
Epigenetic Mechanisms of Endocrine-Disrupting Chemicals in Breast Cancer and Their Impact on Dietary Intake


Abstract
:1. Introduction
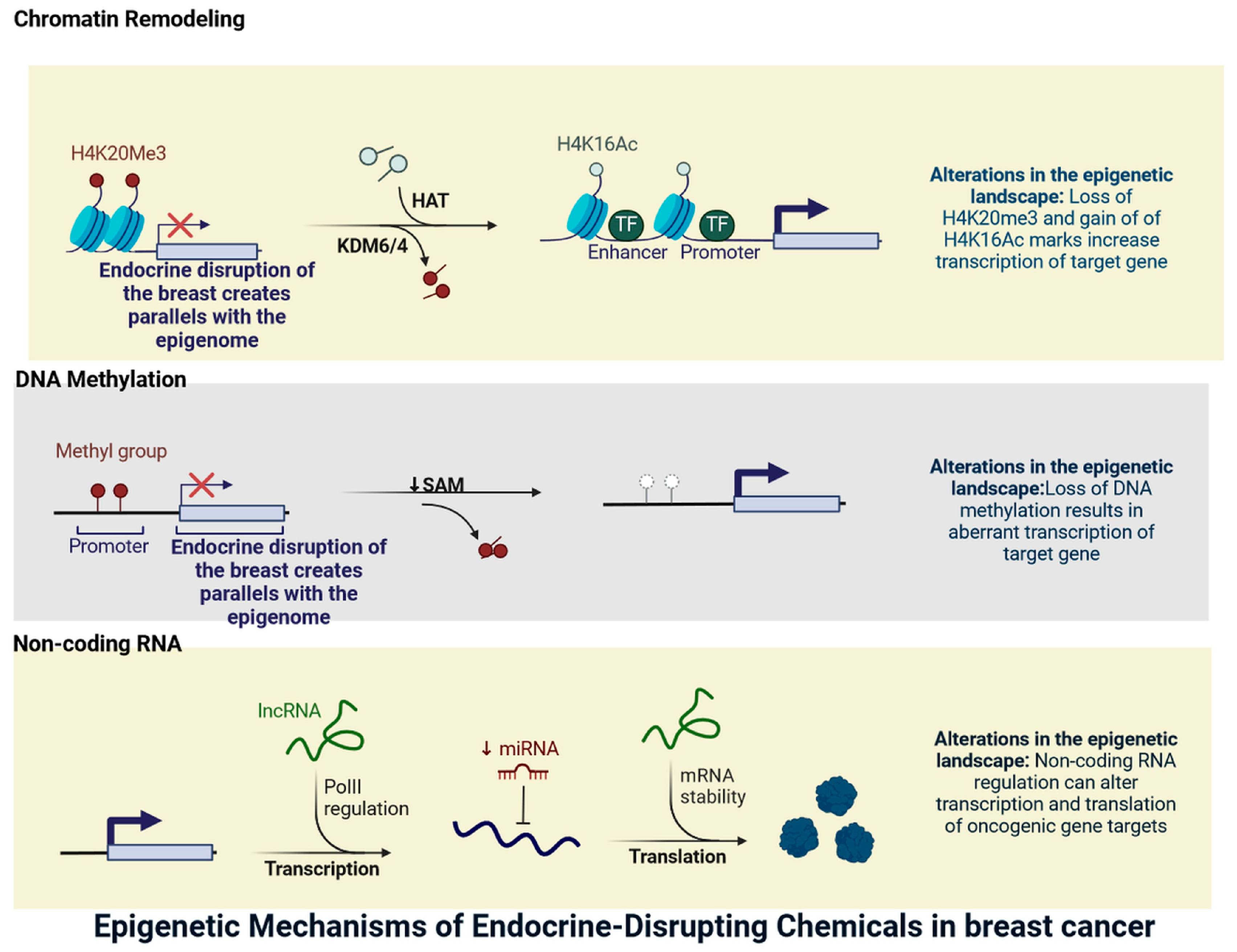
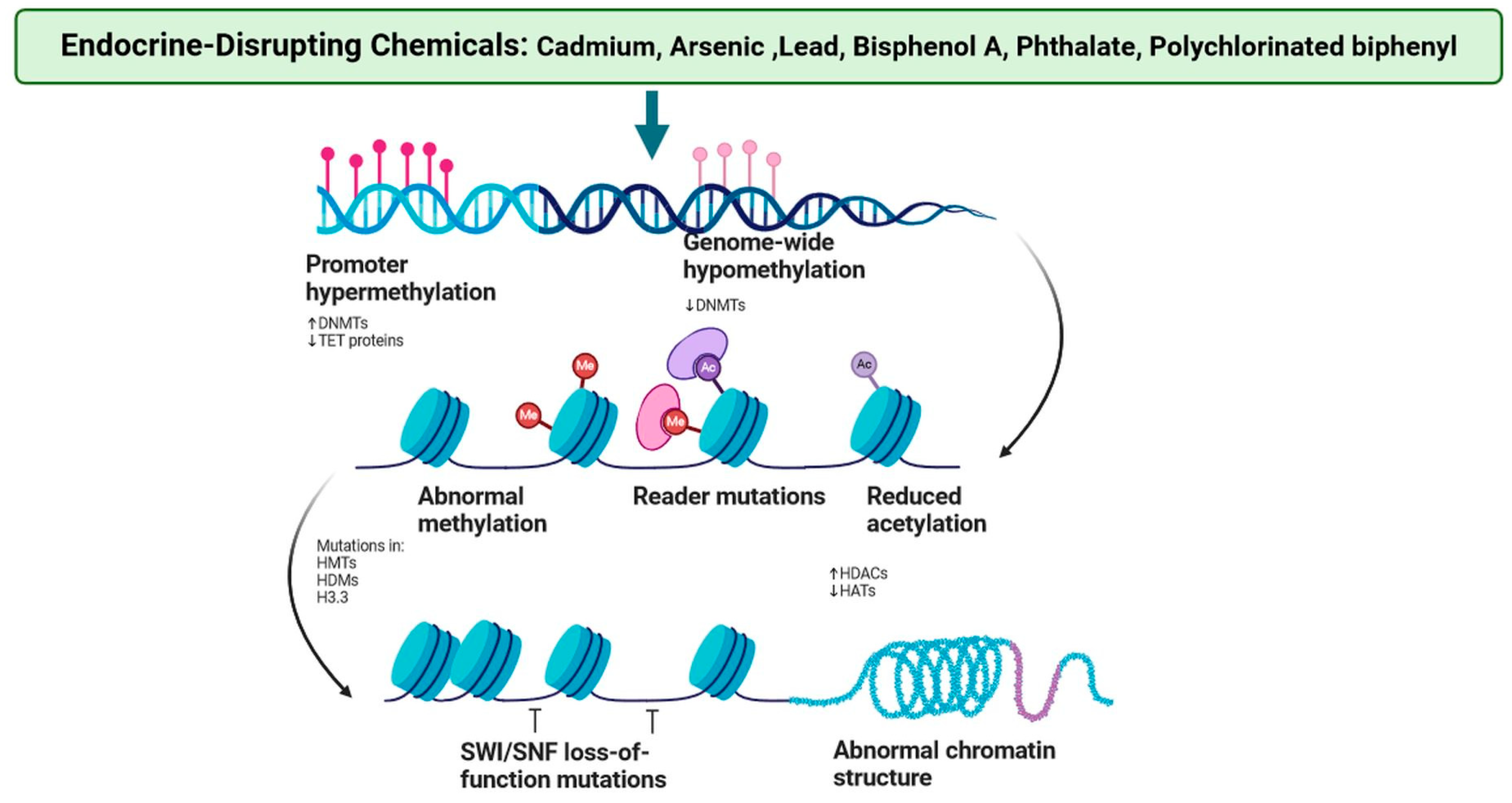
2. Endocrine Disruption in Breast Cancer Parallels the Epigenome
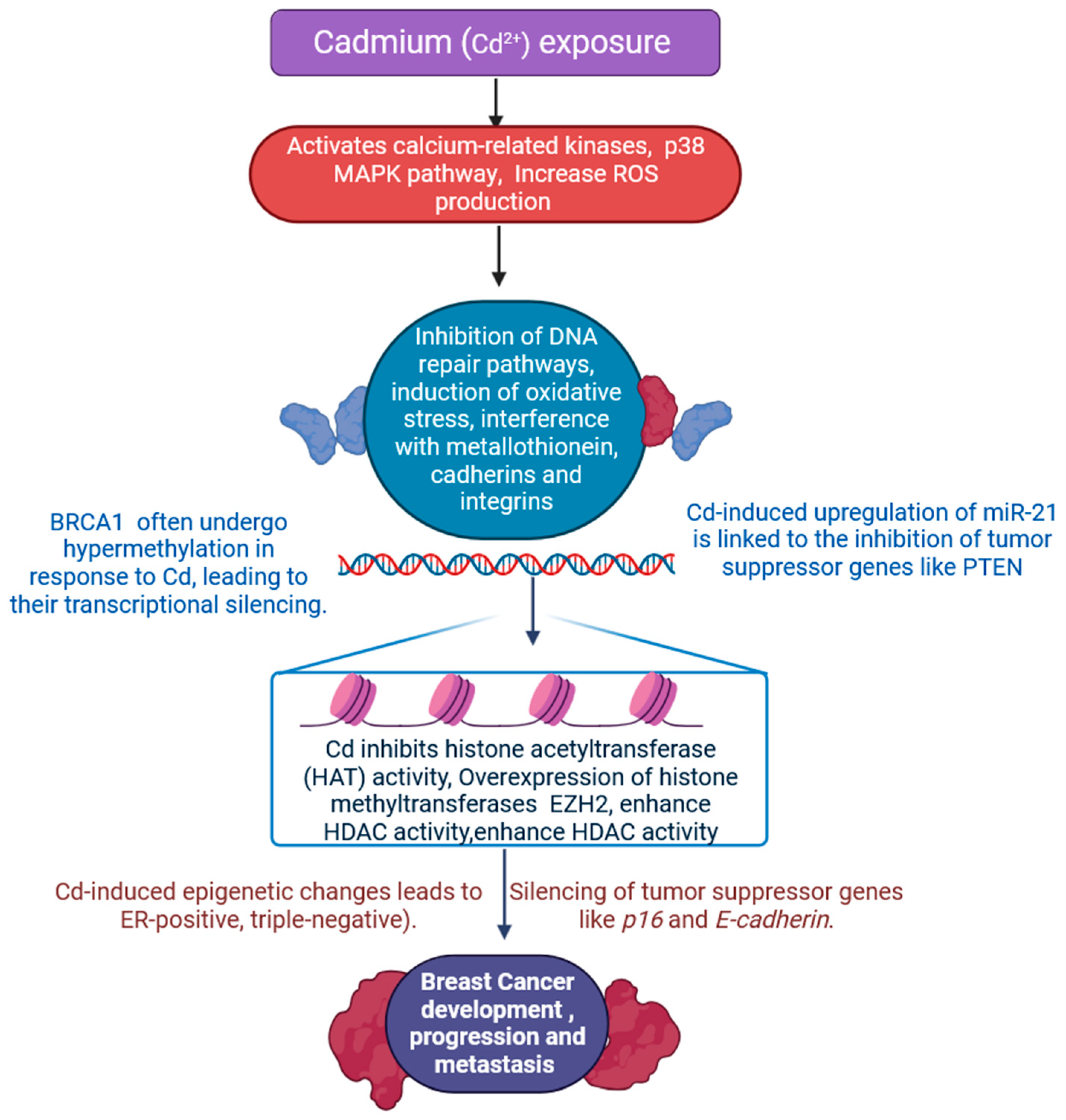
2.1. Cadmium
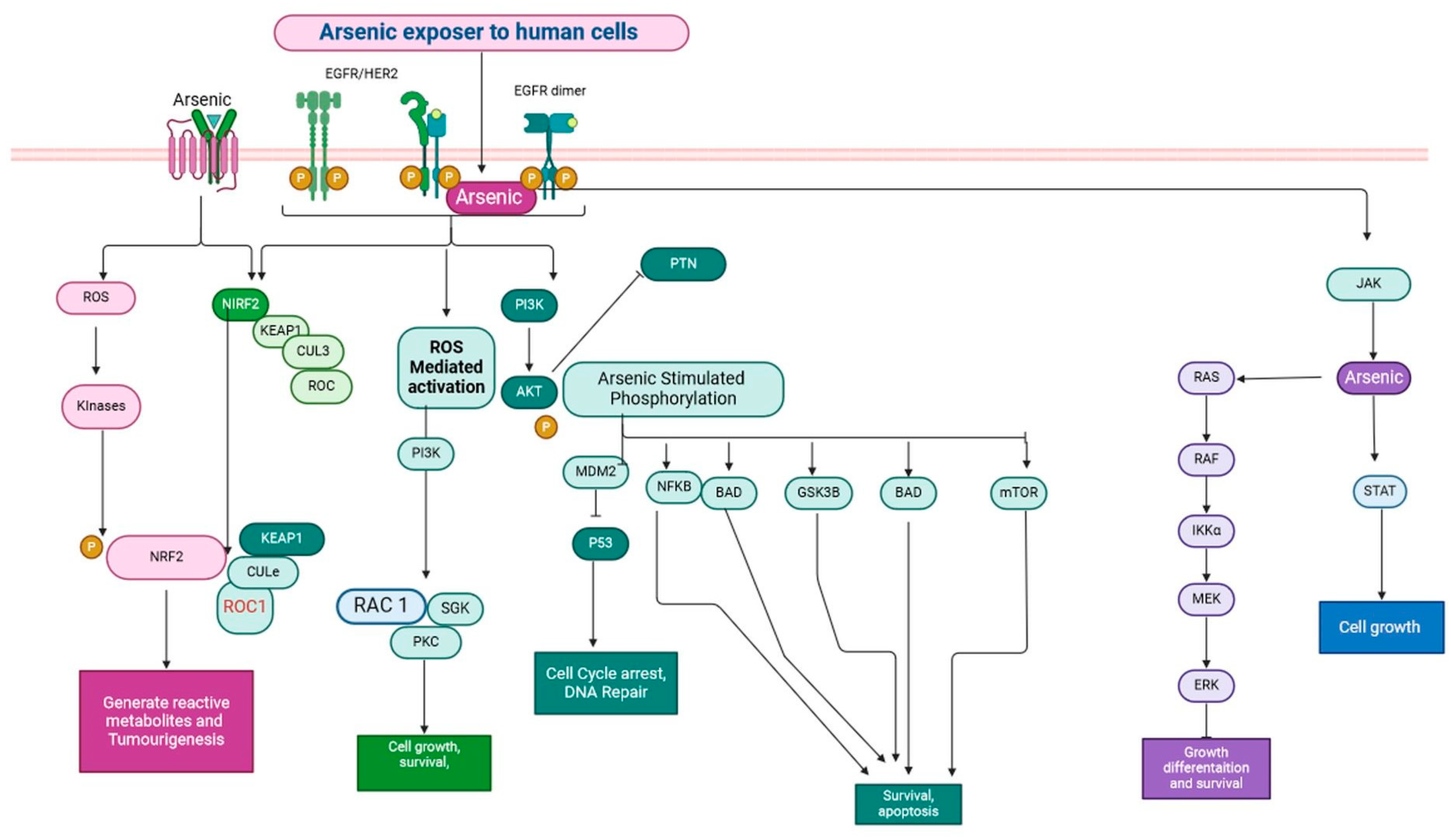
2.2. Arsenic
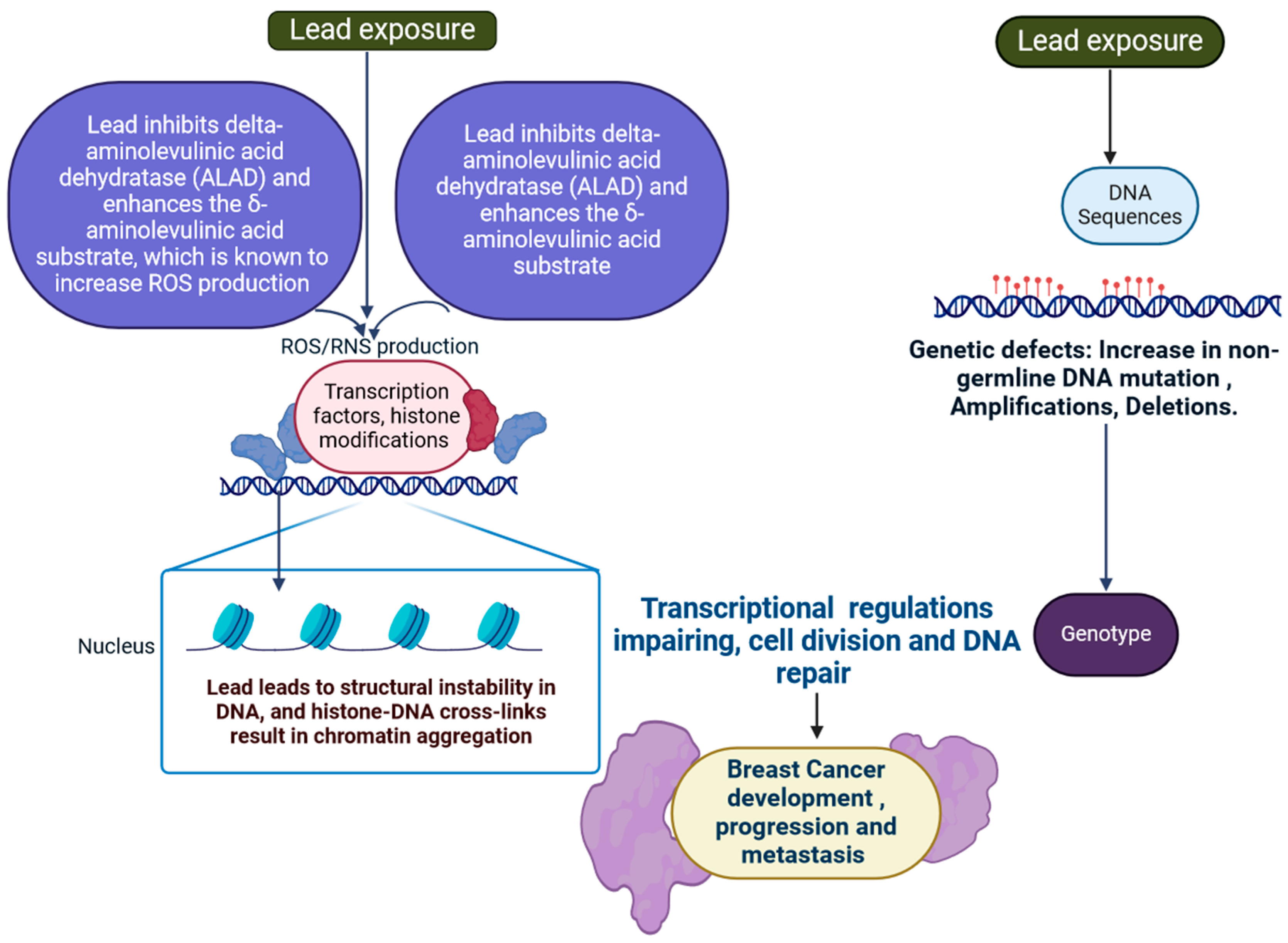
2.3. Lead
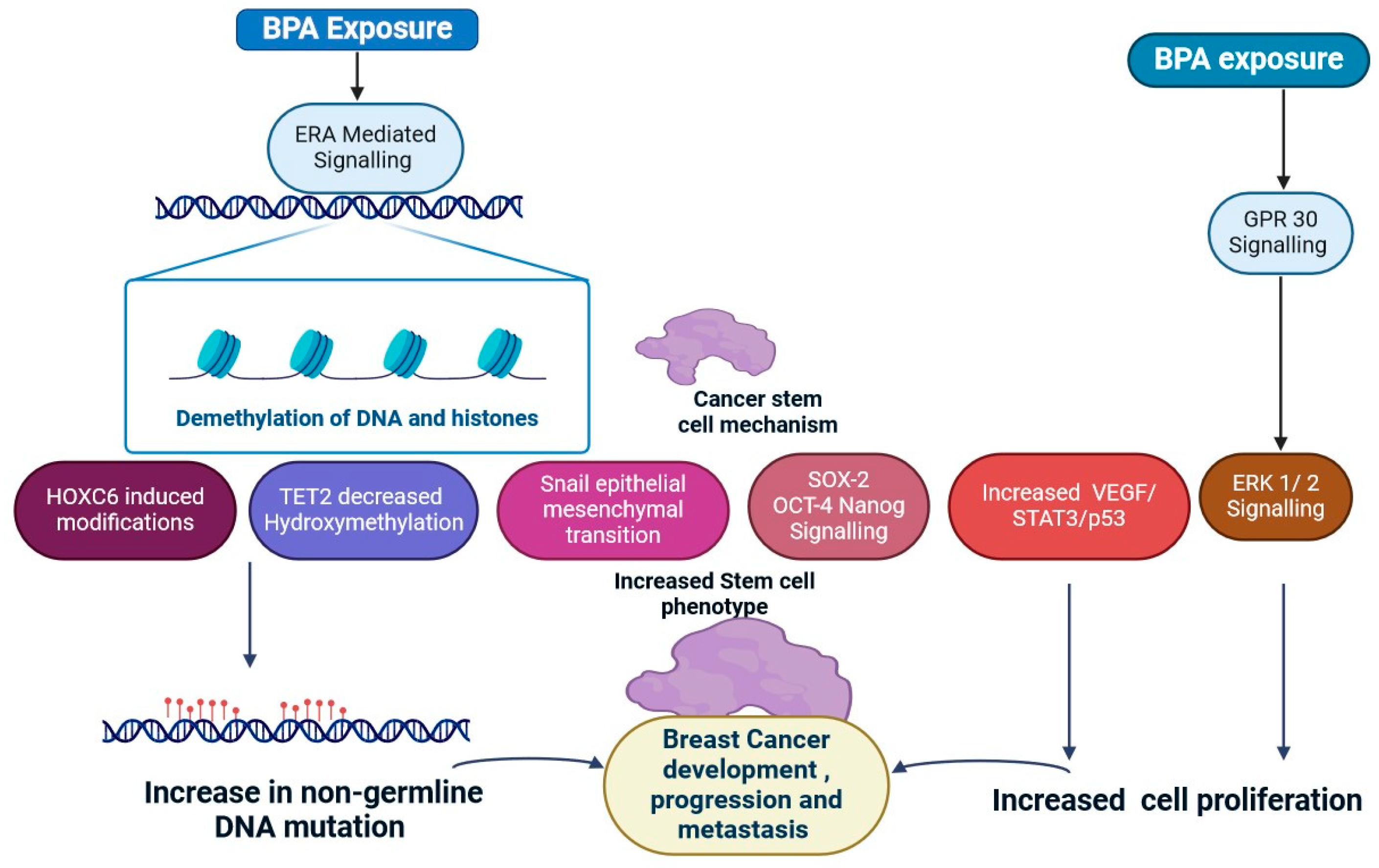
2.4. Bisphenol A
2.5. Phthalates
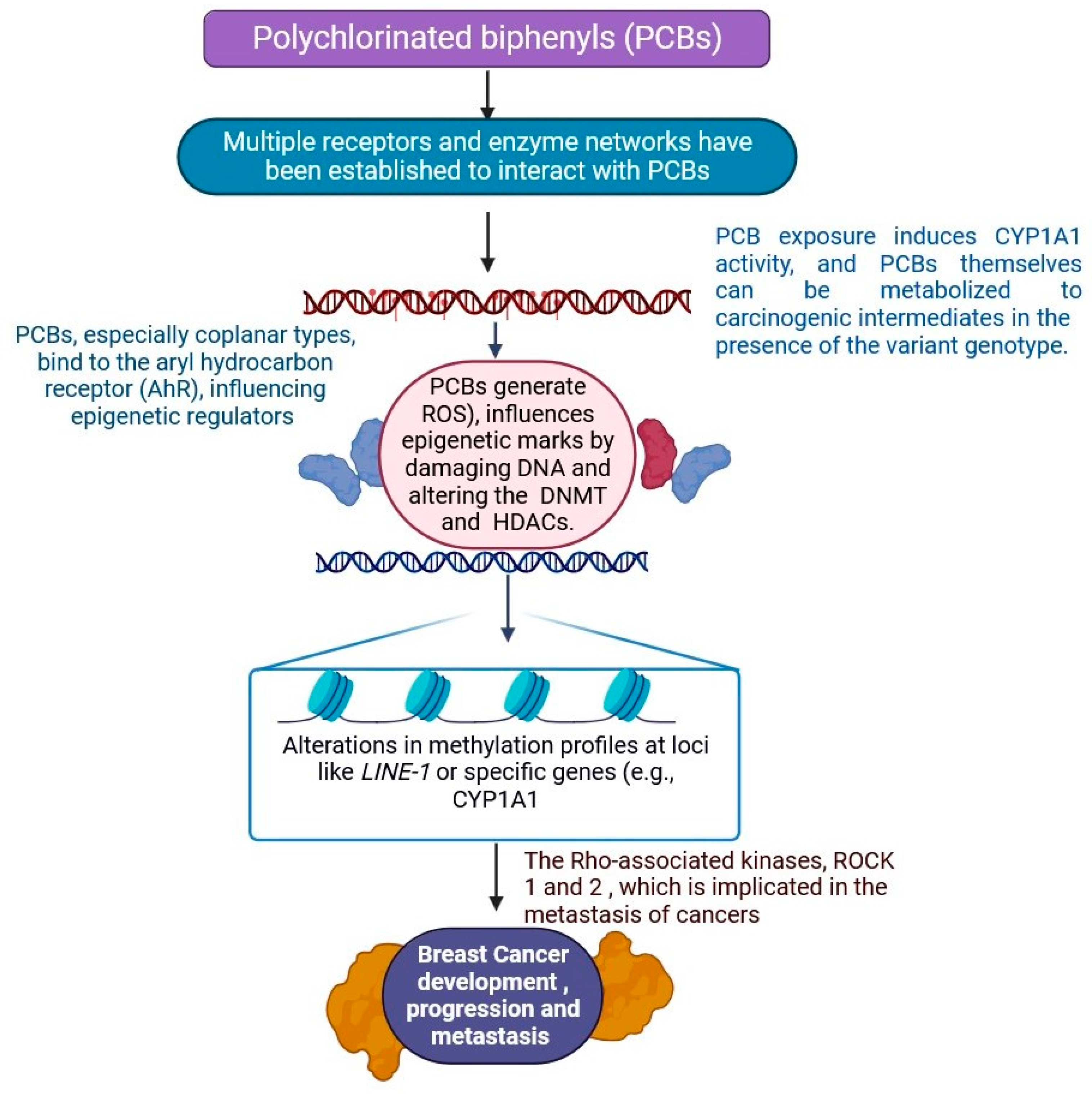
2.6. Polychlorinated Biphenyls
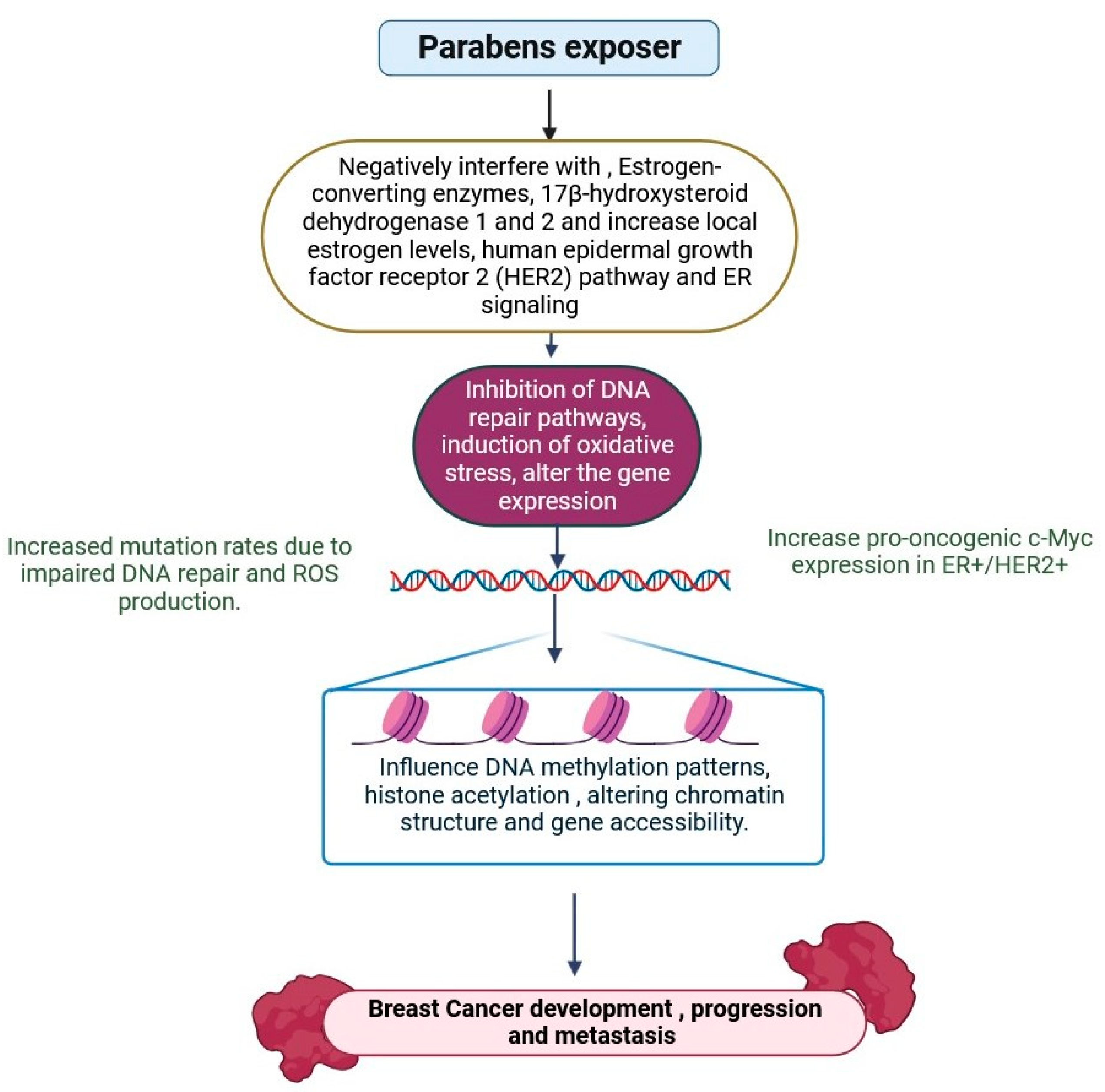
2.7. Parabens
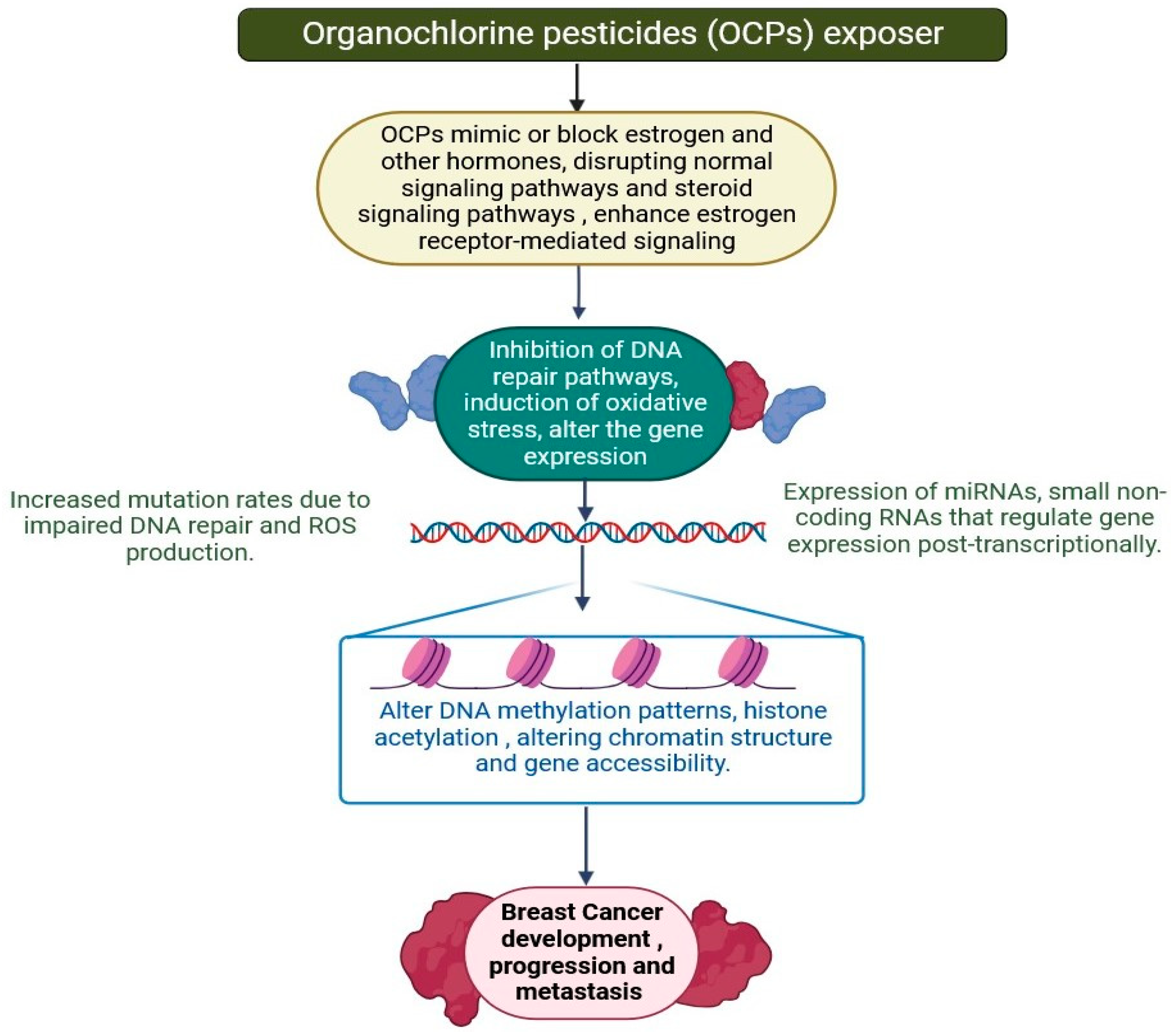
2.8. Organochlorine
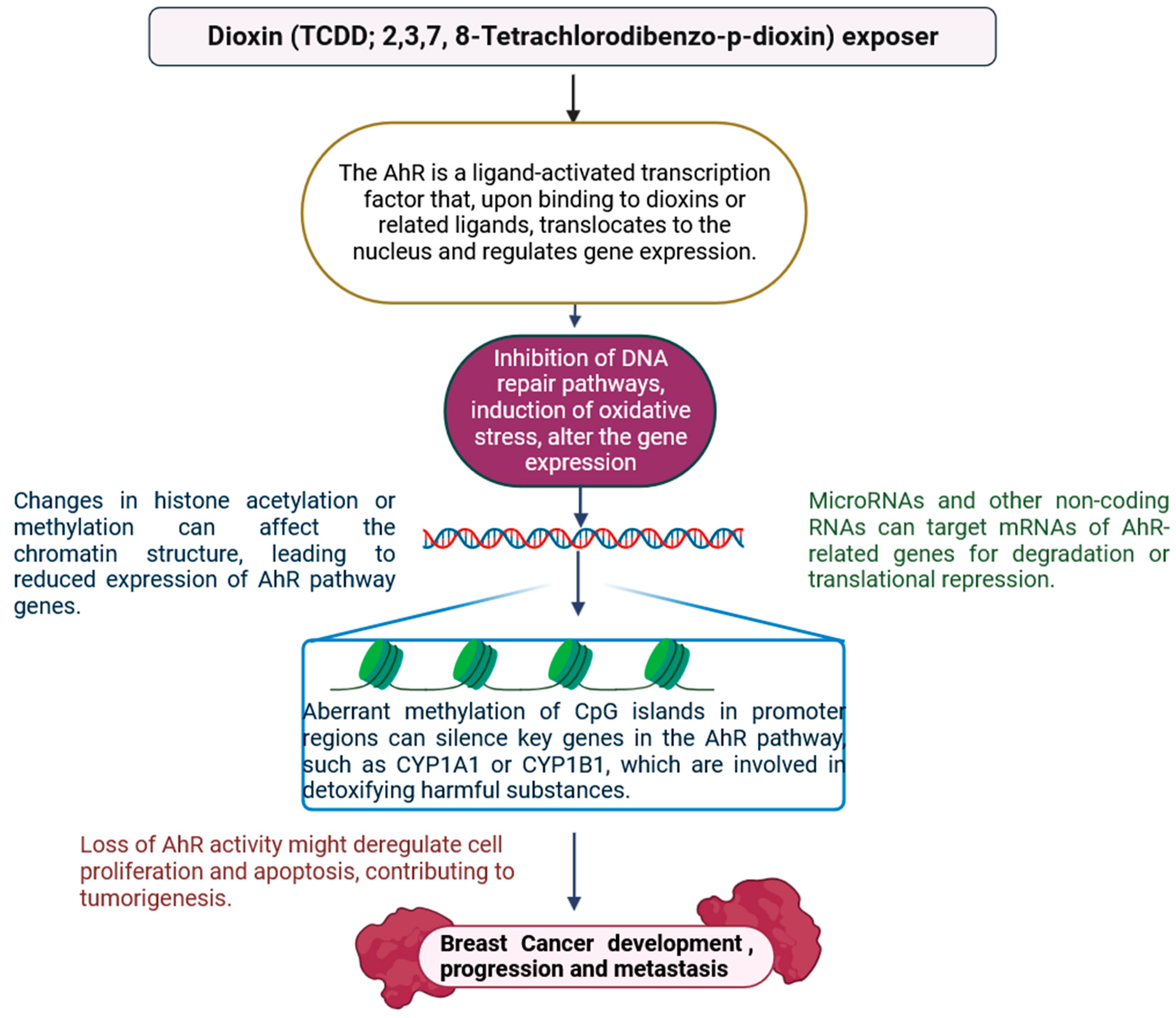
2.9. Dioxins
3. Impact of EDCs on Dietary Intake in Breast Cancer
4. Conclusions and Future Prospective
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed on 23 November 2024).
- Eremici, I.; Borlea, A.; Dumitru, C.; Stoian, D. Breast Cancer Risk Factors among Women with Solid Breast Lesions. Clin. Pract. 2024, 14, 473–485. [Google Scholar] [CrossRef]
- Knower, K.C.; To, S.Q.; Leung, Y.-K.; Ho, S.-M.; Clyne, C.D. Endocrine Disruption of the Epigenome: A Breast Cancer Link. Endocr.-Relat. Cancer 2014, 21, T33–T55. [Google Scholar] [CrossRef]
- Eve, L.; Fervers, B.; Le Romancer, M.; Etienne-Selloum, N. Exposure to Endocrine Disrupting Chemicals and Risk of Breast Cancer. Int. J. Mol. Sci. 2020, 21, 9139. [Google Scholar] [CrossRef] [PubMed]
- Masi, M.; Racchi, M.; Travelli, C.; Corsini, E.; Buoso, E. Molecular Characterization of Membrane Steroid Receptors in Hormone-Sensitive Cancers. Cells 2021, 10, 2999. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y. Nutrition Intervention and Microbiome Modulation in the Management of Breast Cancer. Nutrients 2024, 16, 2644. [Google Scholar] [CrossRef] [PubMed]
- Alavian-Ghavanini, A.; Rüegg, J. Understanding Epigenetic Effects of Endocrine Disrupting Chemicals: From Mechanisms to Novel Test Methods. Basic. Clin. Pharmacol. Toxicol. 2018, 122, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Pitto, L.; Gorini, F.; Bianchi, F.; Guzzolino, E. New Insights into Mechanisms of Endocrine-Disrupting Chemicals in Thyroid Diseases: The Epigenetic Way. Int. J. Environ. Res. Public Health 2020, 17, 7787. [Google Scholar] [CrossRef] [PubMed]
- Bolt, M.J.; Singh, P.; Obkirchner, C.E.; Powell, R.T.; Mancini, M.G.; Szafran, A.T.; Stossi, F.; Mancini, M.A. Endocrine Disrupting Chemicals Differentially Alter Intranuclear Dynamics and Transcriptional Activation of Estrogen Receptor-α. iScience 2021, 24, 103227. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.M.; Greco, C.W. Perturbation of Nuclear Hormone Receptors by Endocrine Disrupting Chemicals: Mechanisms and Pathological Consequences of Exposure. Cells 2020, 9, 13. [Google Scholar] [CrossRef]
- Sabry, R.; Yamate, J.; Favetta, L.; LaMarre, J. MicroRNAs: Potential Targets and Agents of Endocrine Disruption in Female Reproduction. J. Toxicol. Pathol. 2019, 32, 213–221. [Google Scholar] [CrossRef]
- Tsuchida, T.; Kubota, S.; Kamiuezono, S.; Takasugi, N.; Ito, A.; Kumagai, Y.; Uehara, T. Epigenetic Regulation of CXC Chemokine Expression by Environmental Electrophiles Through DNA Methyltransferase Inhibition. Int. J. Mol. Sci. 2024, 25, 11592. [Google Scholar] [CrossRef] [PubMed]
- Derghal, A.; Djelloul, M.; Trouslard, J.; Mounien, L. An Emerging Role of Micro-RNA in the Effect of the Endocrine Disruptors. Front. Neurosci. 2016, 10, 318. [Google Scholar] [CrossRef] [PubMed]
- Shree, N.; Ding, Z.; Flaws, J.; Choudhury, M. Role of microRNA in Endocrine Disruptor-Induced Immunomodulation of Metabolic Health. Metabolites 2022, 12, 1034. [Google Scholar] [CrossRef]
- Alwadi, D.; Felty, Q.; Yoo, C.; Roy, D.; Deoraj, A. Endocrine Disrupting Chemicals Influence Hub Genes Associated with Aggressive Prostate Cancer. Int. J. Mol. Sci. 2023, 24, 3191. [Google Scholar] [CrossRef]
- Wallis, D.; Truong, L.; La Du, J.; Tanguay, R.; Reif, D. Uncovering Evidence for Endocrine-Disrupting Chemicals That Elicit Differential Susceptibility Through Gene-Environment Interactions. Toxics 2021, 9, 77. [Google Scholar] [CrossRef] [PubMed]
- Caserta, D.; De Marco, M.P.; Besharat, A.R.; Costanzi, F. Endocrine Disruptors and Endometrial Cancer: Molecular Mechanisms of Action and Clinical Implications, a Systematic Review. Int. J. Mol. Sci. 2022, 23, 2956. [Google Scholar] [CrossRef] [PubMed]
- Benevolenskaya, E.V.; Islam, A.B.M.M.K.; Ahsan, H.; Kibriya, M.G.; Jasmine, F.; Wolff, B.; Al-Alem, U.; Wiley, E.; Kajdacsy-Balla, A.; Macias, V.; et al. DNA Methylation and Hormone Receptor Status in Breast Cancer. Clin. Epigenet 2016, 8, 17. [Google Scholar] [CrossRef]
- Ennour-Idrissi, K.; Dragic, D.; Issa, E.; Michaud, A.; Chang, S.-L.; Provencher, L.; Durocher, F.; Diorio, C. DNA Methylation and Breast Cancer Risk: An Epigenome-Wide Study of Normal Breast Tissue and Blood. Cancers 2020, 12, 3088. [Google Scholar] [CrossRef]
- Lu, X.; Fraszczyk, E.; Van Der Meer, T.P.; Van Faassen, M.; Bloks, V.W.; Kema, I.P.; Van Beek, A.P.; Li, S.; Franke, L.; Westra, H.-J.; et al. An Epigenome-Wide Association Study Identifies Multiple DNA Methylation Markers of Exposure to Endocrine Disruptors. Environ. Int. 2020, 144, 106016. [Google Scholar] [CrossRef] [PubMed]
- Montjean, D.; Neyroud, A.-S.; Yefimova, M.G.; Benkhalifa, M.; Cabry, R.; Ravel, C. Impact of Endocrine Disruptors upon Non-Genetic Inheritance. Int. J. Mol. Sci. 2022, 23, 3350. [Google Scholar] [CrossRef] [PubMed]
- Buoso, E.; Masi, M.; Racchi, M.; Corsini, E. Endocrine-Disrupting Chemicals’ (EDCs) Effects on Tumour Microenvironment and Cancer Progression: Emerging Contribution of RACK1. Int. J. Mol. Sci. 2020, 21, 9229. [Google Scholar] [CrossRef] [PubMed]
- Maddalon, A.; Masi, M.; Iulini, M.; Linciano, P.; Galbiati, V.; Marinovich, M.; Racchi, M.; Buoso, E.; Corsini, E. Effects of endocrine active contaminating pesticides on RACK1 expression and immunological consequences in THP-1 cells. Environ. Toxicol. Pharmacol. 2022, 202295, 103971. [Google Scholar] [CrossRef] [PubMed]
- Masi, M.; Maddalon, A.; Iulini, M.; Linciano, P.; Galbiati, V.; Marinovich, M.; Racchi, M.; Corsini, E.; Buoso, E. Effects of endocrine disrupting chemicals on the expression of RACK1 and LPS-induced THP-1 cell activation. Toxicology 2022, 480, 153321. [Google Scholar] [CrossRef]
- Buoso, E.; Kenda, M.; Masi, M.; Linciano, P.; Galbiati, V.; Racchi, M.; Dolenc, M.S.; Corsini, E. Effects of Bisphenols on RACK1 Expression and Their Immunological Implications in THP-1 Cells. Front. Pharmacol. 2021, 12, 743991. [Google Scholar] [CrossRef]
- Buoso, E.; Masi, M.; Galbiati, V.; Maddalon, A.; Iulini, M.; Kenda, M.; Sollner Dolenc, M.; Marinovich, M.; Racchi, M.; Corsini, E. Effect of estrogen-active compounds on the expression of RACK1 and immunological implications. Arch. Toxicol. 2020, 94, 2081–2095. [Google Scholar] [CrossRef] [PubMed]
- Sellami, A.; Montes, M.; Lagarde, N. Predicting Potential Endocrine Disrupting Chemicals Binding to Estrogen Receptor α (ERα) Using a Pipeline Combining Structure-Based and Ligand-Based in Silico Methods. Int. J. Mol. Sci. 2021, 22, 2846. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Relloso, A.; Riffo-Campos, A.L.; Haack, K.; Rentero-Garrido, P.; Ladd-Acosta, C.; Fallin, D.M.; Tang, W.Y.; Herreros-Martinez, M.; Gonzalez, J.R.; Bozack, A.K.; et al. Cadmium, Smoking, and Human Blood DNA Methylation Profiles in Adults from the Strong Heart Study. Environ. Health Perspect. 2020, 128, 067005. [Google Scholar] [CrossRef]
- Liu, D.; Shi, Q.; Liu, C.; Sun, Q.; Zeng, X. Effects of Endocrine-Disrupting Heavy Metals on Human Health. Toxics 2023, 11, 322. [Google Scholar] [CrossRef] [PubMed]
- Cirovic, A.; Satarug, S. Toxicity Tolerance in the Carcinogenesis of Environmental Cadmium. Int. J. Mol. Sci. 2024, 25, 1851. [Google Scholar] [CrossRef] [PubMed]
- Islam, R.; Zhao, L.; Wang, Y.; Lu-Yao, G.; Liu, L.-Z. Epigenetic Dysregulations in Arsenic-Induced Carcinogenesis. Cancers 2022, 14, 4502. [Google Scholar] [CrossRef]
- Mani, M.; Kabekkodu, S.; Joshi, M.; Dsouza, H. Ecogenetics of Lead Toxicity and Its Influence on Risk Assessment. Hum. Exp. Toxicol. 2019, 38, 1031–1059. [Google Scholar] [CrossRef] [PubMed]
- Massányi, P.; Massányi, M.; Madeddu, R.; Stawarz, R.; Lukáč, N. Effects of Cadmium, Lead, and Mercury on the Structure and Function of Reproductive Organs. Toxics 2020, 8, 94. [Google Scholar] [CrossRef]
- Xu, F.; Wang, X.; Wu, N.; He, S.; Yi, W.; Xiang, S.; Zhang, P.; Xie, X.; Ying, C. Bisphenol A Induces Proliferative Effects on Both Breast Cancer Cells and Vascular Endothelial Cells Through a Shared GPER-Dependent Pathway in Hypoxia. Environ. Pollut. 2017, 231, 1609–1620. [Google Scholar] [CrossRef]
- Cortes-Ramirez, S.A.; Ho, S.-M.; Leung, Y.-K. Endocrine-Disrupting Chemicals: A Looming Threat to Current and Future Generations. Int. J. Mol. Sci. 2024, 25, 8222. [Google Scholar] [CrossRef]
- Hsieh, T.; Tsai, C.; Hsu, C.; Kuo, P.; Lee, J.; Chai, C.; Wang, S.; Tsai, E. Phthalates Induce Proliferation and Invasiveness of Estrogen Receptor-Negative Breast Cancer Through the AhR/HDAC6/c-Myc Signaling Pathway. FASEB J. 2012, 26, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Fiocchetti, M.; Bastari, G.; Cipolletti, M.; Leone, S.; Acconcia, F.; Marino, M. The Peculiar Estrogenicity of Diethyl Phthalate: Modulation of Estrogen Receptor α Activities in the Proliferation of Breast Cancer Cells. Toxics 2021, 9, 237. [Google Scholar] [CrossRef] [PubMed]
- Thakur, C.; Qiu, Y.; Fu, Y.; Bi, Z.; Zhang, W.; Ji, H.; Chen, F. Epigenetics and Environment in Breast Cancer: New Paradigms for Anti-Cancer Therapies. Front. Oncol. 2022, 12, 971288. [Google Scholar] [CrossRef]
- Leti Maggio, E.; Zucca, C.; Grande, M.; Carrano, R.; Infante, A.; Bei, R.; Lucarini, V.; De Maio, F.; Focaccetti, C.; Palumbo, C.; et al. Polyphenols Regulate the Activity of Endocrine-Disrupting Chemicals, Having Both Positive and Negative Effects. J. Xenobiot. 2024, 14, 1378–1405. [Google Scholar] [CrossRef]
- Hilakivi-Clarke, L. Maternal Exposure to Diethylstilbestrol during Pregnancy and Increased Breast Cancer Risk in Daughters. Breast Cancer Res. 2014, 16, 3394. [Google Scholar] [CrossRef] [PubMed]
- Zamora-León, P. Are the Effects of DES Over? A Tragic Lesson from the Past. Int. J. Environ. Res. Public Health 2021, 18, 10309. [Google Scholar] [CrossRef] [PubMed]
- White, A.J.; Chen, J.; Teitelbaum, S.L.; McCullough, L.E.; Xu, X.; Hee Cho, Y.; Conway, K.; Beyea, J.; Stellman, S.D.; Steck, S.E.; et al. Sources of Polycyclic Aromatic Hydrocarbons Are Associated with Gene-Specific Promoter Methylation in Women with Breast Cancer. Environ. Res. 2016, 145, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Anditi, B.C.; Poór, V.; Szerencsés, D.; Szabó, I.; Wahr, M.; Kőnig-Péter, A.; Dergez, T. Determination of PAH Contamination in Breast Milk Samples from Hungarian Volunteering Mothers, Using HPLC–FLD. Molecules 2024, 29, 5060. [Google Scholar] [CrossRef] [PubMed]
- Strumylaitė, L.; Boguševičius, A.; Ryselis, S.; Pranys, D.; Poškienė, L.; Kregždytė, R.; Abdrachmanovas, O.; Asadauskaitė, R. Association between cadmium and breast cancer. Medicina 2008, 44, 415. [Google Scholar] [CrossRef]
- Strumylaite, L.; Kregzdyte, R.; Bogusevicius, A.; Poskiene, L.; Baranauskiene, D.; Pranys, D. Cadmium Exposure and Risk of Breast Cancer by Histological and Tumor Receptor Subtype in White Caucasian Women: A Hospital-Based Case-Control Study. Int. J. Mol. Sci. 2019, 20, 3029. [Google Scholar] [CrossRef] [PubMed]
- Tarhonska, K.; Lesicka, M.; Janasik, B.; Roszak, J.; Reszka, E.; Braun, M.; Kołacińska-Wow, A.; Jabłońska, E. Cadmium and Breast Cancer—Current State and Research Gaps in the Underlying Mechanisms. Toxicol. Lett. 2022, 361, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Gachowska, M.; Dąbrowska, A.; Wilczyński, B.; Kuźnicki, J.; Sauer, N.; Szlasa, W.; Kobierzycki, C.; Łapińska, Z.; Kulbacka, J. The Influence of Environmental Exposure to Xenoestrogens on the Risk of Cancer Development. Int. J. Mol. Sci. 2024, 25, 12363. [Google Scholar] [CrossRef] [PubMed]
- Brama, M.; Gnessi, L.; Basciani, S.; Cerulli, N.; Politi, L.; Spera, G.; Mariani, S.; Cherubini, S.; d’Abusco, A.S.; Scandurra, R.; et al. Cadmium Induces Mitogenic Signaling in Breast Cancer Cell by an ERα-Dependent Mechanism. Mol. Cell. Endocrinol. 2007, 264, 102–108. [Google Scholar] [CrossRef]
- Wojnarowski, K.; Cholewińska, P.; Palić, D.; Bednarska, M.; Jarosz, M.; Wiśniewska, I. Estrogen Receptors Mediated Negative Effects of Estrogens and Xenoestrogens in Teleost Fishes—Review. Int. J. Mol. Sci. 2022, 23, 2605. [Google Scholar] [CrossRef] [PubMed]
- Aquino, N.B.; Sevigny, M.B.; Sabangan, J.; Louie, M.C. The Role of Cadmium and Nickel in Estrogen Receptor Signaling and Breast Cancer: Metalloestrogens or Not? J. Environ. Sci. Health Part C 2012, 30, 189–224. [Google Scholar] [CrossRef] [PubMed]
- Asara, Y.; Marchal, J.A.; Carrasco, E.; Boulaiz, H.; Solinas, G.; Bandiera, P.; Garcia, M.A.; Farace, C.; Montella, A.; Madeddu, R. Cadmium Modifies the Cell Cycle and Apoptotic Profiles of Human Breast Cancer Cells Treated with 5-Fluorouracil. Int. J. Mol. Sci. 2013, 14, 16600–16616. [Google Scholar] [CrossRef]
- Peana, M.; Pelucelli, A.; Chasapis, C.T.; Perlepes, S.P.; Bekiari, V.; Medici, S.; Zoroddu, M.A. Biological Effects of Human Exposure to Environmental Cadmium. Biomolecules 2023, 13, 36. [Google Scholar] [CrossRef]
- Lordan, R.; Zabetakis, I. Cadmium: A Focus on the Brown Crab (Cancer pagurus) Industry and Potential Human Health Risks. Toxics 2022, 10, 591. [Google Scholar] [CrossRef] [PubMed]
- Luparello, C. Cadmium-Associated Molecular Signatures in Cancer Cell Models. Cancers 2021, 13, 2823. [Google Scholar] [CrossRef]
- Desaulniers, D.; Vasseur, P.; Jacobs, A.; Aguila, M.C.; Ertych, N.; Jacobs, M.N. Integration of Epigenetic Mechanisms into Non-Genotoxic Carcinogenicity Hazard Assessment: Focus on DNA Methylation and Histone Modifications. Int. J. Mol. Sci. 2021, 22, 10969. [Google Scholar] [CrossRef] [PubMed]
- Thévenod, F. Cadmium and Cellular Signaling Cascades: To Be or Not to Be? Toxicol. Appl. Pharmacol. 2009, 238, 221–239. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-H.; Chen, L.-R.; Chen, K.-H. In Vitro and Vivo Identification, Metabolism and Action of Xenoestrogens: An Overview. Int. J. Mol. Sci. 2021, 22, 4013. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Zhang, J.; Qi, X.; Cui, Y.; Yin, K.; Lin, H. Cadmium Induced Inflammation and Apoptosis of Porcine Epididymis via Activating RAF1/MEK/ERK and NF-κB Pathways. Toxicol. Appl. Pharmacol. 2021, 415, 115449. [Google Scholar] [CrossRef]
- Schuler, L.A.; Murdoch, F.E. Endogenous and Therapeutic Estrogens: Maestro Conductors of the Microenvironment of ER+ Breast Cancers. Cancers 2021, 13, 3725. [Google Scholar] [CrossRef] [PubMed]
- Valbonesi, P.; Ricci, L.; Franzellitti, S.; Biondi, C.; Fabbri, E. Effects of Cadmium on MAPK Signalling Pathways and HSP70 Expression in a Human Trophoblast Cell Line. Placenta 2008, 29, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Almatroudi, A.; Allemailem, K.S.; Alwanian, W.M.; Alharbi, B.F.; Alrumaihi, F.; Khan, A.A.; Almatroodi, S.A.; Rahmani, A.H. Effects and Mechanisms of Kaempferol in the Management of Cancers Through Modulation of Inflammation and Signal Transduction Pathways. Int. J. Mol. Sci. 2023, 24, 8630. [Google Scholar] [CrossRef]
- Pakjoo, M.; Ahmadi, S.E.; Zahedi, M.; Jaafari, N.; Khademi, R.; Amini, A.; Safa, M. Interplay Between Proteasome Inhibitors and NF-κB Pathway in Leukemia and Lymphoma: A Comprehensive Review on Challenges Ahead of Proteasome Inhibitors. Cell Commun. Signal 2024, 22, 105. [Google Scholar] [CrossRef]
- Branca, J.J.V.; Fiorillo, C.; Carrino, D.; Paternostro, F.; Taddei, N.; Gulisano, M.; Pacini, A.; Becatti, M. Cadmium-Induced Oxidative Stress: Focus on the Central Nervous System. Antioxidants 2020, 9, 492. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.; Kowald, L.; Van Wijk, S.J.L.; Fulda, S. Differential Involvement of TAK1, RIPK1 and NF-κB Signaling in Smac Mimetic-Induced Cell Death in Breast Cancer Cells. Biol. Chem. 2019, 400, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Qu, F.; Zheng, W. Cadmium Exposure: Mechanisms and Pathways of Toxicity and Implications for Human Health. Toxics 2024, 12, 388. [Google Scholar] [CrossRef]
- Naldi, A.; Larive, R.M.; Czerwinska, U.; Urbach, S.; Montcourrier, P.; Roy, C.; Solassol, J.; Freiss, G.; Coopman, P.J.; Radulescu, O. Reconstruction and Signal Propagation Analysis of the Syk Signaling Network in Breast Cancer Cells. PLoS Comput. Biol. 2017, 13, e1005432. [Google Scholar] [CrossRef] [PubMed]
- Oke, S.L.; Hardy, D.B. The Role of Cellular Stress in Intrauterine Growth Restriction and Postnatal Dysmetabolism. Int. J. Mol. Sci. 2021, 22, 6986. [Google Scholar] [CrossRef]
- Saintilnord, W.N.; Fondufe-Mittendorf, Y. Arsenic-Induced Epigenetic Changes in Cancer Development. Semin. Cancer Biol. 2021, 76, 195–205. [Google Scholar] [CrossRef]
- Virtuoso, S.; Raggi, C.; Maugliani, A.; Baldi, F.; Gentili, D.; Narciso, L. Toxicological Effects of Naturally Occurring Endocrine Disruptors on Various Human Health Targets: A Rapid Review. Toxics 2024, 12, 256. [Google Scholar] [CrossRef] [PubMed]
- Sage, A.P.; Minatel, B.C.; Ng, K.W.; Stewart, G.L.; Dummer, T.J.B.; Lam, W.L.; Martinez, V.D. Oncogenomic Disruptions in Arsenic-Induced Carcinogenesis. Oncotarget 2017, 8, 25736–25755. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Zhang, Y.; Fang, M.; Jehan, S.; Zhou, W. Current Advances of Nanomedicines Delivering Arsenic Trioxide for Enhanced Tumor Therapy. Pharmaceutics 2022, 14, 743. [Google Scholar] [CrossRef] [PubMed]
- Tam, M.; Zhang, C.; Kibriya, M.G.; Jasmine, F.; Roy, S.; Gao, J.; Sabarinathan, M.; Shinkle, J.; Delgado, D.; Ahmed, A.; et al. A study of telomere length, arsenic exposure, and arsenic toxicity in a Bangladeshi cohort. Environ. Res. 2018, 164, 346–355. [Google Scholar] [CrossRef]
- Dratwa-Kuzmin, M.; Hadra, B.A.; Oguz, F.; Ogret, Y.; Constantinescu, I.; Apostol, D.; Talangescu, A.; Constantinescu, A.-E.; Maruntelu, I.; Kościńska, K.; et al. Telomere Length, HLA, and Longevity—Results from a Multicenter Study. Int. J. Mol. Sci. 2024, 25, 9457. [Google Scholar] [CrossRef] [PubMed]
- Tam, L.M.; Price, N.E.; Wang, Y. Molecular Mechanisms of Arsenic-Induced Disruption of DNA Repair. Chem. Res. Toxicol. 2020, 33, 709–726. [Google Scholar] [CrossRef]
- Pasha, Q.; Rain, M.; Tasnim, S.; Kanipakam, H.; Thinlas, T.; Mohammad, G. The Telomere-Telomerase System Is Detrimental to Health at High-Altitude. Int. J. Environ. Res. Public Health 2023, 20, 1935. [Google Scholar] [CrossRef]
- Lin, L.-T.; Liu, S.-Y.; Leu, J.-D.; Chang, C.-Y.; Chiou, S.-H.; Lee, T.-C.; Lee, Y.-J. Arsenic Trioxide-Mediated Suppression of miR-182-5p Is Associated with Potent Anti-Oxidant Effects Through Up-Regulation of SESN2. Oncotarget 2018, 9, 16028–16042. [Google Scholar] [CrossRef] [PubMed]
- Gujrati, H.; Ha, S.; Wang, B.-D. Deregulated microRNAs Involved in Prostate Cancer Aggressiveness and Treatment Resistance Mechanisms. Cancers 2023, 15, 3140. [Google Scholar] [CrossRef] [PubMed]
- Medda, N.; De, S.K.; Maiti, S. Different Mechanisms of Arsenic Related Signaling in Cellular Proliferation, Apoptosis and Neo-Plastic Transformation. Ecotoxicol. Environ. Saf. 2021, 208, 111752. [Google Scholar] [CrossRef] [PubMed]
- De Francisco, P.; Martín-González, A.; Rodriguez-Martín, D.; Díaz, S. Interactions with Arsenic: Mechanisms of Toxicity and Cellular Resistance in Eukaryotic Microorganisms. Int. J. Environ. Res. Public Health 2021, 18, 12226. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Hu, Y.; Li, J.; Lou, B.; Wu, R.; Wang, G.; Lu, C.; Wang, H.; Pi, J.; Xu, Y. The Role of Reactive Oxygen Species in Arsenic Toxicity. Biomolecules 2020, 10, 240. [Google Scholar] [CrossRef] [PubMed]
- Qie, S.; Diehl, J.A. Cyclin D1, Cancer Progression, and Opportunities in Cancer Treatment. J. Mol. Med. 2016, 94, 1313–1326. [Google Scholar] [CrossRef]
- Zargari, F.; Rahaman, M.S.; KazemPour, R.; Hajirostamlou, M. Arsenic, Oxidative Stress and Reproductive System. J. Xenobiot. 2022, 12, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Yu, X. Arsenic-Induced Apoptosis in the P53-Proficient and P53-Deficient Cells Through Differential Modulation of NFkB Pathway. Food Chem. Toxicol. 2018, 118, 849–860. [Google Scholar] [CrossRef]
- Genchi, G.; Lauria, G.; Catalano, A.; Carocci, A.; Sinicropi, M.S. Arsenic: A Review on a Great Health Issue Worldwide. Appl. Sci. 2022, 12, 6184. [Google Scholar] [CrossRef]
- Peng, F.; Liao, M.; Qin, R.; Zhu, S.; Peng, C.; Fu, L.; Chen, Y.; Han, B. Regulated Cell Death (RCD) in Cancer: Key Pathways and Targeted Therapies. Signal Transduct. Target. Ther. 2022, 7, 286. [Google Scholar] [CrossRef] [PubMed]
- Graham-Evans, B.; Tchounwou, P.B.; Cohly, H.H.P. Cytotoxicity and Proliferation Studies with Arsenic in Established Human Cell Lines: Keratinocytes, Melanocytes, Dendritic Cells, Dermal Fibroblasts, Microvascular Endothelial Cells, Monocytes and T-Cells. Int. J. Mol. Sci. 2003, 4, 13–21. [Google Scholar] [CrossRef]
- Atsaves, V.; Leventaki, V.; Rassidakis, G.Z.; Claret, F.X. AP-1 Transcription Factors as Regulators of Immune Responses in Cancer. Cancers 2019, 11, 1037. [Google Scholar] [CrossRef]
- Pittayapruek, P.; Meephansan, J.; Prapapan, O.; Komine, M.; Ohtsuki, M. Role of Matrix Metalloproteinases in Photoaging and Photocarcinogenesis. Int. J. Mol. Sci. 2016, 17, 868. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, M.-C.; Parent, M.-E.; Nadon, L.; Latreille, B.; Siemiatycki, J. Occupational Exposure to Lead Compounds and Risk of Cancer among Men: A Population-Based Case-Control Study. Am. J. Epidemiol. 2007, 166, 1005–1014. [Google Scholar] [CrossRef]
- Mielke, H.W.; Gonzales, C.R.; Powell, E.T.; Egendorf, S.P. Lead in Air, Soil, and Blood: Pb Poisoning in a Changing World. Int. J. Environ. Res. Public Health 2022, 19, 9500. [Google Scholar] [CrossRef]
- Sanders, T.; Liu, Y.; Buchner, V.; Tchounwou, P.B. Neurotoxic Effects and Biomarkers of Lead Exposure: A Review. Rev. Environ. Health 2009, 24, 15–46. [Google Scholar] [CrossRef] [PubMed]
- Olufemi, A.C.; Mji, A.; Mukhola, M.S. Potential Health Risks of Lead Exposure from Early Life Through Later Life: Implications for Public Health Education. Int. J. Environ. Res. Public Health 2022, 19, 16006. [Google Scholar] [CrossRef]
- Liao, L.M.; Friesen, M.C.; Xiang, Y.-B.; Cai, H.; Koh, D.-H.; Ji, B.-T.; Yang, G.; Li, H.-L.; Locke, S.J.; Rothman, N.; et al. Occupational Lead Exposure and Associations with Selected Cancers: The Shanghai Men’s and Women’s Health Study Cohorts. Environ. Health Perspect. 2016, 124, 97–103. [Google Scholar] [CrossRef]
- Kobets, T.; Smith, B.P.C.; Williams, G.M. Food-Borne Chemical Carcinogens and the Evidence for Human Cancer Risk. Foods 2022, 11, 2828. [Google Scholar] [CrossRef] [PubMed]
- Tilghman, S.L.; Bratton, M.R.; Segar, H.C.; Martin, E.C.; Rhodes, L.V.; Li, M.; McLachlan, J.A.; Wiese, T.E.; Nephew, K.P.; Burow, M.E. Endocrine Disruptor Regulation of MicroRNA Expression in Breast Carcinoma Cells. PLoS ONE 2012, 7, e32754. [Google Scholar] [CrossRef] [PubMed]
- Humphries, B.; Wang, Z.; Yang, C. MicroRNA Regulation of Epigenetic Modifiers in Breast Cancer. Cancers 2019, 11, 897. [Google Scholar] [CrossRef] [PubMed]
- Stiefel, C.; Stintzing, F. Endocrine-Active and Endocrine-Disrupting Compounds in Food—Occurrence, Formation and Relevance. NFS J. 2023, 31, 57–92. [Google Scholar] [CrossRef]
- Soldado-Gordillo, A.; Álvarez-Mercado, A.I. Epigenetics, Microbiota, and Breast Cancer: A Systematic Review. Life 2024, 14, 705. [Google Scholar] [CrossRef] [PubMed]
- Scinicariello, F.; Murray, H.E.; Moffett, D.B.; Abadin, H.G.; Sexton, M.J.; Fowler, B.A. Lead and δ-Aminolevulinic Acid Dehydratase Polymorphism: Where Does It Lead? A Meta-Analysis. Environ. Health Perspect. 2007, 115, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-C.; Yang, C.-C.; Liu, T.-Y.; Dai, C.-Y.; Wang, C.-L.; Chuang, H.-Y. Use of Generalized Additive Model to Detect the Threshold of δ-Aminolevulinic Acid Dehydratase Activity Reduced by Lead Exposure. Int. J. Environ. Res. Public Health 2020, 17, 5712. [Google Scholar] [CrossRef] [PubMed]
- Georgiou-Siafis, S.K.; Tsiftsoglou, A.S. The Key Role of GSH in Keeping the Redox Balance in Mammalian Cells: Mechanisms and Significance of GSH in Detoxification via Formation of Conjugates. Antioxidants 2023, 12, 1953. [Google Scholar] [CrossRef]
- Obukhova, L.; Kopytova, T.; Murach, E.; Shchelchkova, N.; Kontorshchikova, C.; Medyanik, I.; Orlinskaya, N.; Grishin, A.; Kontorshchikov, M.; Badanina, D. Relationship Between Glutathione-Dependent Enzymes and the Immunohistochemical Profile of Glial Neoplasms. Biomedicines 2022, 10, 2393. [Google Scholar] [CrossRef] [PubMed]
- Steenland, K.; Boffetta, P. Lead and Cancer in Humans: Where Are We Now? Am. J. Ind. Med. 2000, 38, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Baierle, M.; Charão, M.F.; Göethel, G.; Barth, A.; Fracasso, R.; Bubols, G.; Sauer, E.; Campanharo, S.C.; Rocha, R.C.C.; Saint’Pierre, T.D.; et al. Are Delta-Aminolevulinate Dehydratase Inhibition and Metal Concentrations Additional Factors for the Age-Related Cognitive Decline? Int. J. Environ. Res. Public Health 2014, 11, 10851–10867. [Google Scholar] [CrossRef] [PubMed]
- Cariati, F.; Carbone, L.; Conforti, A.; Bagnulo, F.; Peluso, S.R.; Carotenuto, C.; Buonfantino, C.; Alviggi, E.; Alviggi, C.; Strina, I. Bisphenol A-Induced Epigenetic Changes and Its Effects on the Male Reproductive System. Front. Endocrinol. 2020, 11, 453. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y. Potential Pro-Tumorigenic Effect of Bisphenol A in Breast Cancer via Altering the Tumor Microenvironment. Cancers 2022, 14, 3021. [Google Scholar] [CrossRef] [PubMed]
- Salian-Mehta, S.; Doshi, T.; Vanage, G. Exposure of Neonatal Rats to the Endocrine Disrupter Bisphenol A Affects Ontogenic Expression Pattern of Testicular Steroid Receptors and Their Coregulators. J. Appl. Toxicol. 2014, 34, 307–318. [Google Scholar] [CrossRef]
- Nair, V.A.; Valo, S.; Peltomäki, P.; Bajbouj, K.; Abdel-Rahman, W.M. Oncogenic Potential of Bisphenol A and Common Environmental Contaminants in Human Mammary Epithelial Cells. Int. J. Mol. Sci. 2020, 21, 3735. [Google Scholar] [CrossRef]
- Sugiyama, K.; Kinoshita, M.; Grúz, P.; Kasamatsu, T.; Honma, M. Bisphenol-A Reduces DNA Methylation After Metabolic Activation. Genes. Environ. 2022, 44, 20. [Google Scholar] [CrossRef] [PubMed]
- Besaratinia, A. The State of Research and Weight of Evidence on the Epigenetic Effects of Bisphenol A. Int. J. Mol. Sci. 2023, 24, 7951. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Fu, L.-J.; Zhang, J.; Liu, X.-Q.; Zhang, H.-J.; Zhang, X.; Ma, M.-F.; Chen, X.-M.; He, J.-L.; Li, L.-B.; et al. Bisphenol A Exposure Promotes HTR-8/SVneo Cell Migration and Impairs Mouse Placentation Involving Upregulation of Integrin-Β1 and MMP-9 and Stimulation of MAPK and PI3K Signaling Pathways. Oncotarget 2017, 8, 51507–51521. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Huang, D.; Zhou, P.; Su, X.; Yang, R.; Shao, C.; Ma, A.; Wu, J. Individual and Combined Effect of Bisphenol A and Bisphenol AF on Prostate Cell Proliferation Through NF-κ. Int. J. Mol. Sci. 2022, 23, 12283. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, N.; Weng, S.; Wang, H. Bisphenol A Increases the Migration and Invasion of Triple-Negative Breast Cancer Cells via Oestrogen-related Receptor Gamma. Basic. Clin. Pharmacol. Toxicol. 2016, 119, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Tang, Q.; Lei, B.; Yang, Y.; Xu, L. Bisphenol AF Promoted the Growth of Uterus and Activated Estrogen Signaling Related Targets in Various Tissues of Nude Mice with SK-BR-3 Xenograft Tumor. Int. J. Environ. Res. Public Health 2022, 19, 15743. [Google Scholar] [CrossRef] [PubMed]
- Deb, P.; Bhan, A.; Hussain, I.; Ansari, K.I.; Bobzean, S.A.; Pandita, T.K.; Perrotti, L.I.; Mandal, S.S. Endocrine Disrupting Chemical, Bisphenol-A, Induces Breast Cancer Associated Gene HOXB9 Expression in Vitro and in Vivo. Gene 2016, 590, 234–243. [Google Scholar] [CrossRef]
- Yuan, J.; Yang, J.; Xu, X.; Wang, Z.; Jiang, Z.; Ye, Z.; Ren, Y.; Wang, Q.; Wang, T. Bisphenol A (BPA) Directly Activates the G Protein-Coupled Estrogen Receptor 1 and Triggers the Metabolic Disruption in the Gonadal Tissue of Apostichopus japonicus. Biology 2023, 12, 798. [Google Scholar] [CrossRef]
- Gao, H.; Yang, B.-J.; Li, N.; Feng, L.-M.; Shi, X.-Y.; Zhao, W.-H.; Liu, S.-J. Bisphenol A and Hormone-Associated Cancers: Current Progress and Perspectives. Medicine 2015, 94, e211. [Google Scholar] [CrossRef]
- Tiffon, C. The Impact of Nutrition and Environmental Epigenetics on Human Health and Disease. Int. J. Mol. Sci. 2018, 19, 3425. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.-P.; Chien, M.-H. Lower Concentrations of Phthalates Induce Proliferation in Human Breast Cancer Cells. Climacteric 2014, 17, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Cai, W.; Liu, H.; Jiang, H.; Bi, Y.; Wang, H. The Association of Bisphenol A and Phthalates with Risk of Breast Cancer: A Meta-Analysis. Int. J. Environ. Res. Public Health 2021, 18, 2375. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Li, S.S.-L. Epigenetic Effects of Environmental Chemicals Bisphenol A and Phthalates. Int. J. Mol. Sci. 2012, 13, 10143–10153. [Google Scholar] [CrossRef]
- Huang, W.; Li, H.; Yu, Q.; Xiao, W.; Wang, D.O. LncRNA-Mediated DNA Methylation: An Emerging Mechanism in Cancer and Beyond. J. Exp. Clin. Cancer Res. 2022, 41, 100. [Google Scholar] [CrossRef]
- Stathori, G.; Hatziagapiou, K.; Mastorakos, G.; Vlahos, N.F.; Charmandari, E.; Valsamakis, G. Endocrine-Disrupting Chemicals, Hypothalamic Inflammation and Reproductive Outcomes: A Review of the Literature. Int. J. Mol. Sci. 2024, 25, 11344. [Google Scholar] [CrossRef] [PubMed]
- Abolhasanzadeh, N.; Sarabandi, S.; Dehghan, B.; Karamad, V.; Avci, C.B.; Shademan, B.; Nourazarian, A. Exploring the Intricate Relationship Between miRNA Dysregulation and Breast Cancer Development: Insights into the Impact of Environmental Chemicals. Front. Immunol. 2024, 15, 1333563. [Google Scholar] [CrossRef] [PubMed]
- Sibuh, B.Z.; Quazi, S.; Panday, H.; Parashar, R.; Jha, N.K.; Mathur, R.; Jha, S.K.; Taneja, P.; Jha, A.K. The Emerging Role of Epigenetics in Metabolism and Endocrinology. Biology 2023, 12, 256. [Google Scholar] [CrossRef]
- Dutta, S.; Haggerty, D.K.; Rappolee, D.A.; Ruden, D.M. Phthalate Exposure and Long-Term Epigenomic Consequences: A Review. Front. Genet. 2020, 11, 405. [Google Scholar] [CrossRef] [PubMed]
- Varghese, E.; Liskova, A.; Kubatka, P.; Samuel, S.M.; Büsselberg, D. Anti-Angiogenic Effects of Phytochemicals on miRNA Regulating Breast Cancer Progression. Biomolecules 2020, 10, 191. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, H.; Long, C.; Tsai, C.; Hsieh, T.; Hsu, C.; Tsai, E. Possible Mechanism of Phthalates-induced Tumorigenesis. Kaohsiung J. Med. Sci. 2012, 28, S22–S27. [Google Scholar] [CrossRef] [PubMed]
- Saftić Martinović, L.; Mladenić, T.; Lovrić, D.; Ostojić, S.; Dević Pavlić, S. Decoding the Epigenetics of Infertility: Mechanisms, Environmental Influences, and Therapeutic Strategies. Epigenomes 2024, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Zimeri, A.M.; Robb, S.W.; Hassan, S.M.; Hire, R.R.; Davis, M.B. Assessing Heavy Metal and PCB Exposure from Tap Water by Measuring Levels in Plasma from Sporadic Breast Cancer Patients, a Pilot Study. Int. J. Environ. Res. Public Health 2015, 12, 15683–15691. [Google Scholar] [CrossRef]
- da Mota, T.H.A.; Camargo, R.; Biojone, E.R.; Guimarães, A.F.R.; Pittella-Silva, F.; de Oliveira, D.M. The Relevance of Telomerase and Telomere-Associated Proteins in B-Acute Lymphoblastic Leukemia. Genes 2023, 14, 691. [Google Scholar] [CrossRef] [PubMed]
- Pittman, G.S.; Wang, X.; Campbell, M.R.; Coulter, S.J.; Olson, J.R.; Pavuk, M.; Birnbaum, L.S.; Bell, D.A. Polychlorinated Biphenyl Exposure and DNA Methylation in the Anniston Community Health Survey. Epigenetics 2020, 15, 337–357. [Google Scholar] [CrossRef] [PubMed]
- Łukasiewicz, S.; Czeczelewski, M.; Forma, A.; Baj, J.; Sitarz, R.; Stanisławek, A. Breast Cancer—Epidemiology, Risk Factors, Classification, Prognostic Markers, and Current Treatment Strategies—An Updated Review. Cancers 2021, 13, 4287. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.B.; Miller, C.A. Cooperation of Heat Shock Protein 90 and P23 in Aryl Hydrocarbon Receptor Signaling. Cell Stress. Chaperones 2004, 9, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Montano, L.; Pironti, C.; Pinto, G.; Ricciardi, M.; Buono, A.; Brogna, C.; Venier, M.; Piscopo, M.; Amoresano, A.; Motta, O. Polychlorinated Biphenyls (PCBs) in the Environment: Occupational and Exposure Events, Effects on Human Health and Fertility. Toxics 2022, 10, 365. [Google Scholar] [CrossRef]
- Brown, T.M.; Ross, P.S.; Reimer, K.J.; Veldhoen, N.; Dangerfield, N.J.; Fisk, A.T.; Helbing, C.C. PCB Related Effects Thresholds as Derived Through Gene Transcript Profiles in Locally Contaminated Ringed Seals (Pusa hispida). Environ. Sci. Technol. 2014, 48, 12952–12961. [Google Scholar] [CrossRef] [PubMed]
- Bassal, M.A. The Interplay Between Dysregulated Metabolism and Epigenetics in Cancer. Biomolecules 2023, 13, 944. [Google Scholar] [CrossRef]
- Krasnyi, A.M.; Sadekova, A.A.; Kometova, V.V.; Rodionov, V.V.; Yarotskaya, E.L.; Sukhikh, G.T. Methylation Profile of Small Breast Cancer Tumors Evaluated by Modified MS–HRM. Int. J. Mol. Sci. 2023, 24, 12660. [Google Scholar] [CrossRef] [PubMed]
- Panesar, H.K.; Kennedy, C.L.; Keil Stietz, K.P.; Lein, P.J. Polychlorinated Biphenyls (PCBs): Risk Factors for Autism Spectrum Disorder? Toxics 2020, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Salimi, F.; Asadikaram, G.; Ashrafi, M.R.; Zeynali Nejad, H.; Abolhassani, M.; Abbasi-Jorjandi, M.; Sanjari, M. Organochlorine pesticides and epigenetic alterations in thyroid tumors. Front. Endocrinol. 2023, 14, 1130794. [Google Scholar] [CrossRef]
- Lincho, J.; Martins, R.C.; Gomes, J. Paraben Compounds—Part I: An Overview of Their Characteristics, Detection, and Impacts. Appl. Sci. 2021, 11, 2307. [Google Scholar] [CrossRef]
- Khan, Z.; Zheng, Y.; Jones, T.L.; Delaney, A.A.; Correa, L.F.; Shenoy, C.C.; Khazaie, K.; Daftary, G.S. Epigenetic Therapy: Novel Translational Implications for Arrest of Environmental Dioxin-Induced Disease in Females. Endocrinology 2018, 159, 477–489. [Google Scholar] [CrossRef]
- Zhao, W.; Lu, J.; Lai, Y.; Hou, Y.; Zhao, X.; Wei, Q.; Zou, X.; Gou, Z. Occurrences, Possible Sources, and Risk Impacts of Organochlorine Pesticides in Soil of Changchun Central Urban Area, Northeast China. Sustainability 2023, 15, 16801. [Google Scholar] [CrossRef]
- Li, Y.; Tollefsbol, T.O. Impact on DNA Methylation in Cancer Prevention and Therapy by Bioactive Dietary Components. CMC 2010, 17, 2141–2151. [Google Scholar] [CrossRef]
- Parada, H., Jr.; Sahrai, L.; Wolff, M.S.; Santella, R.M.; Chen, J.; Neugut, A.I.; Teitelbaum, S.L. Urinary parabens and breast cancer risk: Modification by LINE-1 and LUMA global DNA methylation, and associations with breast cancer defined by tumor promoter methylation status. Mol. Carcinog. 2022, 61, 1002–1015. [Google Scholar] [CrossRef] [PubMed]
- Azeredo, D.B.C.; de Sousa Anselmo, D.; Soares, P.; Graceli, J.B.; Magliano, D.C.; Miranda-Alves, L. Environmental Endocrinology: Parabens Hazardous Effects on Hypothalamic–Pituitary–Thyroid Axis. Int. J. Mol. Sci. 2023, 24, 15246. [Google Scholar] [CrossRef]
- Hager, E.; Chen, J.; Zhao, L. Minireview: Parabens Exposure and Breast Cancer. Int. J. Environ. Res. Public Health 2022, 19, 1873. [Google Scholar] [CrossRef]
- Parla, A.; Zormpa, E.; Paloumpis, N.; Kabir, A.; Furton, K.G.; Roje, Ž.; Samanidou, V.; Vinković Vrček, I.; Panderi, I. Determination of Intact Parabens in the Human Plasma of Cancer and Non-Cancer Patients Using a Validated Fabric Phase Sorptive Extraction Reversed-Phase Liquid Chromatography Method with UV Detection. Molecules 2021, 26, 1526. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, Y.; Osaka Twin Research Group; Watanabe, M. Environmental Factor Index (EFI): A Novel Approach to Measure the Strength of Environmental Influence on DNA Methylation in Identical Twins. Epigenomes 2024, 8, 44. [Google Scholar] [CrossRef]
- Ajiboye, T.O.; Kuvarega, A.T.; Onwudiwe, D.C. Recent Strategies for Environmental Remediation of Organochlorine Pesticides. Appl. Sci. 2020, 10, 6286. [Google Scholar] [CrossRef]
- Yao, S.; Huang, J.; Zhou, H.; Cao, C.; Ai, T.; Xing, H.; Sun, J. Levels, Distribution and Health Risk Assessment of Organochlorine Pesticides in Agricultural Soils from the Pearl River Delta of China. Int. J. Environ. Res. Public Health 2022, 19, 13171. [Google Scholar] [CrossRef] [PubMed]
- López-Benítez, A.; Guevara-Lara, A.; Domínguez-Crespo, M.A.; Andraca-Adame, J.A.; Torres-Huerta, A.M. Concentrations of Organochlorine, Organophosphorus, and Pyrethroid Pesticides in Rivers Worldwide (2014–2024): A Review. Sustainability 2024, 16, 8066. [Google Scholar] [CrossRef]
- Bachelet, D.; Verner, M.-A.; Neri, M.; Cordina Duverger, É.; Charlier, C.; Arveux, P.; Haddad, S.; Guénel, P. Breast Cancer and Exposure to Organochlorines in the CECILE Study: Associations with Plasma Levels Measured at the Time of Diagnosis and Estimated during Adolescence. Int. J. Environ. Res. Public Health 2019, 16, 271. [Google Scholar] [CrossRef]
- Lee, E.; Kinninger, A.; Ursin, G.; Tseng, C.; Hurley, S.; Wang, M.; Wang, Y.; Park, J.-S.; Petreas, M.; Deapen, D.; et al. Serum Levels of Commonly Detected Persistent Organic Pollutants and Per- and Polyfluoroalkyl Substances (PFASs) and Mammographic Density in Postmenopausal Women. Int. J. Environ. Res. Public Health 2020, 17, 606. [Google Scholar] [CrossRef] [PubMed]
- Menouni, A.; Duca, R.C.; Berni, I.; Khouchoua, M.; Ghosh, M.; El Ghazi, B.; Zouine, N.; Lhilali, I.; Akroute, D.; Pauwels, S.; et al. The Parental Pesticide and Offspring’s Epigenome Study: Towards an Integrated Use of Human Biomonitoring of Exposure and Effect Biomarkers. Toxics 2021, 9, 332. [Google Scholar] [CrossRef]
- Hoaib, S.; Ansari, M.A.; Ghazwani, M.; Hani, U.; Jamous, Y.F.; Alali, Z.; Wahab, S.; Ahmad, W.; Weir, S.A.; Alomary, M.N.; et al. Prospective Epigenetic Actions of Organo-Sulfur Compounds against Cancer: Perspectives and Molecular Mechanisms. Cancers 2023, 15, 697. [Google Scholar] [CrossRef]
- Furue, M.; Ishii, Y.; Tsukimori, K.; Tsuji, G. Aryl Hydrocarbon Receptor and Dioxin-Related Health Hazards—Lessons from Yusho. Int. J. Mol. Sci. 2021, 22, 708. [Google Scholar] [CrossRef]
- Choi, H.; Ha, K.; Kim, J.T.; Moon, M.K.; Joung, H.; Lee, H.K.; Pak, Y.K. Relationships among Dioxin-like Mitochondria Inhibitor Substances (MIS)-Mediated Mitochondria Dysfunction, Obesity, and Lung Function in a Korean Cohort. Toxics 2024, 12, 735. [Google Scholar] [CrossRef] [PubMed]
- Tue, N.M.; Kimura, E.; Maekawa, F.; Goto, A.; Uramaru, N.; Kunisue, T.; Suzuki, G. Uptake, Elimination and Metabolism of Brominated Dibenzofurans in Mice. Toxics 2024, 12, 656. [Google Scholar] [CrossRef] [PubMed]
- Ennour-Idrissi, K.; Ayotte, P.; Diorio, C. Persistent Organic Pollutants and Breast Cancer: A Systematic Review and Critical Appraisal of the Literature. Cancers 2019, 11, 1063. [Google Scholar] [CrossRef]
- Brown, L.J.; Achinger-Kawecka, J.; Portman, N.; Clark, S.; Stirzaker, C.; Lim, E. Epigenetic Therapies and Biomarkers in Breast Cancer. Cancers 2022, 14, 474. [Google Scholar] [CrossRef]
- Kim, A.; Mo, K.; Kwon, H.; Choe, S.; Park, M.; Kwak, W.; Yoon, H. Epigenetic Regulation in Breast Cancer: Insights on Epidrugs. Epigenomes 2023, 7, 6. [Google Scholar] [CrossRef]
- Roy, D.; Morgan, M.; Yoo, C.; Deoraj, A.; Roy, S.; Yadav, V.K.; Garoub, M.; Assaggaf, H.; Doke, M. Integrated Bioinformatics, Environmental Epidemiologic and Genomic Approaches to Identify Environmental and Molecular Links Between Endometriosis and Breast Cancer. Int. J. Mol. Sci. 2015, 16, 25285–25322. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, S.; Arora, I.; Tollefsbol, T.O. Impact of Stilbenes as Epigenetic Modulators of Breast Cancer Risk and Associated Biomarkers. Int. J. Mol. Sci. 2021, 22, 10033. [Google Scholar] [CrossRef]
- Bhat, S.S.; Prasad, S.K.; Shivamallu, C.; Prasad, K.S.; Syed, A.; Reddy, P.; Cull, C.A.; Amachawadi, R.G. Genistein: A Potent Anti-Breast Cancer Agent. Curr. Issues Mol. Biol. 2021, 43, 1502–1517. [Google Scholar] [CrossRef] [PubMed]
- Sohel, M.; Biswas, P.; Al Amin, M.; Hossain, M.A.; Sultana, H.; Dey, D.; Aktar, S.; Setu, A.; Khan, M.S.; Paul, P.; et al. Genistein, a Potential Phytochemical against Breast Cancer Treatment-Insight into the Molecular Mechanisms. Processes 2022, 10, 415. [Google Scholar] [CrossRef]
- Donovan, M.G.; Selmin, O.I.; Doetschman, T.C.; Romagnolo, D.F. Epigenetic Activation of BRCA1 by Genistein In Vivo and Triple Negative Breast Cancer Cells Linked to Antagonism toward Aryl Hydrocarbon Receptor. Nutrients 2019, 11, 2559. [Google Scholar] [CrossRef] [PubMed]
- Filippone, A.; Rossi, C.; Rossi, M.M.; Di Micco, A.; Maggiore, C.; Forcina, L.; Natale, M.; Costantini, L.; Merendino, N.; Di Leone, A.; et al. Endocrine Disruptors in Food, Estrobolome and Breast Cancer. J. Clin. Med. 2023, 12, 3158. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Osipo, C. Targeting Breast Cancer Stem Cells Using Naturally Occurring Phytoestrogens. Int. J. Mol. Sci. 2022, 23, 6813. [Google Scholar] [CrossRef]
- Salmerón-Bárcenas, E.G.; Zacapala-Gómez, A.E.; Torres-Rojas, F.I.; Antonio-Véjar, V.; Ávila-López, P.A.; Baños-Hernández, C.J.; Núñez-Martínez, H.N.; Dircio-Maldonado, R.; Martínez-Carrillo, D.N.; Ortiz-Ortiz, J.; et al. TET Enzymes and 5hmC Levels in Carcinogenesis and Progression of Breast Cancer: Potential Therapeutic Targets. Int. J. Mol. Sci. 2024, 25, 272. [Google Scholar] [CrossRef]
- Zheng, K.; Lyu, Z.; Chen, J.; Chen, G. 5-Hydroxymethylcytosine: Far Beyond the Intermediate of DNA Demethylation. Int. J. Mol. Sci. 2024, 25, 11780. [Google Scholar] [CrossRef] [PubMed]
- Theinel, M.H.; Nucci, M.P.; Alves, A.H.; Dias, O.F.M.; Mamani, J.B.; Garrigós, M.M.; Oliveira, F.A.; Rego, G.N.A.; Valle, N.M.E.; Cianciarullo, G.; et al. The Effects of Omega-3 Polyunsaturated Fatty Acids on Breast Cancer as a Preventive Measure or as an Adjunct to Conventional Treatments. Nutrients 2023, 15, 1310. [Google Scholar] [CrossRef]
- Bobin-Dubigeon, C.; Nazih, H.; Croyal, M.; Bard, J.-M. Link Between Omega 3 Fatty Acids Carried by Lipoproteins and Breast Cancer Severity. Nutrients 2022, 14, 2461. [Google Scholar] [CrossRef] [PubMed]
- Blewitt, M.; Whitelaw, E. The Use of Mouse Models to Study Epigenetics. Cold Spring Harb. Perspect. Biol. 2013, 5, a017939. [Google Scholar] [CrossRef] [PubMed]
- Kiyama, R. Estrogenic Flavonoids and Their Molecular Mechanisms of Action. J. Nutr. Biochem. 2023, 114, 109250. [Google Scholar] [CrossRef]
- Pejčić, T.; Zeković, M.; Bumbaširević, U.; Kalaba, M.; Vovk, I.; Bensa, M.; Popović, L.; Tešić, Ž. The Role of Isoflavones in the Prevention of Breast Cancer and Prostate Cancer. Antioxidants 2023, 12, 368. [Google Scholar] [CrossRef] [PubMed]
- Romagnolo, D.F.; Daniels, K.D.; Grunwald, J.T.; Ramos, S.A.; Propper, C.R.; Selmin, O.I. Epigenetics of Breast Cancer: Modifying Role of Environmental and Bioactive Food Compounds. Mol. Nutr. Food Res. 2016, 60, 1310–1329. [Google Scholar] [CrossRef]
- Da Cruz, R.S.; Chen, E.; Smith, M.; Bates, J.; De Assis, S. Diet and Transgenerational Epigenetic Inheritance of Breast Cancer: The Role of the Paternal Germline. Front. Nutr. 2020, 7, 93. [Google Scholar] [CrossRef]
- Patrizi, B.; Siciliani de Cumis, M. TCDD Toxicity Mediated by Epigenetic Mechanisms. Int. J. Mol. Sci. 2018, 19, 4101. [Google Scholar] [CrossRef] [PubMed]
- Harandi-Zadeh, S.; Boycott, C.; Beetch, M.; Yang, T.; Martin, B.J.E.; Ren, K.; Kwasniak, A.; Dupuis, J.H.; Lubecka, K.; Yada, R.Y.; et al. Pterostilbene Changes Epigenetic Marks at Enhancer Regions of Oncogenes in Breast Cancer Cells. Antioxidants 2021, 10, 1232. [Google Scholar] [CrossRef]
- Lee, R.S.; Sad, K.; Fawwal, D.V.; Spangle, J.M. Emerging Role of Epigenetic Modifiers in Breast Cancer Pathogenesis and Therapeutic Response. Cancers 2023, 15, 4005. [Google Scholar] [CrossRef] [PubMed]
- Aldeli, N.; Murphy, D.; Hanano, A. Impact of Dioxins on Reproductive Health in Female Mammals. Front. Toxicol. 2024, 6, 1392257. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-N.; Hung, C.-H.; Hou, C.-Y.; Chang, C.-I.; Tain, Y.-L. Perinatal Resveratrol Therapy to Dioxin-Exposed Dams Prevents the Programming of Hypertension in Adult Rat Offspring. Antioxidants 2021, 10, 1393. [Google Scholar] [CrossRef] [PubMed]
- Overhof, D.R.; Kwekel, J.C.; Humes, D.G.; Burgoon, L.D.; Zacharewski, T.R. Dioxin Induces an Estrogen-Like, Estrogen Receptor-Dependent Gene Expression Response in the Murine Uterus. Mol. Pharmacol. 2006, 69, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Ellsworth, L.; McCaffery, H.; Chernyak, S.; Lam, S.; Sargis, R.M.; Padmanabhan, V.; Gregg, B. Lactational Exposure to Polychlorinated Biphenyls Is Higher in Overweight /Obese Women and Associated with Altered Infant Growth Trajectory: A Pilot Study. Curr. Res. Toxicol. 2020, 1, 133–140. [Google Scholar] [CrossRef]
- Hang, B. Formation and Repair of Tobacco Carcinogen-Derived Bulky DNA Adducts. J. Nucleic Acids 2010, 2010, 709521. [Google Scholar] [CrossRef]
- White, S.S.; Fenton, S.E.; Hines, E.P. Endocrine Disrupting Properties of Perfluorooctanoic Acid. J. Steroid Biochem. Mol. Biol. 2011, 127, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Group on Hormonal Factors in Breast Cancer. Menarche, Menopause, and Breast Cancer Risk: Individual Participant Meta-Analysis, Including 118 964 Women with Breast Cancer from 117 Epidemiological Studies. Lancet Oncol. 2012, 13, 1141–1151. [Google Scholar] [CrossRef]
- Pierozan, P.; Jerneren, F.; Karlsson, O. Perfluorooctanoic Acid (PFOA) Exposure Promotes Proliferation, Migration and Invasion Potential in Human Breast Epithelial Cells. Arch. Toxicol. 2018, 92, 1729–1739. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Mirji, N.; Irudayaraj, J. Epigenetic Toxicity of PFOA and GenX in HepG2 Cells and Their Role in Lipid Metabolism. Toxicol. In Vitro 2020, 65, 104797. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.-Y.; Xu, X.-L.; Ma, W.-Z.; Deng, S.-L.; Lian, Z.-X.; Yu, K. Effects of Organochlorine Pesticide Residues in Maternal Body on Infants. Front. Endocrinol. 2022, 13, 890307. [Google Scholar] [CrossRef]
- Aubé, M.; Larochelle, C.; Ayotte, P. 1,1-Dichloro-2,2-Bis(p-Chlorophenyl)Ethylene (p,p’-DDE) Disrupts the Estrogen-Androgen Balance Regulating the Growth of Hormone-Dependent Breast Cancer Cells. Breast Cancer Res. 2008, 10, R16. [Google Scholar] [CrossRef]
- Cocco, P.; Fadda, D.; Billai, B.; D’Atri, M.; Melis, M.; Blair, A. Cancer Mortality among Men Occupationally Exposed to Dichlorodiphenyltrichloroethane. Cancer Res. 2005, 65, 9588–9594. [Google Scholar] [CrossRef]
- Strong, A.L.; Shi, Z.; Strong, M.J.; Miller, D.F.B.; Rusch, D.B.; Buechlein, A.M.; Flemington, E.K.; McLachlan, J.A.; Nephew, K.P.; Burow, M.E.; et al. Effects of the Endocrine-Disrupting Chemical DDT on Self-Renewal and Differentiation of Human Mesenchymal Stem Cells. Environ. Health Perspect. 2015, 123, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Esposito, E.; Indolfi, C.; Bello, I.; Smimmo, M.; Vellecco, V.; Schettino, A.; Montanaro, R.; Morroni, F.; Sita, G.; Graziosi, A.; et al. The Endocrine Disruptor Vinclozolin Causes Endothelial Injury via eNOS/Nox4/IRE1α Signaling. Eur. J. Pharmacol. 2024, 977, 176758. [Google Scholar] [CrossRef]
- Van Maele-Fabry, G.; Lombaert, N.; Lison, D. Dietary Exposure to Cadmium and Risk of Breast Cancer in Postmenopausal Women: A Systematic Review and Meta-Analysis. Environ. Int. 2016, 86, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pullella, K.; Kotsopoulos, J. Arsenic Exposure and Breast Cancer Risk: A Re-Evaluation of the Literature. Nutrients 2020, 12, 3305. [Google Scholar] [CrossRef] [PubMed]
- Peivasteh-roudsari, L.; Barzegar-bafrouei, R.; Sharifi, K.A.; Azimisalim, S.; Karami, M.; Abedinzadeh, S.; Asadinezhad, S.; Tajdar-oranj, B.; Mahdavi, V.; Alizadeh, A.M.; et al. Origin, Dietary Exposure, and Toxicity of Endocrine-Disrupting Food Chemical Contaminants: A Comprehensive Review. Heliyon 2023, 9, e18140. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, H.; Liu, S. Low-Dose Bisphenol A Exposure: A Seemingly Instigating Carcinogenic Effect on Breast Cancer. Adv. Sci. 2017, 4, 1600248. [Google Scholar] [CrossRef]
- Zuccarello, P.; Oliveri Conti, G.; Cavallaro, F.; Copat, C.; Cristaldi, A.; Fiore, M.; Ferrante, M. Implication of Dietary Phthalates in Breast Cancer. A Systematic Review. Food Chem. Toxicol. 2018, 118, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Fiolet, T.; Casagrande, C.; Nicolas, G.; Horvath, Z.; Frenoy, P.; Weiderpass, E.; Katzke, V.; Kaaks, R.; Rodriguez-Barranco, M.; Panico, S.; et al. Dietary Intakes of Dioxins and Polychlorobiphenyls (PCBs) and Breast Cancer Risk in 9 European Countries. Environ. Int. 2022, 163, 107213. [Google Scholar] [CrossRef]
- González, N.; Domingo, J.L. Polychlorinated Dibenzo-p-Dioxins and Dibenzofurans (PCDD/Fs) in Food and Human Dietary Intake: An Update of the Scientific Literature. Food Chem. Toxicol. 2021, 157, 112585. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. N. | Endocrine Disruptor | Mode of Exposer | Epigenetic Effect on Breast Cancer | Epigenetic Effect on Other Tissues | Affected Signalling Mechanism | References |
|---|---|---|---|---|---|---|
| 1 | Cadmium | As a naturally occurring element of the Earth’s crust, tobacco smoke and food contain cadmium. | Modified methylation of CCT3 chaperonin containing TCP1 subunit 3 (CCT3) and Thioredoxin Reductase (TXNRD1). | Thyroid, pancreas, prostate, and kidney. | Important signalling pathways such the NF-κB, p53, and MAPK (Mitogen-activated protein kinase) pathways are impacted by Cd2+, either directly or indirectly. | [30,33] |
| 2 | Arsenic | Arsenic is a common component in semiconductor circuits and lead alloys that are employed in batteries and arms. | Matrix metalloproteinas-2 (MMP2) and MMP-9 (Matrix metalloproteinase) are TNFR (tumour necrosis factor receptor) family members. | The highest associations between long-term exposure to arsenic and cancer are seen in bladder, lung, and skin cancers. Liver and kidney. | Rac, Rho, and MEKK 1–4 (mitogen-activated protein kinase kinase kinase-1) mediate JNK activation, whereas Ras/Raf/Mek signalling activates ERKs and p38 signalling. The MAPK cascade is responsible for arsenic poisoning. | [30] |
| 3 | Lead | Lead (Pb) is found naturally in the Earth’s crust. | WNT signalling epigenetic modification in ER+ breast cancer, SFRP and DKK are two examples of WNT antagonist genes whose epigenetic silencing leads to breast cancer. | Blood, digestive organs, brain, nerves, and more. | Lead causes chromatin aggregation through histone-DNA cross-links and DNA structural instability. | [32,33] |
| 4 | Bisphenol A (BPA) | Dental sealants, thermal paper, epoxy resins, and plastics. | Modifications in CDNK2A (cyclin-dependent kinase inhibitor 2A), THBS1 (Thrombospondin 1), BRCA1, CCNA1, LAMP3, TNFRSF10C, and TNFRSF10D methylation. | Increased brain tissue DNMT activity. | The activation of AKT and ERK1/2 is necessary for the proliferative and prosurvival effects of BPA in breast cancer. | [34,35] |
| 5 | Phthalates | Utilised as liquid plasticisers, which are used to make wall covers, tiles for the floor, pacifiers for teethers, toys, furniture fabric and mattresses, textiles, household goods, and medical equipment. | Kcnk5, COX-2/PGE2, PPARa, HER2/neu, v-myc, and c-myc. | The male reproductive system may be harmed by phthalate. | PKA is short for cyclin-dependent kinase (CDK). AhR-HDAC6-c-Myc and COX-2/PGE2 pathways are activated. | [36,37] |
| 6 | Polychlorinated biphenyls (PCBs) | Agents for heat transfer and coolants. | The AHRR gene’s methylation | Increased DNMT and SAM content, which raises methylation in rat liver cells | PCBs cause dysregulatory thyroid hormone and impairment of intracellular calcium signalling. | [38,39] |
| 7 | Diethylstilbestrol (DES) | A nonsteroidal oestrogen drug is diethylstilbestrol, often known as stilboestrol or stilboestrol. | Elevation of H3 trimethylation through EZH2 expression increases. | Hox gene methylation pattern in mouse endometrium; higher expression of Dnmt1 in mouse uterus. | DES and E2 share a number of pathways, such as the oestrogen receptor pathway and mammary gland development. | [40,41] |
| 8 | Polycyclic aromatic hydrocarbons (PAHs) | Incomplete combustion of materials such as coal, wood, cigarettes, and petroleum oil. | Generates DNA adducts close to breast epithelial methylation sites. | Lung cancer. | The stimulation of cytochrome P450 to produce DNA. The main factor contributing to PAH-induced carcinogenesis is epigenetic changes. | [42,43] |
| S.N. | Group of Chemicals | Agent | Origin | Main Source of Dietary Exposure | References |
|---|---|---|---|---|---|
| 1 | Cd2+ | Cadmium and cadmium compounds | Erosion, weathering, river transport, volcanic activity, and human actions including burning trash, smoking cigarettes, burning metal ore, using fossil fuels, and industrial pollution | Products from agriculture, seafood, shellfish | [197] |
| 2 | As | Agricultural products, especially rice | Occurring naturally in the soil, highly released by forest fires, volcanic eruptions, rock erosion, human activities, and chemicals such as soap, paint, dye, metal, medication, semi-conductors, fertilisers, and pesticides | Agricultural goods, particularly rice | [198] |
| 3 | Pb | Methylmercury compounds | Cans of food, water pipes, polluted water, paint, cosmetics, batteries, fuel, traditional medicine, Pb-crystal, kids’ toys, vinyl lunchboxes, and cigarettes | Seafood, poultry | [199] |
| 4 | Bisphenol | Tetrabromobisphenol A | Epoxy resins and polycarbonate plastics | Canned food | [200] |
| 5 | Phthalate | Di (2-ethylhexyl) phthalate | Detergents, plasticisers, and pesticides | Cereals, veggies, and legumes | [201] |
| 6 | Biphenyls | Polychlorinated biphenyls that resemble dioxin | Development, application, and removal of items treated with PCBs, accidental emissions resulting from burning operations, and re-emission of PCBs from water, sediment, and soil in environmental reservoirs | Seafood, meat, dairy, and fats | [202] |
| 7 | Dibenzofurans and dioxins | 2,3,7,8-Tetrachlorodibenzo-para-dioxin | Byproducts in the processes of production and disposal (organochlorides) Manufacturing, bleaching of paper, and burning of materials containing chloride | Eggs, seafood, dairy products, and adipose tissue from cows | [203] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, D.D. Epigenetic Mechanisms of Endocrine-Disrupting Chemicals in Breast Cancer and Their Impact on Dietary Intake. J. Xenobiot. 2025, 15, 1. https://doi.org/10.3390/jox15010001
Singh DD. Epigenetic Mechanisms of Endocrine-Disrupting Chemicals in Breast Cancer and Their Impact on Dietary Intake. Journal of Xenobiotics. 2025; 15(1):1. https://doi.org/10.3390/jox15010001
Chicago/Turabian StyleSingh, Desh Deepak. 2025. "Epigenetic Mechanisms of Endocrine-Disrupting Chemicals in Breast Cancer and Their Impact on Dietary Intake" Journal of Xenobiotics 15, no. 1: 1. https://doi.org/10.3390/jox15010001
APA StyleSingh, D. D. (2025). Epigenetic Mechanisms of Endocrine-Disrupting Chemicals in Breast Cancer and Their Impact on Dietary Intake. Journal of Xenobiotics, 15(1), 1. https://doi.org/10.3390/jox15010001