
A Novel MSH6 Gene Variant in a Lynch Syndrome Patient with Lipomas



{kind=link}
{kind=link}
Abstract
1. Introduction
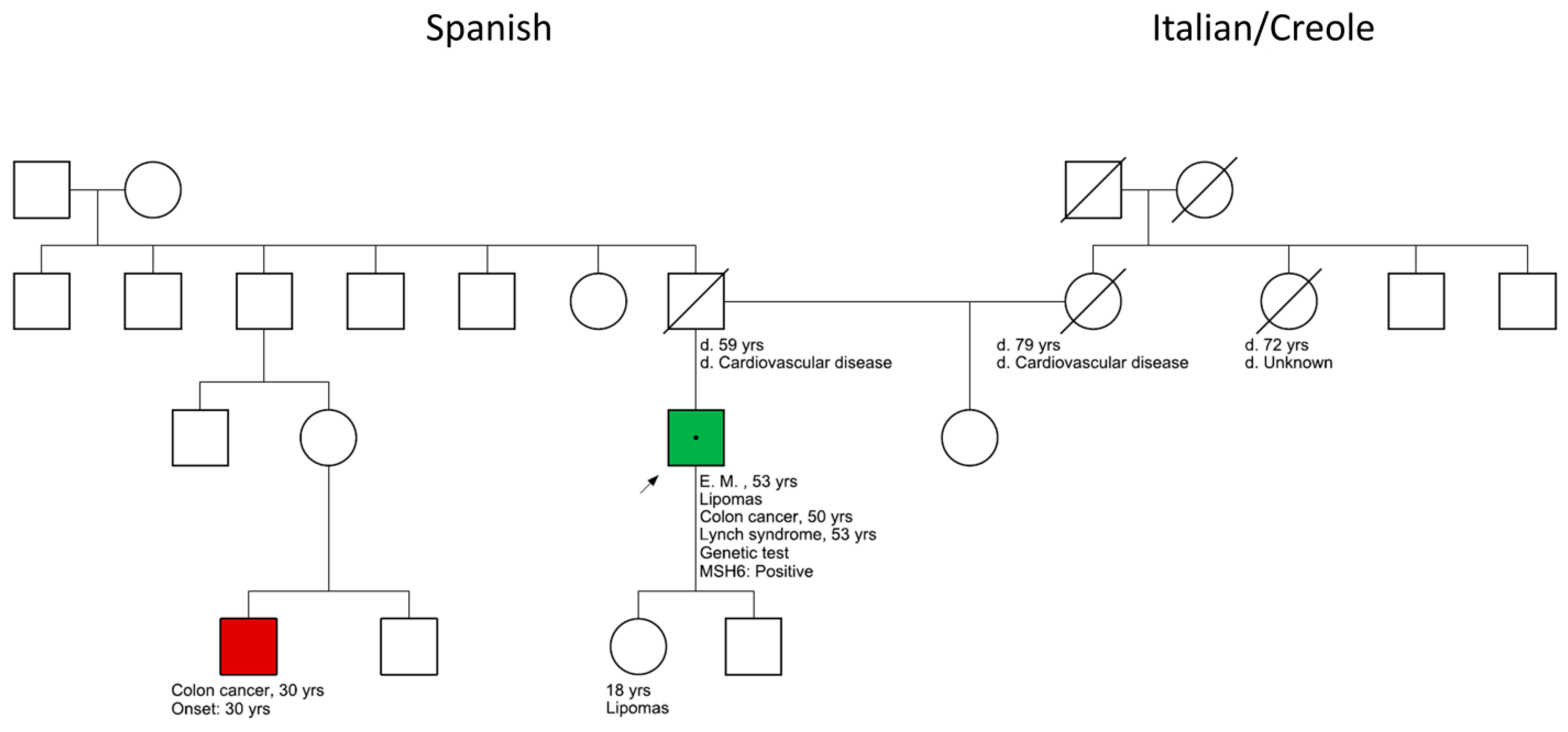
2. Case Presentation
2.1. Tumor Biopsy
2.2. PREMM5 Risk Prediction
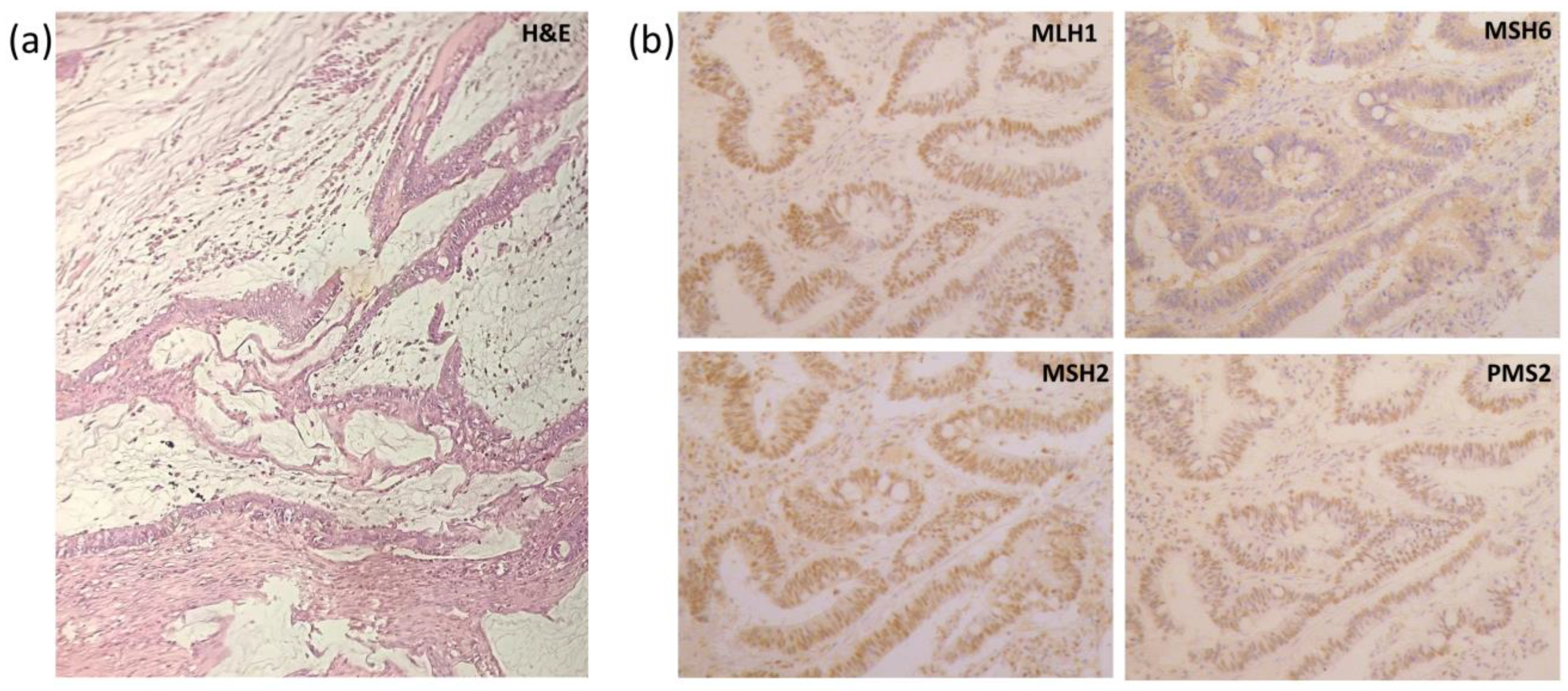
2.3. Histological Results

2.4. Genetic Panel Results
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.H.; Robinson, P.S.; Coorens, T.H.H.; Yan, H.H.N.; Olafsson, S.; Lee-Six, H.; Sanders, M.A.; Siu, H.C.; Hewinson, J.; Yue, S.S.K.; et al. Mutational landscape of normal epithelial cells in Lynch Syndrome patients. Nat. Commun. 2022, 13, 2710. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, J.; Chan, P.; Hogden, E.; Tiernan, G.; Morrow, A.; Kang, Y.-J.; He, E.; Venchiarutti, R.; Titterton, L.; Sankey, L.; et al. Lynch syndrome testing of colorectal cancer patients in a high-income country with universal healthcare: A retrospective study of current practice and gaps in seven australian hospitals. Hered. Cancer Clin. Pract. 2022, 20, 18. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Huang, T.; Lin, X.; Zhang, Y.; Yang, X.; Ye, Z.; Ren, X.; Teng, L.; Li, J.; Kong, M.; et al. Long-Term Survival of a Lynch Syndrome Patient with Eight Primary Tumors: A Case Report. Front. Oncol. 2022, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yurgelun, M.B.; Uno, H.; Furniss, C.S.; Ukaegbu, C.; Horiguchi, M.; Yussuf, A.; LaDuca, H.; Chittenden, A.; Garber, J.E.; Syngal, S. Development and Validation of the PREMMplus Model for Multigene Hereditary Cancer Risk Assessment. J. Clin. Oncol. 2022, 40, 4083–4094. [Google Scholar] [CrossRef] [PubMed]
- Kastrinos, F.; Uno, H.; Ukaegbu, C.; Alvero, C.; McFarland, A.; Yurgelun, M.B.; Kulke, M.H.; Schrag, D.; Meyerhardt, J.A.; Fuchs, C.S.; et al. Development and Validation of the PREMM5 Model for Comprehensive Risk Assessment of Lynch Syndrome. J. Clin. Oncol. 2017, 35, 2165–2172. [Google Scholar] [CrossRef] [PubMed]
- Aoun, R.J.N.; Kalady, M.F. The importance of genetics for timing and extent of surgery in inherited colorectal cancer syndromes. Surg. Oncol. 2022, 43, 101765. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhao, Z.; Dong, L.; Jia, J.; Su, K.; Bai, H.; Wang, J. Case Report: A New Subtype of Lynch Syndrome Associated with MSH2 c.1024_1026 Identified in a Chinese Family. Front. Med. 2022, 9, 811368. [Google Scholar] [CrossRef] [PubMed]
- Einarsson, H.; Runarsdottir, J.R.; Tryggvason, T.; Snaebjornsson, P.; Smaradottir, A.; Stefansdottir, V.; Thoroddsen, A.; Arngrimsson, R.; Jonasson, J.G.; Haraldsdottir, S. Universal tumor screening in a population with MSH6- and PMS2-associated Lynch syndrome. Genet. Med. 2022, 24, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Kamburova, Z.B.; Popovska, S.L.; Kovacheva, K.S.; Petrov, K.T.; Nikolova, S.E. Familial Lynch syndrome with early age of onset and confirmed splice site mutation in MSH2: A case report. Biomed. Rep. 2022, 16, 39. [Google Scholar] [CrossRef] [PubMed]
- López, V.H.Á.; Lima, M.S. Muir-Torre Syndrome with Loss of Expression msh2 and msh6: A Case Report. Dermatología 2021, 19, 24–27. [Google Scholar]
- Burris, C.K.; Rodriguez, M.E.; Raven, M.L.; Reddy, D.N.; Xu, Y.G.; Wiggs, J.L.; Potter, H.D.; Albert, D.M. Muir-Torre Syndrome: The Importance of a Detailed Family History. Case Rep. Ophthalmol. 2019, 10, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Flaum, N.; Crosbie, E.J.; Edmondson, R.; Woodward, E.R.; Lalloo, F.; Smith, M.J.; Schlecht, H.; Evans, D.G. High detection rate from genetic testing in BRCA-negative women with familial epithelial ovarian cancer. Genet. Med. 2022, 24, 2578–2586. [Google Scholar] [CrossRef] [PubMed]
- Navarro García, M.; Grau, E.; Munté, E.; Solanes, A.; Pineda, M.; Dueñas, N.; Carrato, C.; Teulé, À.; Salinas, M.; Iglesias, S.; et al. Cáncer de tracto urotelial alto en Síndrome de Lynch: La experiencia del Institut Català d’Oncologia (ICO). In Proceedings of the Sociedad Española de Oncología Médica (SEOM) 2020, Virtual, 19–23 October 2020. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giannoni, A.P.; Sevic, I.; Parenti, F.; Alaniz, L. A Novel MSH6 Gene Variant in a Lynch Syndrome Patient with Lipomas. Clin. Pract. 2023, 13, 515-519. https://doi.org/10.3390/clinpract13020047
Giannoni AP, Sevic I, Parenti F, Alaniz L. A Novel MSH6 Gene Variant in a Lynch Syndrome Patient with Lipomas. Clinics and Practice. 2023; 13(2):515-519. https://doi.org/10.3390/clinpract13020047
Chicago/Turabian StyleGiannoni, Ana Paula, Ina Sevic, Fernanda Parenti, and Laura Alaniz. 2023. "A Novel MSH6 Gene Variant in a Lynch Syndrome Patient with Lipomas" Clinics and Practice 13, no. 2: 515-519. https://doi.org/10.3390/clinpract13020047
APA StyleGiannoni, A. P., Sevic, I., Parenti, F., & Alaniz, L. (2023). A Novel MSH6 Gene Variant in a Lynch Syndrome Patient with Lipomas. Clinics and Practice, 13(2), 515-519. https://doi.org/10.3390/clinpract13020047