
Influence of Alkalis on Natural Carbonation of Limestone Calcined Clay Cement Pastes

Abstract
:1. Introduction
2. Materials and Methods
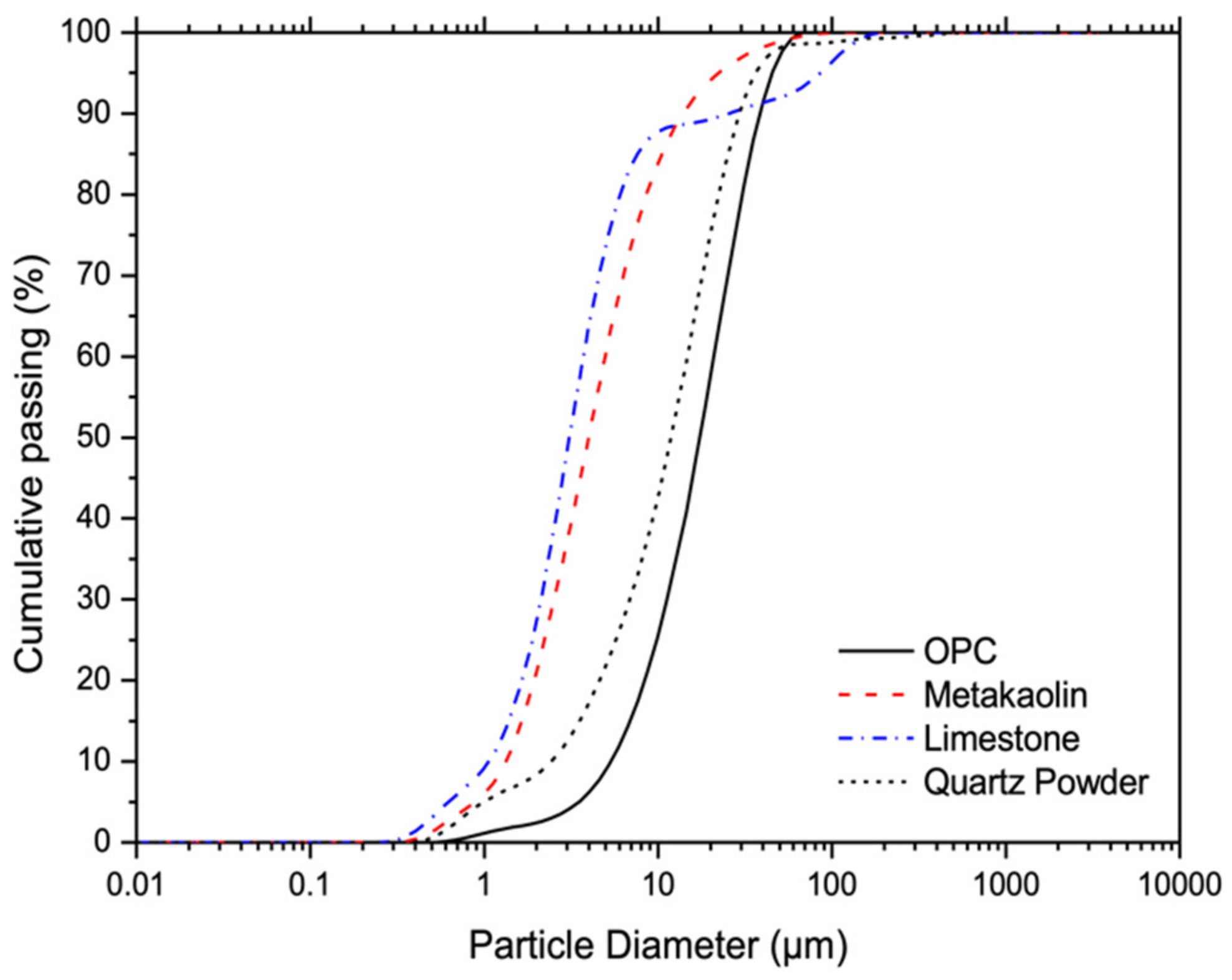
2.1. Materials and Mixture
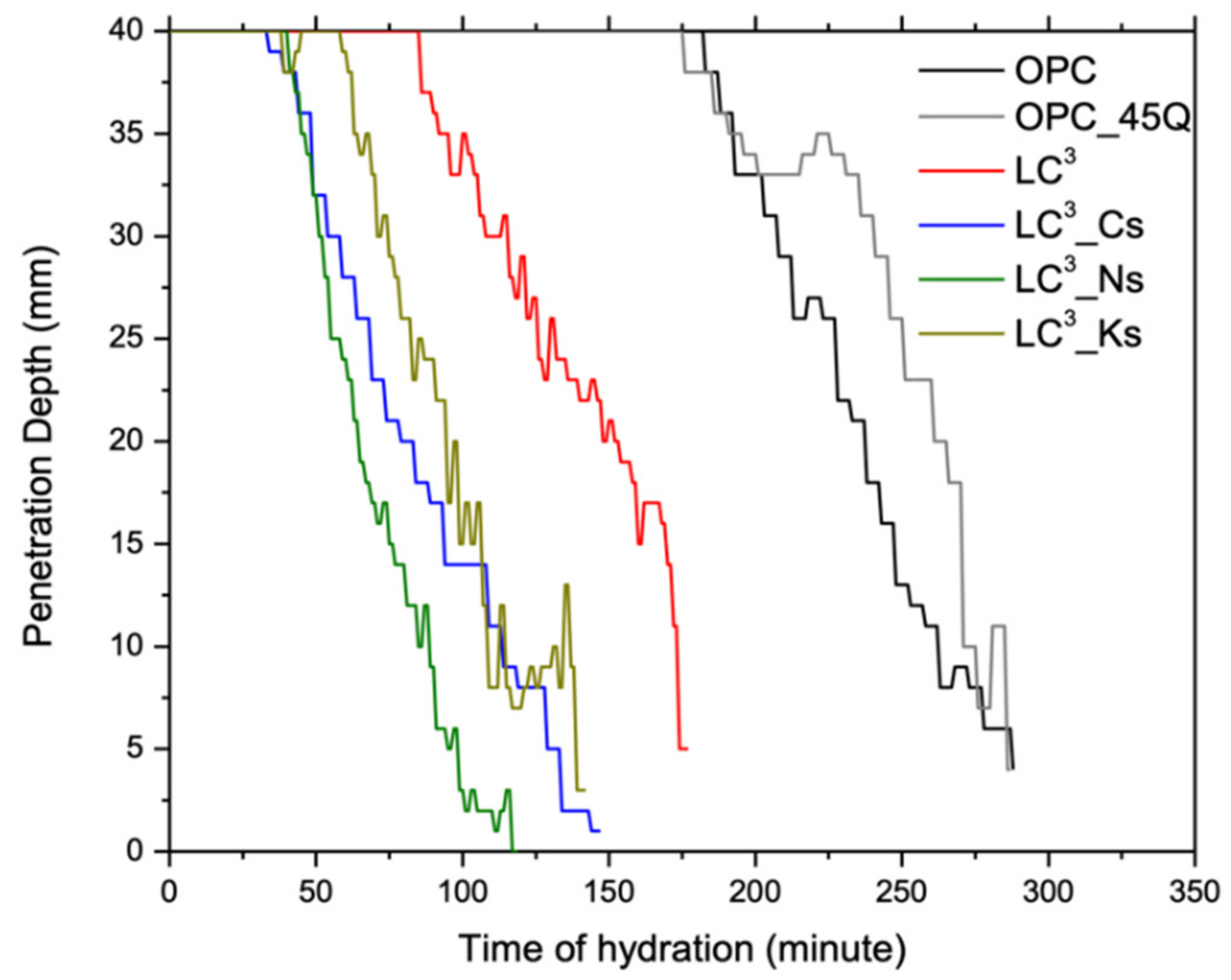
2.2. Vicat Setting Test
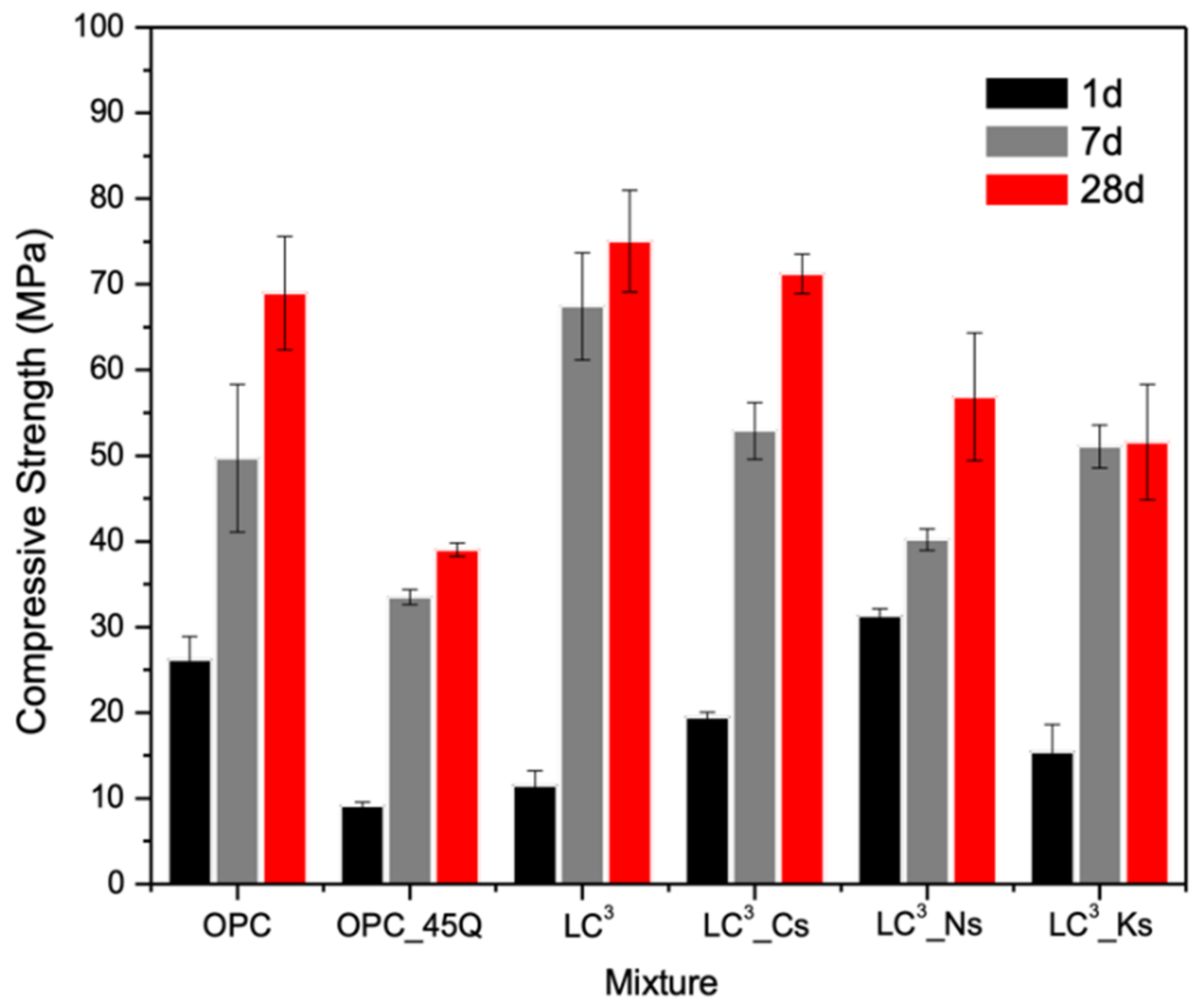
2.3. Compressive Strength
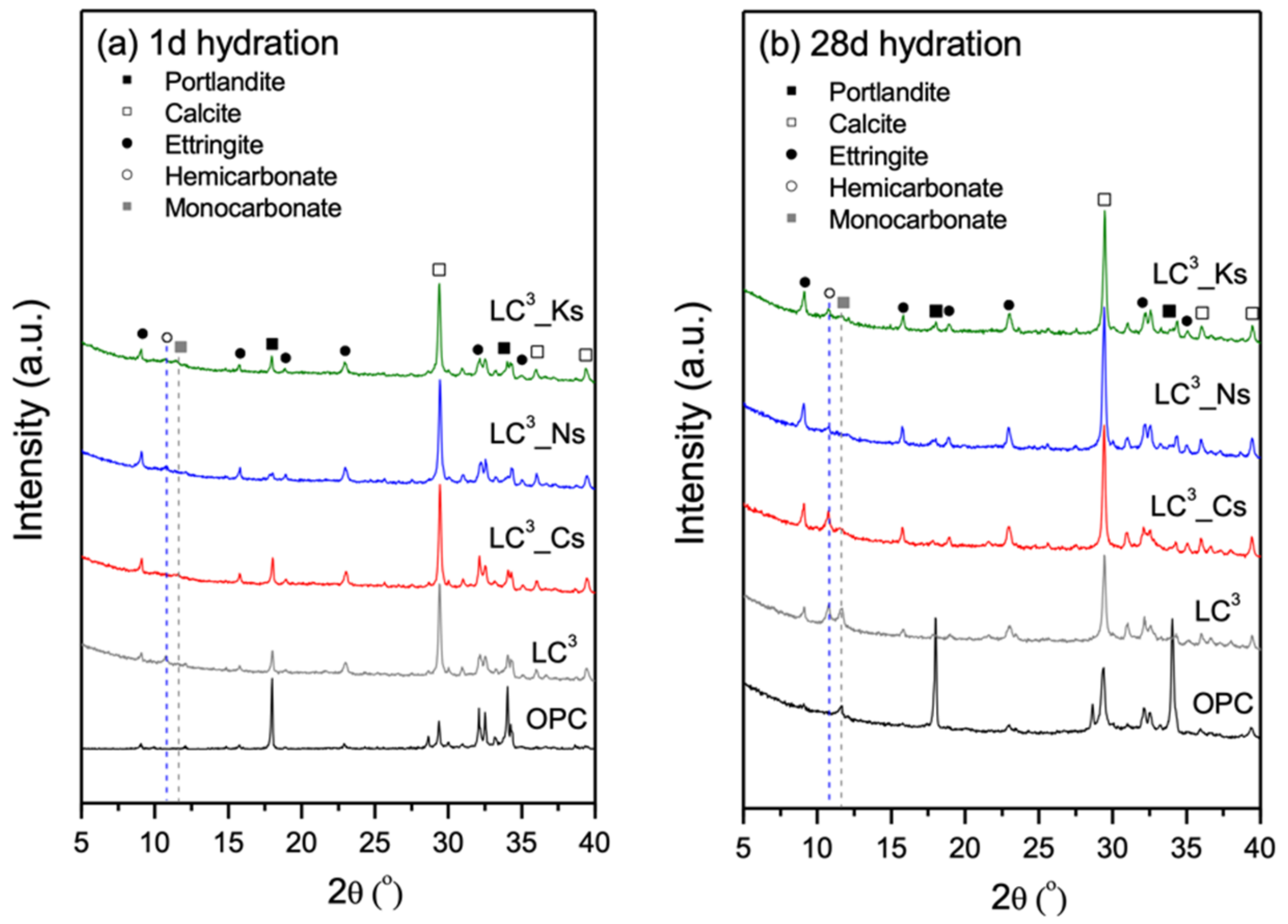
2.4. Phase Analysis Prior to Carbonation
2.5. Natural Carbonation
2.6. Pore Size Distribution Analysis
3. Results and Discussion
3.1. Setting Times
3.2. Strength Development
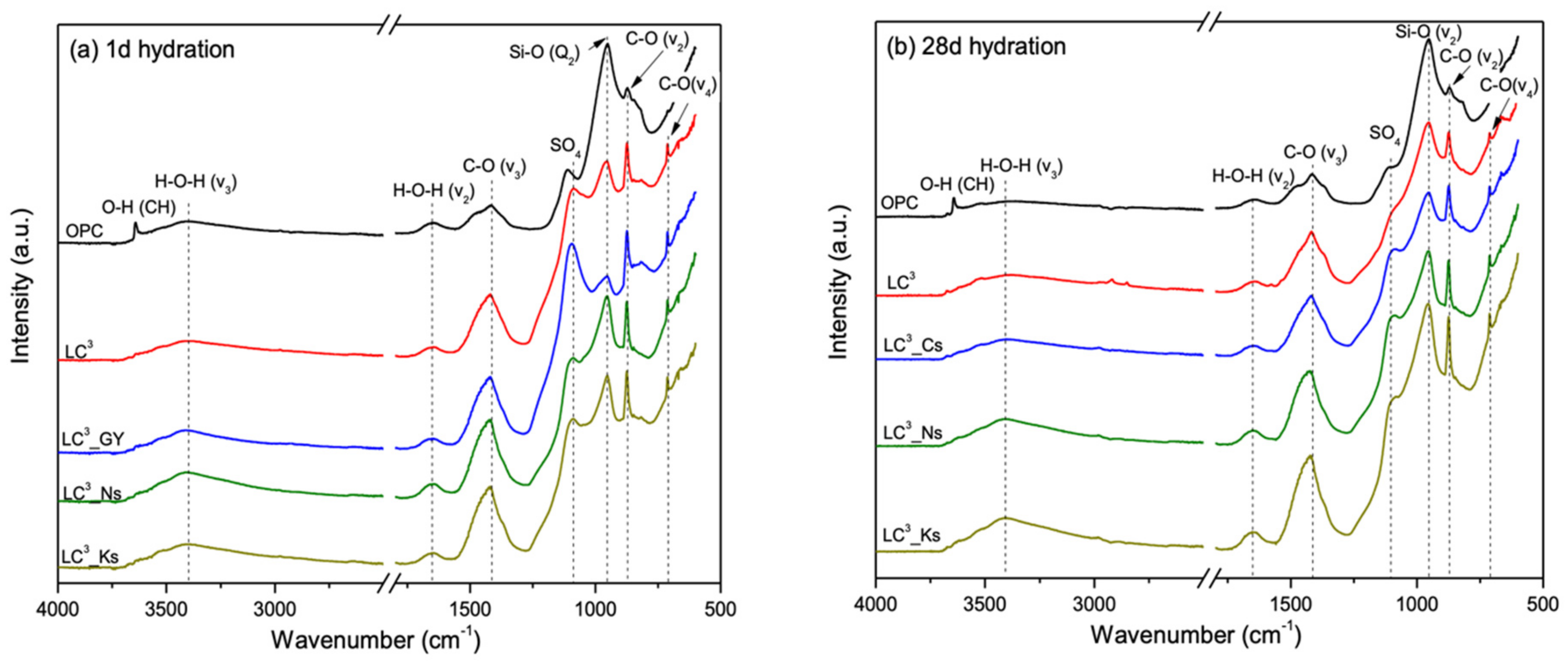
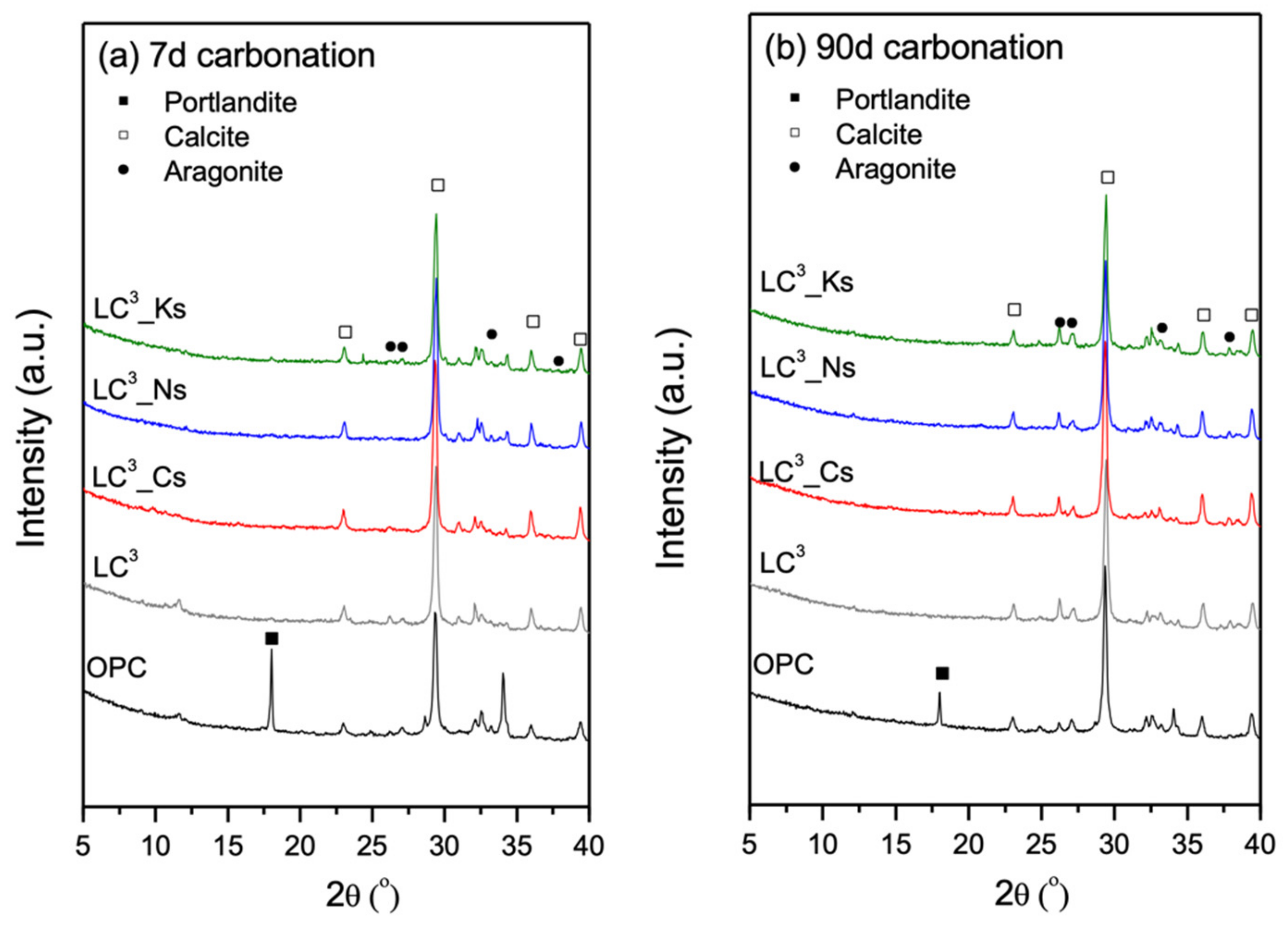
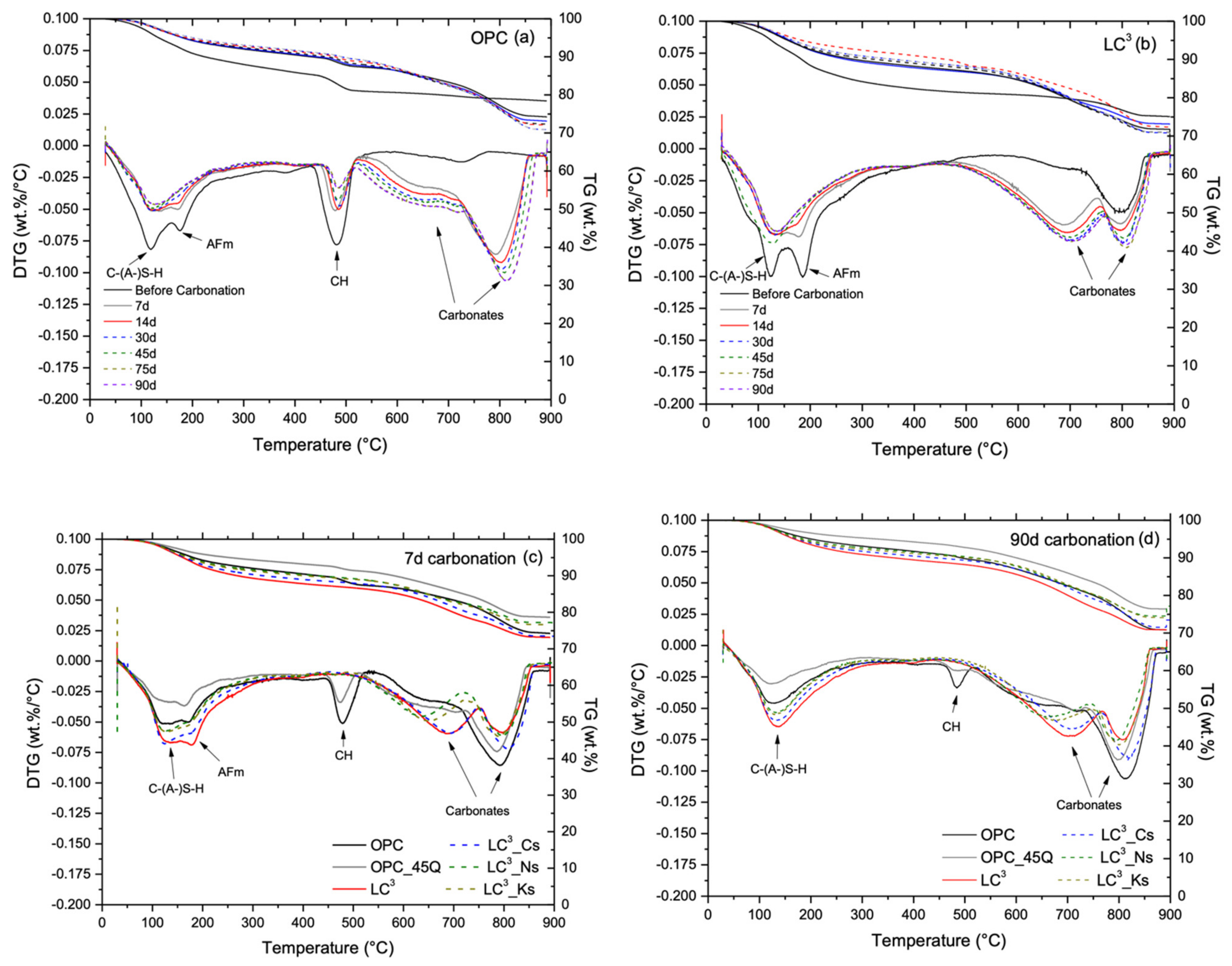
3.3. Phase Assemblage Prior to Carbonation
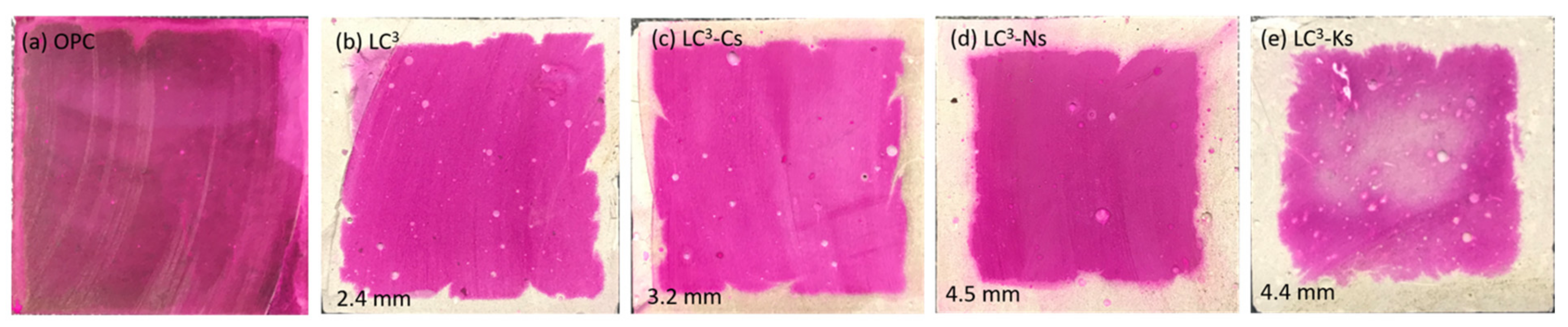
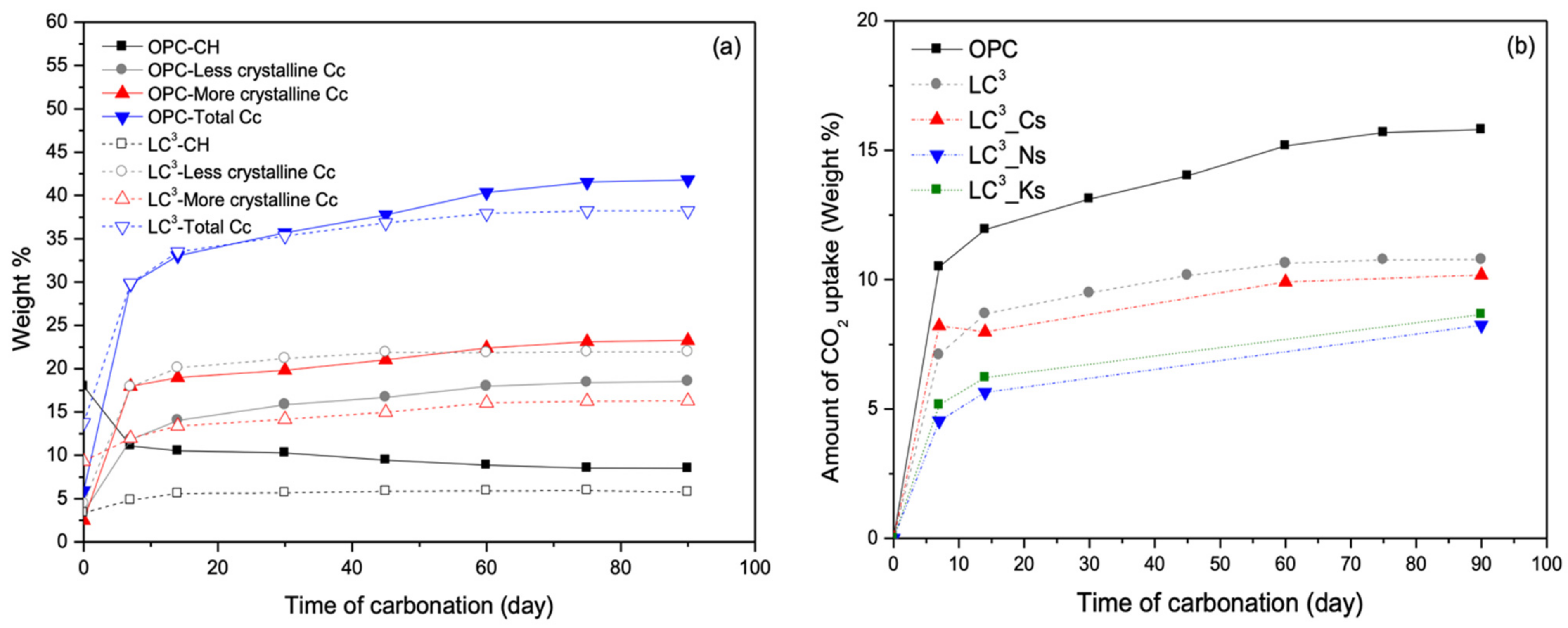
3.4. Carbonation Resistance
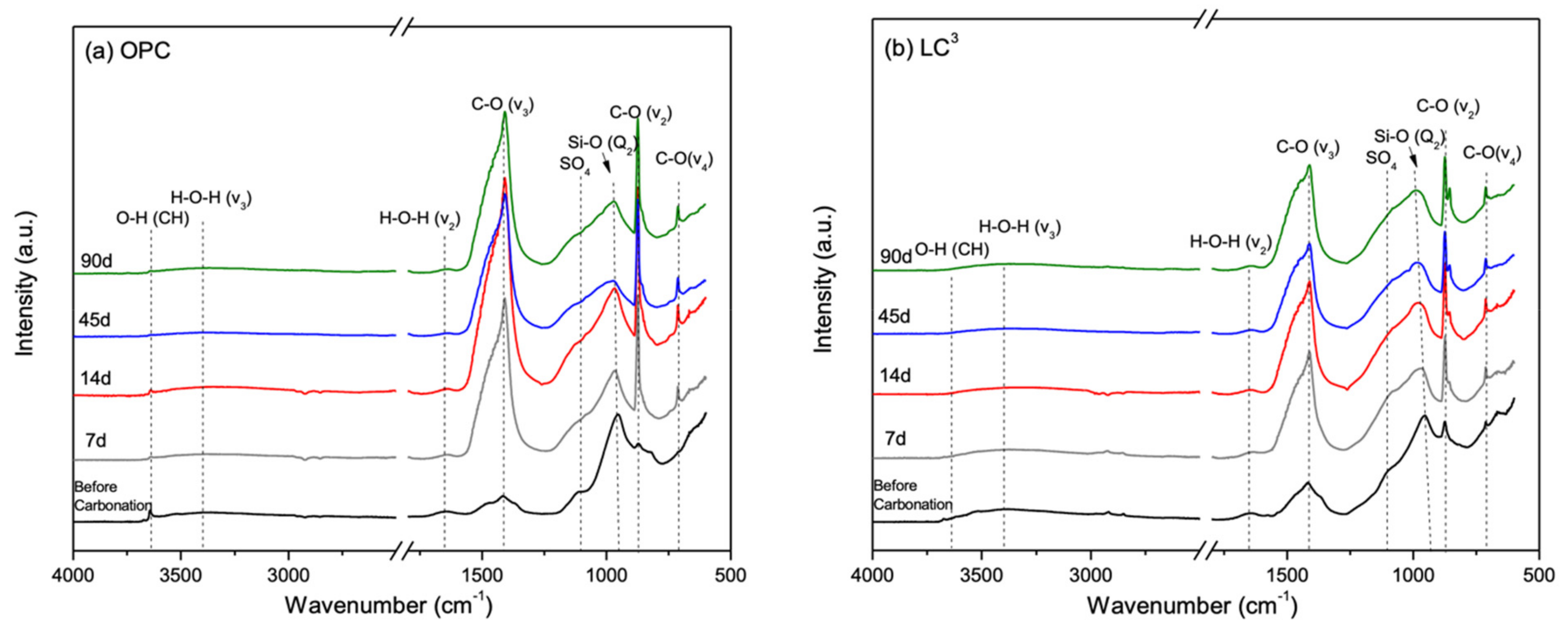
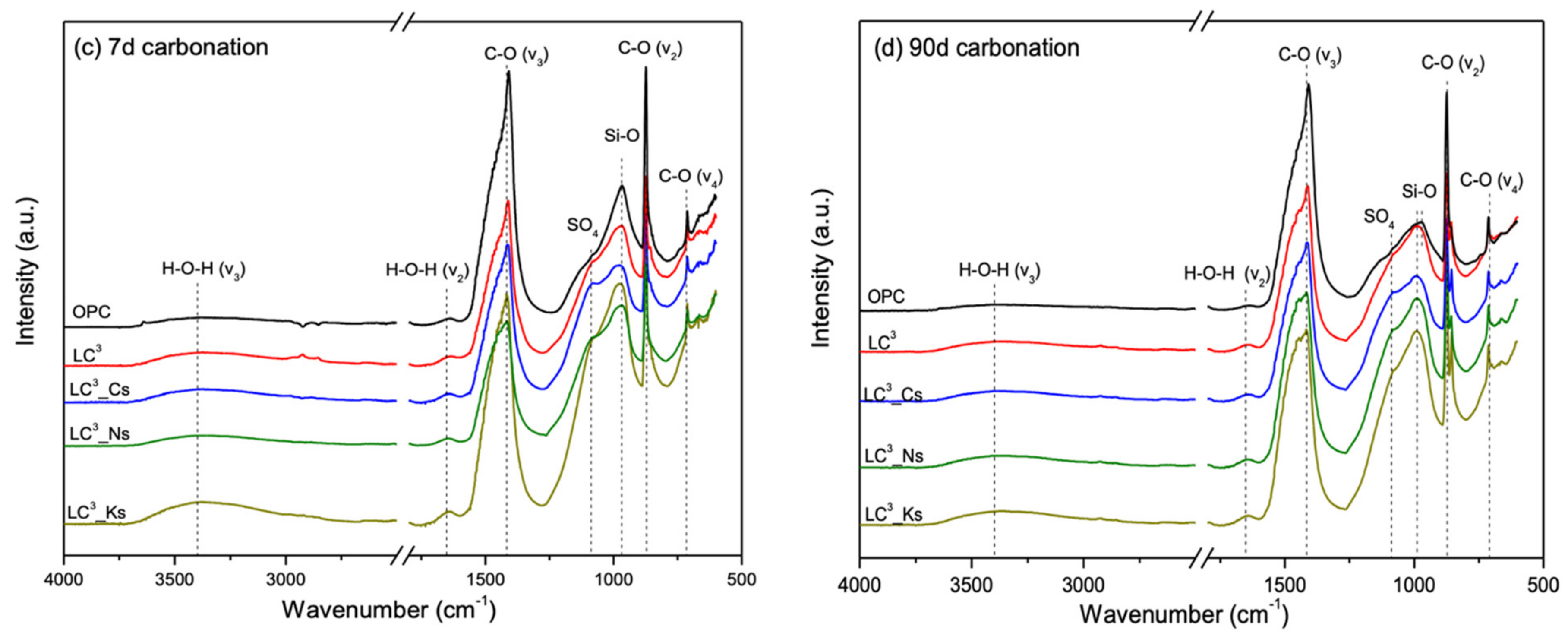
3.5. Phase Alternations Post-Carbonation
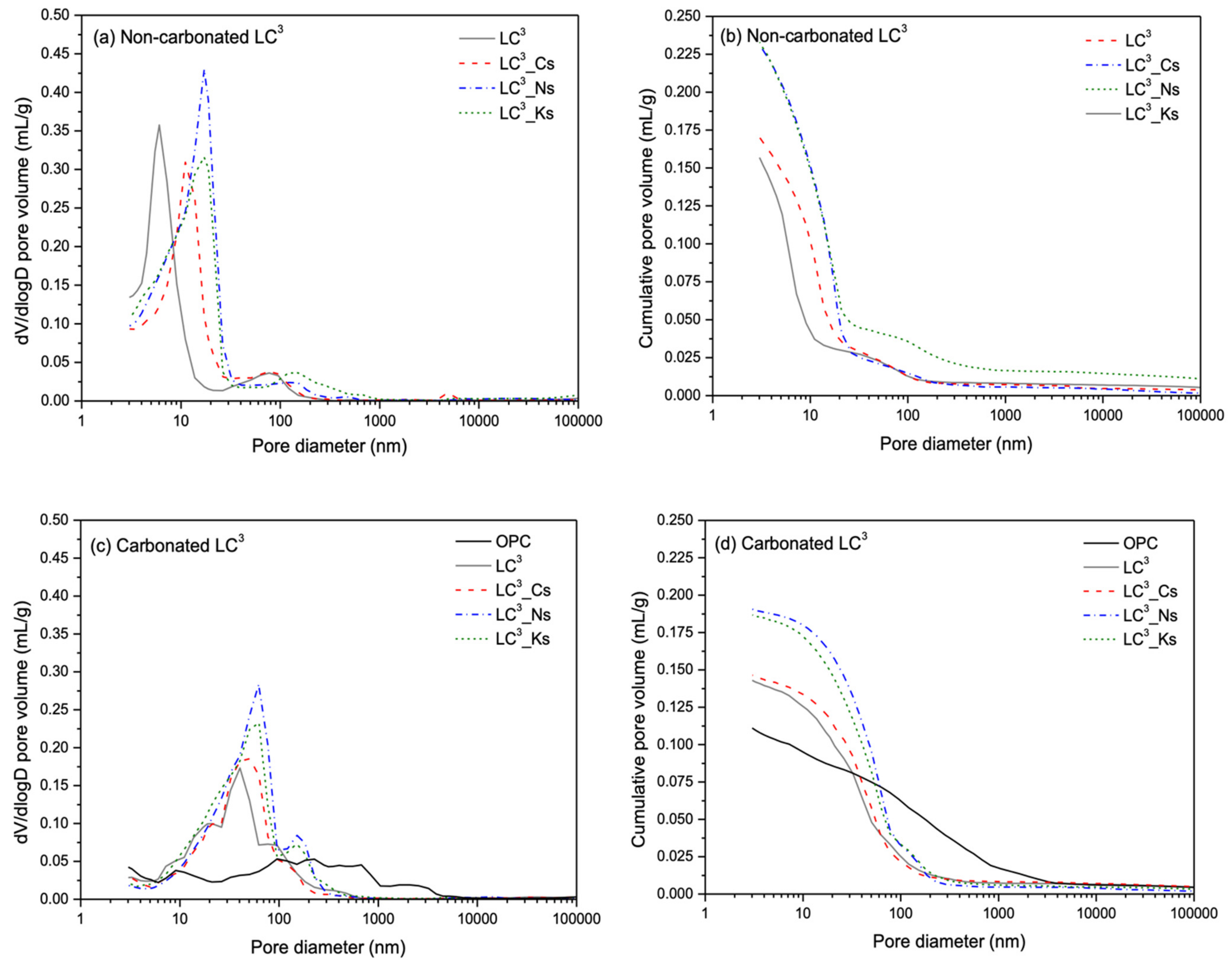
3.6. Pore Structure Alterations
3.7. Microstructure Analyses at the Carbonation Skin
4. Conclusions
- (1)
- The incorporation of sulfate salts considerably accelerates setting and strength gain of LC3 systems, but undermines its later age strength. The early strength acceleration in LC3 by sulfate salts is likely due to enhanced ettringite formation with supplemented sulfate; however, the reduced later age strength is likely due to the deleterious effects of alkali ions (i.e., Na+ and K+) on the diffusion-controlled OPC hydration process.
- (2)
- The carbonation resistance of LC3 systems is noticeably compromised with the incorporation of sodium or potassium sulfate salts, mainly due to the pore structure coarsening effect and reduced CO2-binding capacity.
- (3)
- In comparison to calcium sulfate, the negative effects of Na2SO4, and K2SO4 on carbonation resistance of LC3 are more significant. As such, limiting the alkali content in LC3 systems is recommended, concerning the durability requirement against carbonation-induced materials degradations.
- (4)
- Carbonation of OPC and LC3 pastes containing various alkalis leads to a considerable reduction of porosity and coarsening of pore structure, as revealed by MIP results.
- (5)
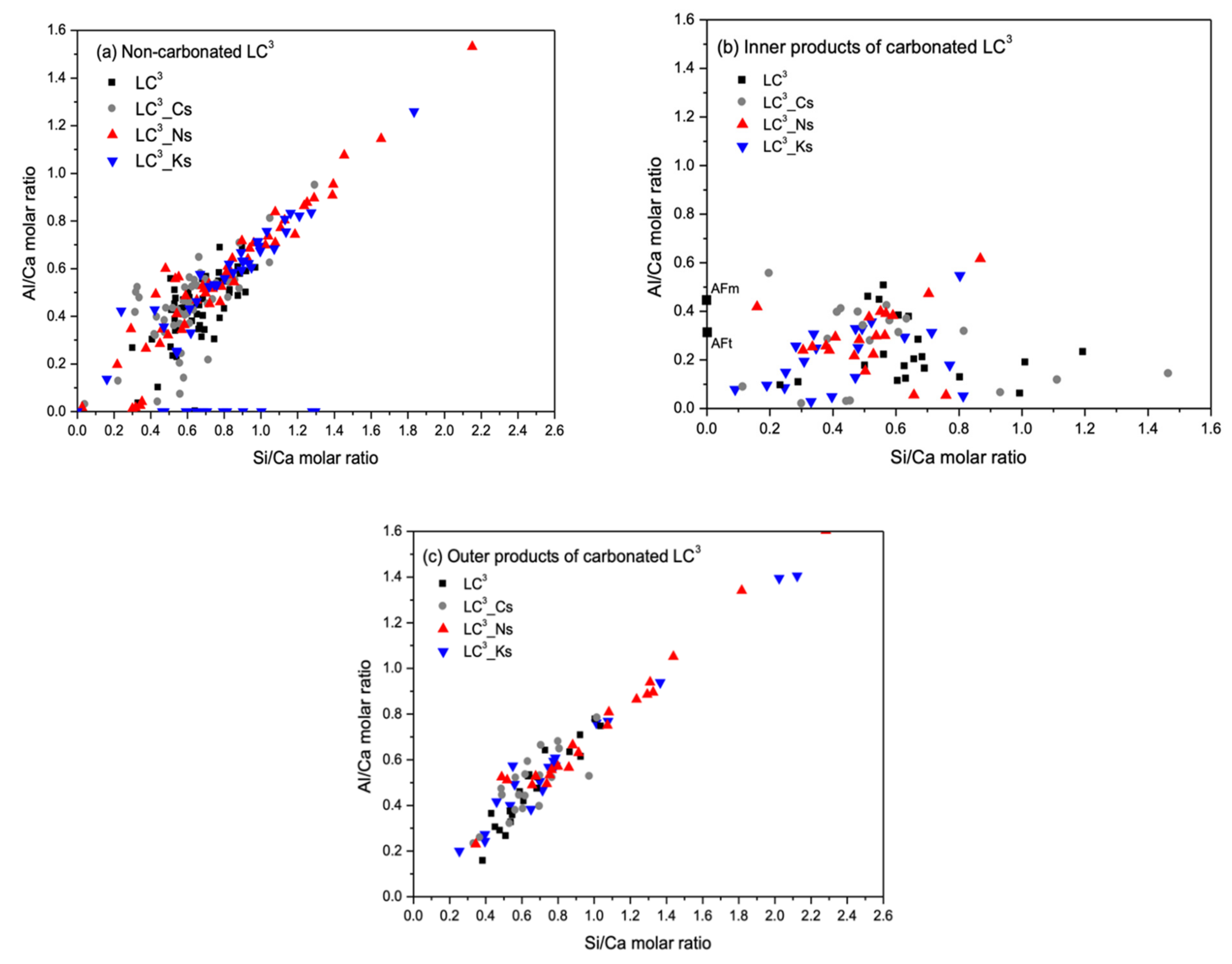
- Reference OPC and LC3 pastes show different microstructure characteristics at carbonation layers. Distinctive carbonated products (inner and outer products) can be identified in the microstructure of LC3 pastes, implying a highly heterogeneous distribution of hydrates phases prior to carbonation. The inner product is mainly composed of C-S-H and CH from hydration of OPC clinkers, while the outer product is composed of C-A-S-H and AFm phases from LS and MK hydration in an alkaline condition created by hydrating OPC. The carbonation of outer product results in considerable decalcification via calcium carbonate formation, but has little influence on the Al/Si ratio of the carbonated phases.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cancio Díaz, Y.; Sánchez Berriel, S.; Heierli, U.; Favier, A.R.; Sánchez Machado, I.R.; Scrivener, K.L.; Martirena Hernández, J.F.; Habert, G. Limestone calcined clay cement as a low-carbon solution to meet expanding cement demand in emerging economies. Dev. Eng. 2017, 2, 82–91. [Google Scholar] [CrossRef]
- Sharma, M.; Bishnoi, S.; Martirena, F.; Scrivener, K. Limestone calcined clay cement and concrete: A state-of-the-art review. Cem. Concr. Res. 2021, 149, 106564. [Google Scholar] [CrossRef]
- Antoni, M.; Rossen, J.; Martirena, F.; Scrivener, K. Cement substitution by a combination of metakaolin and limestone. Cem. Concr. Res. 2012, 42, 1579–1589. [Google Scholar] [CrossRef]
- Scrivener, K.; Martirena, F.; Bishnoi, S.; Maity, S. Calcined clay limestone cements (LC3). Cem. Concr. Res. 2018, 114, 49–56. [Google Scholar] [CrossRef]
- Nguyen, Q.D.; Khan, M.S.H.; Castel, A. Chloride Diffusion in Limestone Flash Calcined Clay Cement Concrete. ACI Mater. J. 2020, 117, 165–175. [Google Scholar] [CrossRef]
- Shi, Z.; Geiker, M.R.; De Weerdt, K.; Lothenbach, B.; Kaufmann, J.; Kunther, W.; Ferreiro, S.; Herfort, D.; Skibsted, J. Durability of Portland Cement Blends Including Calcined Clay and Limestone: Interactions with Sulfate, Chloride and Carbonate Ions. In Calcined Clays for Sustainable Concrete; Scrivener, K., Favier, A., Eds.; Springer: Singapore, 2015; pp. 133–141. [Google Scholar]
- Díaz, E.; González, R.; Rocha, D.; Alujas, A.; Martirena, F. Carbonation of concrete with low carbon cement LC3 exposed to dif-ferent environmental conditions. In Calcined Clays for Sustainable Concrete; Springer: Dordrecht, The Netherlands, 2018; pp. 141–146. [Google Scholar]
- Shi, Z.; Lothenbach, B.; Geiker, M.R.; Kaufmann, J.; Leemann, A.; Ferreiro, S.; Skibsted, J. Experimental studies and thermody-namic modeling of the carbonation of Portland cement, metakaolin and limestone mortars. Cem. Concr. Res. 2016, 88, 60–72. [Google Scholar] [CrossRef]
- Fu, C.; Fang, D.; Ye, H.; Huang, L.; Wang, J. Bond degradation of non-uniformly corroded steel rebars in concrete. Eng. Struct. 2021, 226, 111392. [Google Scholar] [CrossRef]
- Nguyen, Q.D.; Castel, A. Reinforcement corrosion in limestone flash calcined clay cement-based concrete. Cem. Concr. Res. 2020, 132, 106051. [Google Scholar] [CrossRef]
- Ye, H.; Jin, X.; Fu, C.; Jin, N.; Xu, Y.; Huang, T. Chloride penetration in concrete exposed to cyclic drying-wetting and carbonation. Constr. Build. Mater. 2016, 112, 457–463. [Google Scholar] [CrossRef]
- Ye, H.; Radlińska, A. Effect of Alkalis on Cementitious Materials:Understanding the Relationship between Composition, Structure, and Volume Change Mechanism. J. Adv. Concr. Technol. 2017, 15, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Jawed, I.; Skalny, J. Alkalies in cement: A review: II. Effects of alkalies on hydration and performance of Portland cement. Cem. Concr. Res. 1978, 8, 37–51. [Google Scholar] [CrossRef]
- Maslehuddin, M.; Page, C.L. Rasheeduzzafar Effect of Temperature and Salt Contamination on Carbonation of Cements. J. Mater. Civ. Eng. 1996, 8, 63–69. [Google Scholar] [CrossRef]
- Dow, C.; Glasser, F. Calcium carbonate efflorescence on Portland cement and building materials. Cem. Concr. Res. 2003, 33, 147–154. [Google Scholar] [CrossRef]
- Krishnan, S.; Singh, A.; Bishnoi, S. Impact of Alkali Salts on the Hydration of Ordinary Portland Cement and Limestone–Calcined Clay Cement. J. Mater. Civ. Eng. 2021, 33, 04021223. [Google Scholar] [CrossRef]
- Schulze, S.E.; Rickert, J. Suitability of natural calcined clays as supplementary cementitious material. Cem. Concr. Compos. 2019, 95, 92–97. [Google Scholar] [CrossRef]
- Rajabipour, F.; Giannini, E.; Dunant, C.; Ideker, J.; Thomas, M.D. Alkali–silica reaction: Current understanding of the reaction mechanisms and the knowledge gaps. Cem. Concr. Res. 2015, 76, 130–146. [Google Scholar] [CrossRef]
- Zunino, F.; Scrivener, K. Factors influencing the sulfate balance in pure phase C3S/C3A systems. Cem. Concr. Res. 2020, 133, 106085. [Google Scholar] [CrossRef]
- ASTM Standard C191. Standard Test Method for Time of Setting of Hydraulic Cement by Vicat Needle; ASTM International: West Conshohocken, PA, USA, 2019. [Google Scholar]
- Hidalgo, A.; Domingo, C.; García-González, C.A.; Petit, S.; Andrade, C.; Alonso, C. Microstructural changes induced in Portland cement-based materials due to natural and supercritical carbonation. J. Mater. Sci. 2008, 43, 3101–3111. [Google Scholar] [CrossRef]
- Ye, H.; Cai, R.; Tian, Z. Natural carbonation-induced phase and molecular evolution of alkali-activated slag: Effect of activator composition and curing temperature. Constr. Build. Mater. 2020, 248, 118726. [Google Scholar] [CrossRef]
- Choi, J.-I.; Lee, Y.; Kim, Y.Y.; Lee, B.Y. Image-processing technique to detect carbonation regions of concrete sprayed with a phenolphthalein solution. Constr. Build. Mater. 2017, 154, 451–461. [Google Scholar] [CrossRef]
- Scrivener, K.; Snellings, R.; Lothenbach, B. A Practical Guide to Microstructural Analysis of Cementitious Materials; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Aligizaki, K.K. Pore Structure of Cement-Based Materials: Testing, Interpretation and Requirements; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Gallé, C. Effect of drying on cement-based materials pore structure as identified by mercury intrusion porosimetry: A comparative study between oven-, vacuum-, and freeze-drying. Cem. Concr. Res. 2001, 31, 1467–1477. [Google Scholar] [CrossRef]
- Dhandapani, Y.; Sakthivel, T.; Santhanam, M.; Gettu, R.; Pillai, R.G. Mechanical properties and durability performance of con-cretes with Limestone Calcined Clay Cement (LC3). Cem. Concr. Res. 2018, 107, 136–151. [Google Scholar] [CrossRef]
- Wang, L.; Rehman, N.U.; Curosu, I.; Zhu, Z.; Beigh, M.A.B.; Liebscher, M.; Chen, L.; Tsang, D.C.; Hempel, S.; Mechtcherine, V. On the use of limestone calcined clay cement (LC3) in high-strength strain-hardening cement-based composites (HS-SHCC). Cem. Concr. Res. 2021, 144, 106421. [Google Scholar] [CrossRef]
- Emmanuel, A.C.; Haldar, P.; Maity, S.; Bishnoi, S. Second pilot production of limestone calcined clay cement in India: The expe-rience. Indian Concr. J 2016, 90, 57–64. [Google Scholar]
- Ye, H. Autogenous formation and smart behaviors of nitrite- and nitrate-intercalated layered double hydroxides (LDHs) in Port-land cement-metakaolin-dolomite blends. Cem. Concr. Res. 2021, 139, 106267. [Google Scholar] [CrossRef]
- Mota, B.; Matschei, T.; Scrivener, K. Impact of NaOH and Na2SO4 on the kinetics and microstructural development of white cement hydration. Cem. Concr. Res. 2018, 108, 172–185. [Google Scholar] [CrossRef]
- Ye, H.; Radlińska, A.; Neves, J. Drying and carbonation shrinkage of cement paste containing alkalis. Mater. Struct. 2017, 50, 50. [Google Scholar] [CrossRef]
- Tironi, A.; Scian, A.N.; Irassar, E.F. Blended Cements with Limestone Filler and Kaolinitic Calcined Clay: Filler and Pozzolanic Effects. J. Mater. Civ. Eng. 2017, 29, 04017116. [Google Scholar] [CrossRef]
- Huang, L.; Yan, P. Effect of alkali content in cement on its hydration kinetics and mechanical properties. Constr. Build. Mater. 2019, 228, 116833. [Google Scholar] [CrossRef]
- Zajac, M.; Skocek, J.; Lothenbach, B.; Mohsen, B.H. Late hydration kinetics: Indications from thermodynamic analysis of pore solution data. Cem. Concr. Res. 2020, 129, 105975. [Google Scholar] [CrossRef]
- Zunino, F.; Scrivener, K. The reaction between metakaolin and limestone and its effect in porosity refinement and mechanical properties. Cem. Concr. Res. 2021, 140, 106307. [Google Scholar] [CrossRef]
- Matschei, T.; Lothenbach, B.; Glasser, F. The AFm phase in Portland cement. Cem. Concr. Res. 2007, 37, 118–130. [Google Scholar] [CrossRef]
- Lodeiro, I.G.; Macphee, D.E.; Palomo, A.; Fernández-Jiménez, A. Effect of alkalis on fresh C–S–H gels. FTIR analysis. Cem. Concr. Res. 2009, 39, 147–153. [Google Scholar] [CrossRef]
- Yu, P.; Kirkpatrick, R.J.; Poe, B.; McMillan, P.F.; Cong, X. Structure of calcium silicate hydrate (C-S-H): Near-, Mid-, and Far-infrared spectroscopy. J. Am. Ceram. Soc. 1999, 82, 742–748. [Google Scholar] [CrossRef]
- Ghosh, S.N.; Handoo, S.K. Infrared and Raman spectral studies in cement and concrete (review). Cem. Concr. Res. 1980, 10, 771–782. [Google Scholar] [CrossRef]
- Hughes, T.L.; Methven, C.M.; Jones, T.G.; Pelham, S.E.; Fletcher, P.; Hall, C. Determining cement composition by Fourier transform infrared spectroscopy. Adv. Cem. Based Mater. 1995, 2, 91–104. [Google Scholar] [CrossRef]
- Villain, G.; Thiery, M.; Platret, G. Measurement methods of carbonation profiles in concrete: Thermogravimetry, chemical analysis and gammadensimetry. Cem. Concr. Res. 2007, 37, 1182–1192. [Google Scholar] [CrossRef]
- Chang, C.-F.; Chen, J.-W. The experimental investigation of concrete carbonation depth. Cem. Concr. Res. 2006, 36, 1760–1767. [Google Scholar] [CrossRef]
- Shah, V.; Scrivener, K.; Bhattacharjee, B.; Bishnoi, S. Changes in microstructure characteristics of cement paste on carbonation. Cem. Concr. Res. 2018, 109, 184–197. [Google Scholar] [CrossRef]
- Dhandapani, Y.; Santhanam, M. Assessment of pore structure evolution in the limestone calcined clay cementitious system and its implications for performance. Cem. Concr. Compos. 2017, 84, 36–47. [Google Scholar] [CrossRef]
- Cai, R.; Tian, Z.; Ye, H.; He, Z.; Tang, S. The role of metakaolin in pore structure evolution of Portland cement pastes revealed by an impedance approach. Cem. Concr. Compos. 2021, 119, 103999. [Google Scholar] [CrossRef]
- Ngala, V.; Page, C. Effects of carbonation on pore structure and diffusional properties of hydrated cement pastes. Cem. Concr. Res. 1997, 27, 995–1007. [Google Scholar] [CrossRef]
- Chindaprasirt, P.; Rukzon, S. Pore Structure Changes of Blended Cement Pastes Containing Fly Ash, Rice Husk Ash, and Palm Oil Fuel Ash Caused by Carbonation. J. Mater. Civ. Eng. 2009, 21, 666–671. [Google Scholar] [CrossRef]
- Kutchko, B.G.; Strazisar, B.R.; Dzombak, D.A.; Lowry, G.V.; Thaulow, N. Degradation of Well Cement by CO2 under Geologic Sequestration Conditions. Environ. Sci. Technol. 2007, 41, 4787–4792. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxide Composition (wt.%) | OPC | Metakaolin | Limestone |
|---|---|---|---|
| CaO | 66.48 | - | 54.57 |
| SiO2 | 20.17 | 55.31 | - |
| Al2O3 | 4.01 | 43.55 | 0.08 |
| MgO | 0.85 | - | 0.95 |
| SO3 | 5.13 | - | 0.07 |
| Fe2O3 | 2.77 | 0.44 | 0.03 |
| Na2O | - | - | - |
| K2O | 0.58 | - | - |
| MnO | - | - | - |
| TiO2 | - | 0.7 | - |
| LOI at 900 °C | 1.43 | 1.15 | 45.43 |
| Mixture ID | OPC (%) | MK (%) | LS (%) | QP (%) | CaSO4 (%) | Na2SO4 (%) | K2SO4 (%) | w/p a |
|---|---|---|---|---|---|---|---|---|
| OPC | 100.0 | - | - | - | - | - | - | 0.4 |
| OPC_45Q | 55.0 | - | - | 45.0 | - | - | - | |
| LC3 | 55.0 | 30.0 | 15.0 | - | - | - | - | |
| LC3_Cs | 52.5 | 30.0 | 15.0 | - | 2.5 | - | - | |
| LC3_Ns | 52.5 | 30.0 | 15.0 | - | - | 2.5 | - | |
| LC3_Ks | 52.5 | 30.0 | 15.0 | - | - | - | 2.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Ye, H. Influence of Alkalis on Natural Carbonation of Limestone Calcined Clay Cement Pastes. Sustainability 2021, 13, 12833. https://doi.org/10.3390/su132212833
Li R, Ye H. Influence of Alkalis on Natural Carbonation of Limestone Calcined Clay Cement Pastes. Sustainability. 2021; 13(22):12833. https://doi.org/10.3390/su132212833
Chicago/Turabian StyleLi, Ruoying, and Hailong Ye. 2021. "Influence of Alkalis on Natural Carbonation of Limestone Calcined Clay Cement Pastes" Sustainability 13, no. 22: 12833. https://doi.org/10.3390/su132212833
APA StyleLi, R., & Ye, H. (2021). Influence of Alkalis on Natural Carbonation of Limestone Calcined Clay Cement Pastes. Sustainability, 13(22), 12833. https://doi.org/10.3390/su132212833