.; Veloz, M.A.; González, I. Galvanic interactions between galena–sphalerite and their reactivity. Int. J. Miner. Process. 2007, 82, 148–155. [Google Scholar] [CrossRef]
- Karimia, S.; Ghahreman, A.; Rashchia, F.; Moghaddam, J. The mechanism of electrochemical dissolution of sphalerite in sulfuric acid media. Electrochim. Acta 2017, 253, 47–58. [Google Scholar] [CrossRef]
- Plaksin, I.N.; Shafeev, R.S. Effect of electrochemical inhomogeneity of the surface of sulfide minerals on the distribution of xanthate under flotation. Dokl. Akad. Nauk. USSR 1958, 121, 145–146. (In Russian) [Google Scholar]
- Plaksin, I.N.; Shafeev, R.S. Mechanism of occurrence of electrochemical inhomogeneity in the surface of sulfide minerals. Dokl. Akad. Nauk. USSR 1959, 125, 599–600. (In Russian) [Google Scholar]
- Plaksin, I.N.; Shafeev, R.S. Quantitative assessment of xanthate attachment depending on the surface properties of sulfide minerals. Dokl. Akad. Nauk. USSR 1959, 128, 777–780. (In Russian) [Google Scholar]
- Plaksin, I.N.; Shafeev, R.S. Hydrophobic effect of oxygen on the surface of sulfide minerals. Dokl. Akad. Nauk. USSR 1960, 135, 140–142. (In Russian) [Google Scholar]
- Chander, S. Electrochemistry of sulfide mineral flotation. Min. Metall. Explor. 1988, 5, 104–114. [Google Scholar] [CrossRef]
- Biegler, T. Oxygen reduction on sulphide minerals. 2. Relation between activity and semiconducting properties of pyrite electrodes. J. Electroanal. Chem. 1976, 70, 265–275. [Google Scholar] [CrossRef]
- Biegler, T.; Rand, D.A.J.; Woods, R. Oxygen reduction on sulphide minerals. 1. Kinetics and mechanism at rotated pyrite electrodes. J. Electroanal. Chem. 1975, 60, 151–162. [Google Scholar] [CrossRef]
- Biegler, T.; Rand, D.A.J.; Woods, R. Oxygen reduction on sulphide minerals. In Trends in Electrochemistry; Bockris, J.O.M., Ed.; Plenum Press: New York, NY, USA, 1977; pp. 291–302. [Google Scholar]
- Rand, D.A.J. Oxygen reduction on sulphide minerals. 3. Comparison of activities of various copper, iron, lead and nickel mineral electrodes. J. Electroanal. Chem. 1977, 83, 19–32. [Google Scholar] [CrossRef]
- Doo, S.G.; Sohn, H.J. Electrochemical reduction of oxygen on a pyrite surface. Min. Metall. Explor. 1989, 6, 201–205. [Google Scholar] [CrossRef]
- Wigdergauz, V.E.; Radyushkina, K.A. Mechanism and kinetics of oxygen reduction on pyrite. In Achieving Complete Recovery of Valuable Components in Mineral Processing; IPKON, Academy of Sciences of the USSR: Moscow, Russia, 1986; pp. 8–20. (In Russian) [Google Scholar]
- Tarasevich, M.R.; Kudaikulova, G.A.; Radyushkina, K.A. Electroreduction of oxygen on copper-containing sulfide minerals. Russ. J. Electrochem. 2000, 36, 49–53. [Google Scholar] [CrossRef]
- Kudaykulova, G.A. Electrochemical Processes of Oxidation on the Surface of Sulfide Minerals in Aqueous and Aprotic Environments. Extended Abstract. Ph.D. Thesis, Institute of Electrochemistry, Academy of Sciences of the USSR, Moscow, Russia, 1990; 22p. (In Russian). [Google Scholar]
- Pozzo, R.L.; Malicsi, A.S.; Iwasaki, I. Pyrite-pyrrhotite-grinding media contact and its effect on flotation. Min. Metall. Explor. 1990, 7, 16–21. [Google Scholar] [CrossRef]
- Rao, S.R.; Finch, J.A. Galvanic interaction studies on sulphide minerals. Can. Metall. Q. 1988, 27, 253–259. [Google Scholar] [CrossRef]
- Hu, Y.; Wu, M.; Liu, R.; Sun, W. A review on the electrochemistry of galena flotation. Miner. Eng. 2020, 150, 106272. [Google Scholar] [CrossRef]
- Ke, B.; Chen, J.; Li, Y.; Chen, Y. Galvanic contacting effect of pyrite on xanthate adsorption on galena surface: DFT simulation and cyclic voltammetric measurements. Physicochem. Probl. Miner. Process. 2018, 54, 826–836. [Google Scholar]
- Qi, C.; Khalkhali, M.; Grundy, J.S.; Liu, B.; Wang, A.; Liu, Q. Unraveling H2O2-stimulated surface oxidation of non-magnetic pyrrhotite and pentlandite by underlying electronic structures. Miner. Eng. 2021, 170, 106964. [Google Scholar] [CrossRef]
- Mu, Y.; Peng, Y. The role of sodium metabisulphite in depressing pyrite in chalcopyrite flotation using saline water. Miner. Eng. 2019, 142, 105921. [Google Scholar] [CrossRef]
- Mu, Y.; Cheng, Y.; Peng, Y. The interaction between grinding media and collector in pyrite flotation at neutral and slightly acidic pH. Miner. Eng. 2020, 145, 106063. [Google Scholar] [CrossRef]
- Mu, Y.; Cheng, Y.; Peng, Y. The interaction of grinding media and collector in pyrite flotation at alkaline pH. Miner. Eng. 2020, 152, 106344. [Google Scholar] [CrossRef]
- Xuemin, Q.; Hongying, Y.; Guobao, C.; Shuiping, Z.; Chuangkai, C.; Bibo, L. Inhibited mechanism of carboxymethyl cellulose as a galena depressant in chalcopyrite and galena separation flotation. Miner. Eng. 2020, 150, 106273. [Google Scholar] [CrossRef]
- Aikawa, K.; Ito, M.; Segawa, T.; Jeon, S.; Park, I.; Tabelin, C.B.; Hiroyoshi, N. Depression of lead-activated sphalerite by pyrite via galvanic interactions: Implications to the selective flotation of complex sulfide ores. Miner. Eng. 2020, 152, 106367. [Google Scholar] [CrossRef]
- Mhonde, N.; Johansson, L.-S.; Corin, K.; Schreithofer, N. The effect of sodium isobutyl xanthate on galena and chalcopyrite flotation in the presence of dithionite ions. Miner. Eng. 2021, 169, 106985. [Google Scholar] [CrossRef]
- Suyantara, G.P.W.; Hirajima, T.; Miki, H.; Sasaki, K.; Kuroiwa, S.; Aoki, Y. Effect of H2O2 and potassium amyl xanthate on separation of enargite and tennantite from chalcopyrite and bornite using flotation. Miner. Eng. 2020, 152, 106371. [Google Scholar] [CrossRef]
- Wang, X.; Qin, W.; Jiao, F.; Wu, J. The influence of galvanic interaction on the dissolution and surface composition of galena and pyrite in flotation system. Miner. Eng. 2020, 156, 106525. [Google Scholar] [CrossRef]
- Huai, Y.; Peng, Y. The formation of iron sulphide on oxidised pyrite during sulphidisation and subsequent interactions with xanthate. Miner. Eng. 2020, 157, 106564. [Google Scholar] [CrossRef]
- Kaksonen, A.H.; Deng, X.; Bohu, T.; Zea, L.; Khaleque, H.N.; Gumulya, Y.; Boxall, N.J.; Morris, C.; Cheng, K.Y. Prospective directions for biohydrometallurgy. Hydrometallurgy 2020, 195, 105376. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, H.; Qian, L.; Sun, M.; Lv, X.; Zhang, L.; Petersen, J.; Qiu, G. A brief overview on the dissolution mechanisms of sulfide minerals in acidic sulfate environments at low temperatures: Emphasis on electrochemical cyclic voltammetry analysis. Miner. Eng. 2020, 158, 106586. [Google Scholar] [CrossRef]
- Tanne, C.K.; Schippers, A. Electrochemical investigation of chalcopyrite (bio)leaching residues. Hydrometallurgy 2019, 187, 8–17. [Google Scholar] [CrossRef]
- Tobolik, M.; Krinik, L. Electrolyzer for Anodic Dissolution of Conductive Materials. Patent 93102 PPR, MKI S25S7/00, 6 December 1973. [Google Scholar]
- Morozov, A.F.; Morozova, V.P.; Mutylin, Y.V. Electrochemical treatment in the gravity flotation of tin-containing products. Ore Benef. Irkutsk. Polytech. Inst. 1988, 4, 88–92. (In Russian) [Google Scholar]
- Zhao, H.; Zhang, Y.; Zhang, X.; Qian, L.; Sun, M.; Yang, Y.; Zhang, Y.; Wang, J.; Kim, H.; Qiu, G. The dissolution and passivation mechanism of chalcopyrite in bioleaching: An overview. Miner. Eng. 2019, 136, 140–154. [Google Scholar] [CrossRef]
- O’Connor, G.M.; Eksteen, J.J. A critical review of the passivation and semiconductor mechanisms of chalcopyrite leaching. Miner. Eng. 2020, 154, 106401. [Google Scholar] [CrossRef]
- Tapera, T.; Nikoloski, A.N. The effect of silver on the acidic ferric sulfate leaching of primary copper sulfides under recycle solution conditions observed in heap leaching. Part 3: Surface characterization. Hydrometallurgy 2019, 183, 130–141. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, Y.; Sun, M.; Ou, P.; Zhang, Y.; Liao, R.; Qiu, G. Catalytic mechanism of silver in the oxidative dissolution process of chalcopyrite: Experiment and DFT calculation. Hydrometallurgy 2019, 187, 18–29. [Google Scholar]
- Zeng, W.; Peng, Y.; Peng, T.; Nan, M.; Chen, M.; Qiub, G.; Shen, L. Electrochemical studies on dissolution and passivation behavior of low temperature bioleaching of chalcopyrite by Acidithiobacillus ferrivorans YL15. Miner. Eng. 2020, 155, 106416. [Google Scholar] [CrossRef]
- Tanne, C.; Schippers, A. Electrochemical investigation of microbially and galvanically leached chalcopyrite. Hydrometallurgy 2021, 202, 105603. [Google Scholar] [CrossRef]
- Hong, M.; Huang, X.; Gan, X.; Qiu, G.; Wang, J. The use of pyrite to control redox potential to enhance chalcopyrite bioleaching in the presence of Leptospirillum ferriphilum. Miner. Eng. 2021, 172, 107145. [Google Scholar] [CrossRef]
- Saavedra, A.; García-Meza, J.V.; Cortón, E.; González, I. Understanding galvanic interactions between chalcopyrite and magnetite in acid medium to improve copper (Bio)Leaching. Electrochim. Acta 2018, 265, 569–576. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, H.; Zhang, Y.; Liu, H.; Yin, H.; Deng, J.; Qiu, G. Interaction mechanism between marmatite and chalcocite in acidic (microbial) environments. Hydrometallurgy 2020, 191, 105217. [Google Scholar] [CrossRef]
- Hong, M.; Wang, X.; Yang, B.; Liu, S.; Lin, H.; Qiu, G.; Qin, W.; Wang, J. Effect of pyrite with different semiconducting properties on bornite bioleaching in the presence of Leptospirillum ferriphilum. Hydrometallurgy 2020, 196, 105414. [Google Scholar] [CrossRef]
- Rivera-Vasquez, B.F.; Dixon, D. Rapid atmospheric leaching of enargite in acidic ferric sulfate media. Hydrometallurgy 2015, 152, 149–158. [Google Scholar]
- Ma, Y.; Yang, Y.; Gao, X.; Fan, R.; Chen, M. The galvanic effect of pyrite enhanced (bio)leaching of enargite (Cu3AsS4). Hydrometallurgy 2021, 202, 105613. [Google Scholar] [CrossRef]
- Azizi, D.; Larachi, F. DFT simulations of pyrite galvanic interactions with bulk, solid-solution and nanoparticle Au occurrences–Insights into gold cyanidation. Miner. Eng. 2020, 149, 106239. [Google Scholar]
- Hofmann, T.; Schuwirth, N. Zn and Pb release of sphalerite (ZnS)-bearing mine waste tailings. J. Soils Sediments 2008, 8, 433–441. [Google Scholar] [CrossRef]
- Pabst, T.; Aubertin, M.; Bussière, B.; Molson, J. Experimental and numerical evaluation of single-layer covers placed on acid-generating tailings. Geotech. Geol. Eng. 2017, 35, 1421–1438. [Google Scholar] [CrossRef]
- Benzaazoua, M.; Bouzahzah, H.; Taha, Y.; Kormos, L.; Kabombo, D.; Lessard, F.; Bussière, B.; Demers, I.; Kongolo, M. Integrated environmental management of pyrrhotite tailings at Raglan Mine: Part 1 challenges of desulphurization process and reactivity prediction. J. Clean. Prod. 2017, 162, 86–95. [Google Scholar] [CrossRef]
- Rey, N.J.; Demers, I.; Bussière, B.; Mbonimpa, M.; Gagnon, M. A geochemical evaluation of a monolayer cover with an elevated water table for the reclamation of the Doyon-Westwood tailings ponds, Canada. Environ. Earth Sci. 2020, 79, 58. [Google Scholar] [CrossRef]
- Rakotonimaro, T.V.; Guittony, M.; Neculita, C.M. Compaction of peat cover over desulfurized gold mine tailings changes: Arsenic speciation and mobility. Appl. Geochem. 2021, 128, 104923. [Google Scholar] [CrossRef]
- Perlatti, F.; Martins, E.P.; Pontes de Oliveira, D.; Ruiz, F.; Asensio, V.; Rezende, C.F.; Otero, X.L.; Ferreira, T.O. Copper release from waste rocks in an abandoned mine (NE, Brazil) and its impacts on ecosystem environmental quality. Chemosphere 2021, 262, 127843. [Google Scholar] [CrossRef]
- Qian, G.; Fan, R.; Huang, J.; Pring, A.; Harmer, S.L.; Zhang, H.D.; Rea, M.A.; Brugger, J.; Teasdale, P.R.; Gibson, C.T.; et al. Oxidative dissolution of sulfide minerals in single and mixed sulfide systems under simulated acid and metalliferous drainage conditions. Environ. Sci. Technol. 2021, 55, 2369–2380. [Google Scholar] [CrossRef]
- Qian, Y.; Li, G.; Brown, P.L.; Gerson, A.R. Chalcopyrite dissolution: Scanning photoelectron microscopy examination of the evolution of sulfur species with and without added iron or pyrite. Geochim. Et Cosmochim. Acta 2017, 212, 33–47. [Google Scholar]
- Ferreira, P.M.; Majuste, D.; Freitas, E.T.F.; Caldeira, C.L.; Dantas, M.S.S.; Ciminelli, V.S.T. Galvanic effect of pyrite on arsenic release from arsenopyrite dissolution in oxygen-depleted and oxygen-saturated circumneutral solutions. J. Hazard. Mater. 2021, 412, 125236. [Google Scholar] [CrossRef]
- Fuchida, S.; Kawachi, M.; Koshikawa, H. Kinetic investigation of galvanic dissolution of ZnS and PbS with FeS2 from hydrothermal sulfides in seawater. Appl. Geochem. 2021, 129, 104963. [Google Scholar] [CrossRef]
- Qian, G.; Fan, R.; Short, M.D.; Schumann, R.C.; Pring, A.; Gerson, A.R. The combined effects of galvanic interaction and silicate addition on the oxidative dissolution of pyrite: Implications for acid and metalliferous drainage control. Environ. Sci. Technol. 2019, 53, 11922–11931. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chanturiya, V.A.; Krasavtseva, E.A.; Makarov, D.V. Electrochemistry of Sulfides: Process and Environmental Aspects. Sustainability 2022, 14, 11285. https://doi.org/10.3390/su141811285
Chanturiya VA, Krasavtseva EA, Makarov DV. Electrochemistry of Sulfides: Process and Environmental Aspects. Sustainability. 2022; 14(18):11285. https://doi.org/10.3390/su141811285
Chicago/Turabian StyleChanturiya, Valentine A., Eugenia A. Krasavtseva, and Dmitriy V. Makarov. 2022. "Electrochemistry of Sulfides: Process and Environmental Aspects" Sustainability 14, no. 18: 11285. https://doi.org/10.3390/su141811285
APA StyleChanturiya, V. A., Krasavtseva, E. A., & Makarov, D. V. (2022). Electrochemistry of Sulfides: Process and Environmental Aspects. Sustainability, 14(18), 11285. https://doi.org/10.3390/su141811285