Cannabis- and Substance-Related Carcinogenesis in Europe: A Lagged Causal Inferential Panel Regression Study




Abstract
1. Background
- (1)
- Is there evidence for a link between cannabinoid exposure and patterns of cancer incidence in Europe?
- (2)
- How do these findings compare with similar data from elsewhere?
- (3)
- How do the putative carcinogenic effects of cannabis compare to those of the known carcinogens, tobacco and alcohol?
- (4)
- Was there evidence of inheritable tumourigenicity or cancerogenicity?
2. Methods
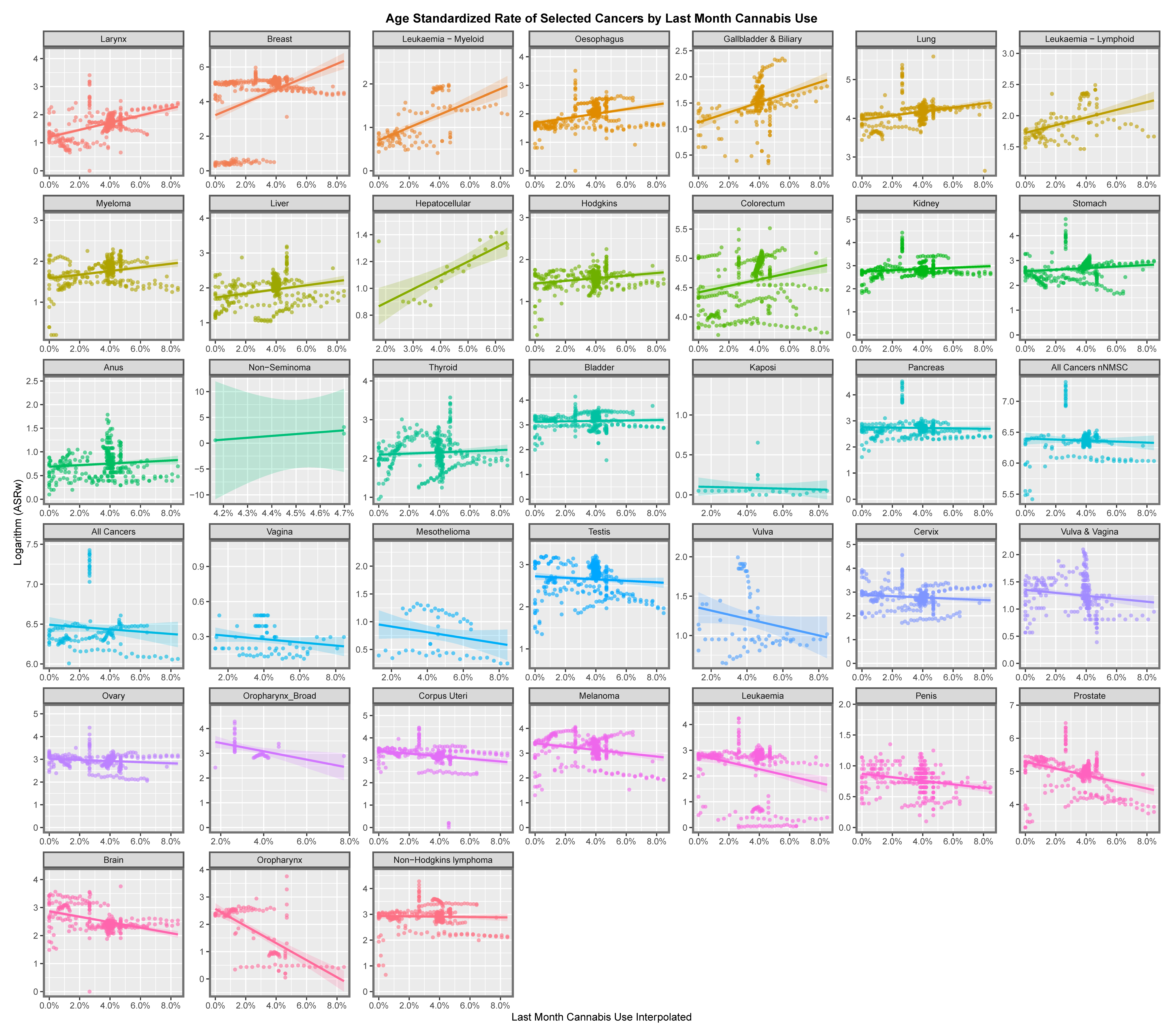
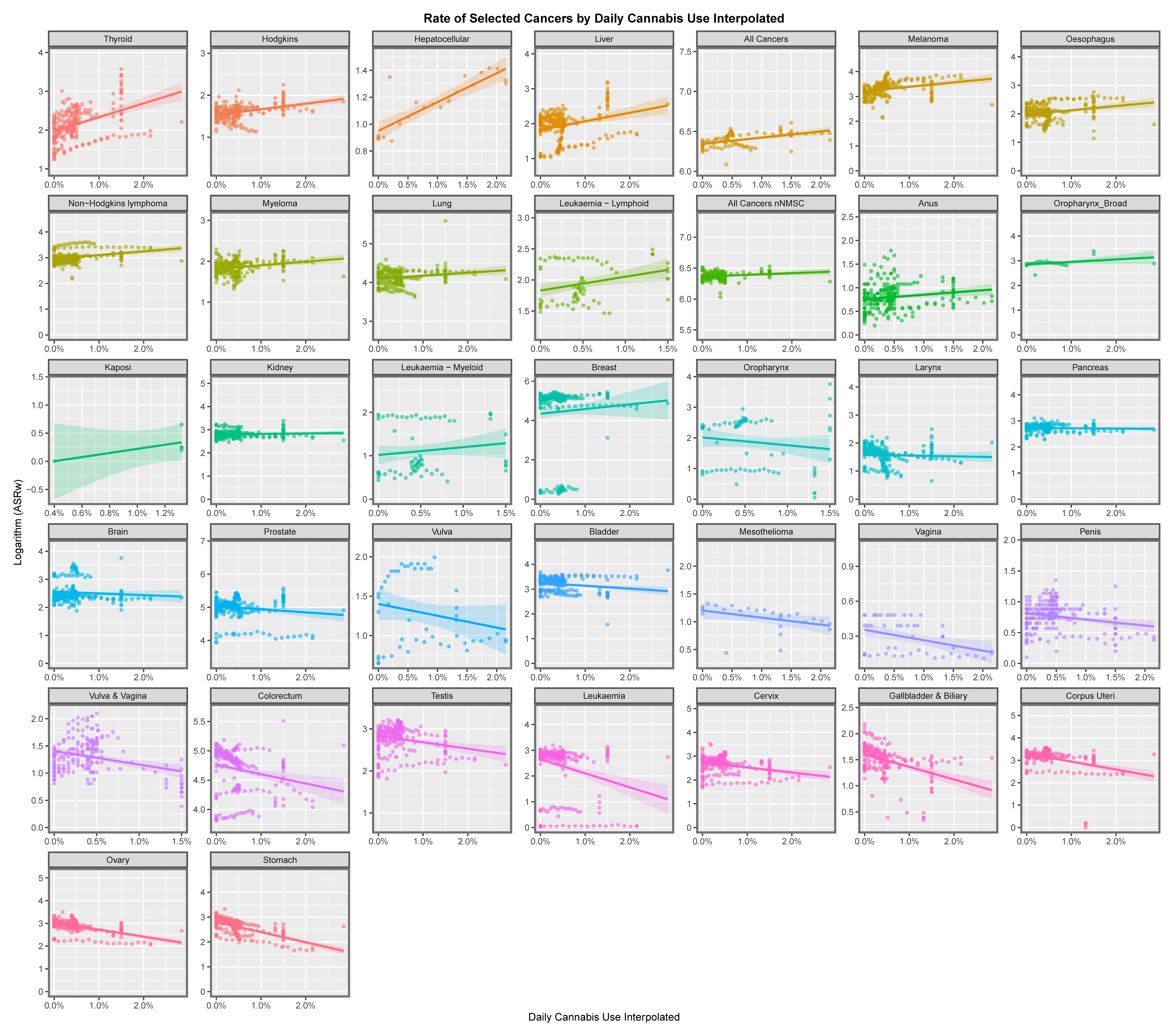
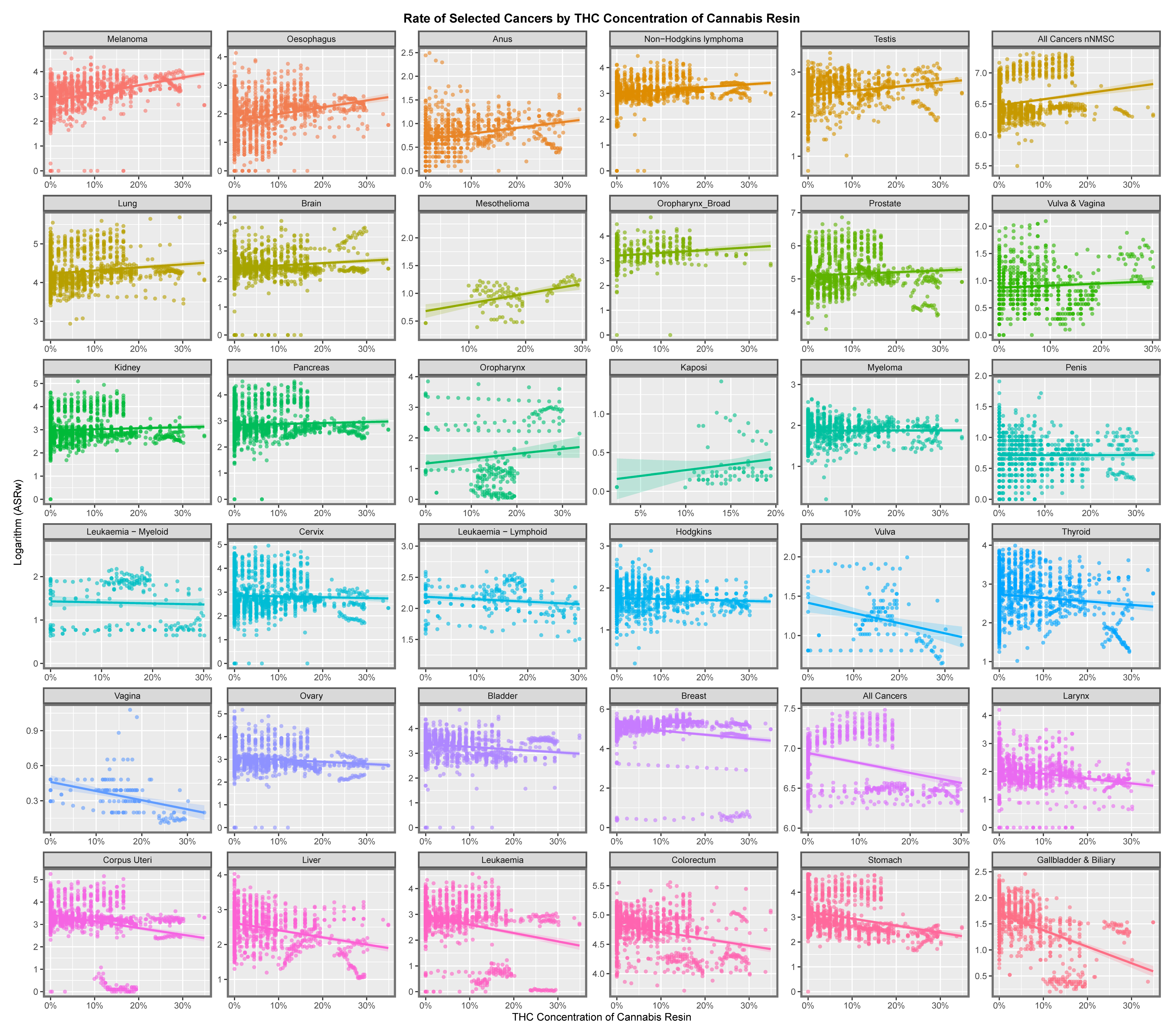
2.1. Data: Cancer—Annual Country Rates
2.2. Substances—Annual Country Estimates
2.3. Household Income
2.4. Data Analysis
2.5. Missing Data: Interpolation
2.6. Causal Inference
2.7. Ethics
3. Results
- 3.1
- Data
- 3.2
- Bivariate Analysis
- 3.2.1
- Continuous
- Graphical
- Tabular analysis
- Bivariate conclusions
- Correlation analysis
- Mapping review
- 3.2.2
- Categorical
- Tabular analysis
- Graphical analysis
- 3.3
- Multivariable panel regression analysis
- 3.3.1
- Additive
- Mixed-effects model
- Panel model—additive
- 3.3.2
- Interactive panel modelling
- No temporal lags (unlagged)
- Two-year temporal lags
- Four-year temporal lags
- Six-year temporal lags
- 3.3.3
- Multivariable conclusions
3.1. Data
3.2. Bivariate Analysis
3.2.1. Continuous Analysis
Graphical Analysis
Tabular Analysis
Bivariate Conclusions
Correlation Analysis
Mapping Analysis
3.2.2. Categorical Analysis
Tabular Analysis
Graphical Analysis
3.3. Multivariable Regression Analysis
3.3.1. Additive
Mixed-Effects Model
Panel Model—Additive
3.3.2. Interactive Panel Modelling
No Temporal Lags (Unlagged)
Two-Year Temporal Lags
Four-Year Temporal Lags
Six-Year Temporal Lags
3.3.3. Multivariable Conclusions
4. Discussion
4.1. Main Results and Interpretation
4.2. Cannabis-Linked Cancers
4.3. Specific Cancers
4.4. Reproductive Cancers
4.5. Cannabis Herb THC Concentration
4.6. Comparison with USA Data
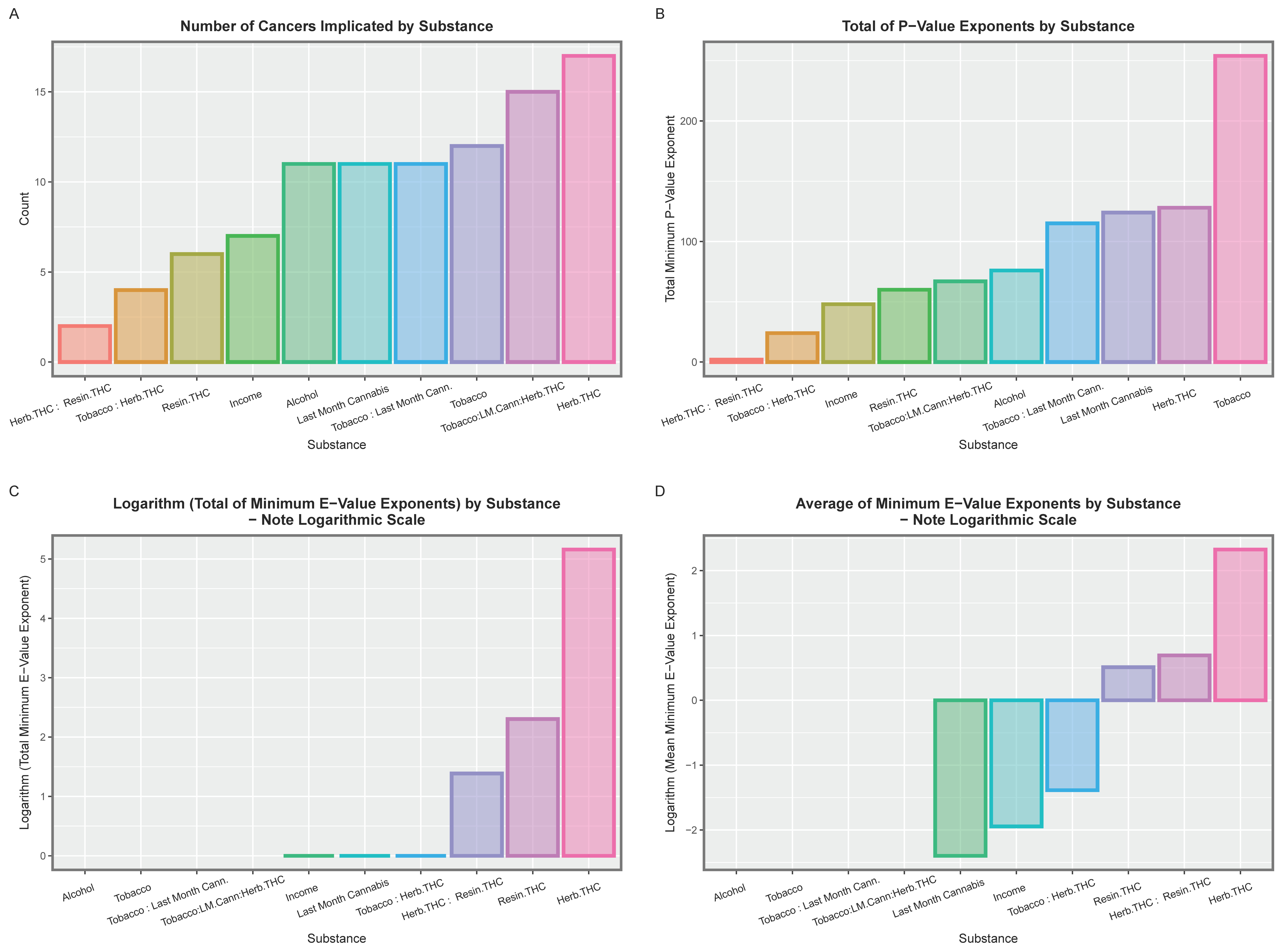
4.7. Causality
4.8. Specific Cannabinoids
4.9. Mechanisms
4.10. Carcinoma of the Testis
4.11. Structural Observations
4.12. Mechanistic Observations
4.13. Major Errors of Mitosis and Meiosis
4.14. Scope of Chromosomal Involvement
4.15. Epigenomic Effects
4.16. Comparison to Tobacco and Alcohol
4.17. Cocaine
4.18. Generalizability
4.19. Strengths and Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Acronym | Meaning |
| AFE | Attributable Fraction in the Exposed |
| AIC | Akaike Information Criterion |
| ALL | Acute Lymphoid Leukaemia |
| AME | Average Marginal Effect |
| ASRe | Age Standardised Rate of cancer—European 2013 Population |
| ASRw | Age Standardised Rate of cancer—World Population, 1973 |
| CI5 | Cancer in Five Continents Report of IARC by the WHO |
| ECIS | European Cancer Information System |
| EMCDDA | European Monitoring Centre for Drugs and Drug Addiction |
| E-value | Expected Value |
| IARC | International Association Against Cancer |
| IPW | Inverse-Probability Weighting |
| mEV | Minimum E-value |
| PAF | Population Attributable Fraction |
| P-FDR | p-value Corrected for False Discovery Rate |
| PR | Prevalence Ratio |
| THC | Δ9-Tetrahydrocannabinol |
References
- Ghasemiesfe, M.; Barrow, B.; Leonard, S.; Keyhani, S.; Korenstein, D. Association Between Marijuana Use and Risk of Cancer: A Systematic Review and Meta-analysis. JAMA Netw. Open 2019, 2, e1916318–e1916332. [Google Scholar] [CrossRef]
- Volkow, N.D.; Compton, W.M.; Weiss, S.R. Adverse health effects of marijuana use. N. Engl. J. Med. 2014, 371, 878–879. [Google Scholar] [CrossRef]
- Rehm, J.; Manthey, J. Cannabis and public health: A global experiment without control. World Psychiatry 2020, 19, 192–194. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, R.C.; Allebeck, P.; Akre, O.; McGlynn, K.A.; Sidorchuk, A. Cannabis Use and Incidence of Testicular Cancer: A 42-Year Follow-up of Swedish Men between 1970 and 2011. Cancer Epidemiol. Biomark. Prev. 2017, 26, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Daling, J.R.; Doody, D.R.; Sun, X.; Trabert, B.L.; Weiss, N.S.; Chen, C.; Biggs, M.L.; Starr, J.R.; Dey, S.K.; Schwartz, S.M. Association of marijuana use and the incidence of testicular germ cell tumors. Cancer 2009, 115, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Lacson, J.C.; Carroll, J.D.; Tuazon, E.; Castelao, E.J.; Bernstein, L.; Cortessis, V.K. Population-based case-control study of recreational drug use and testis cancer risk confirms an association between marijuana use and nonseminoma risk. Cancer 2012, 118, 5374–5383. [Google Scholar] [CrossRef] [PubMed]
- Trabert, B.; Sigurdson, A.J.; Sweeney, A.M.; Strom, S.S.; McGlynn, K.A. Marijuana use and testicular germ cell tumors. Cancer 2011, 117, 848–853. [Google Scholar] [CrossRef]
- Cheng, L.; Albers, P.; Berney, D.M.; Feldman, D.R.; Daugaard, G.; Gilligan, T.; Looijenga, L.H.J. Testicular cancer. Nat. Rev. Dis. Prim. 2018, 4, 29. [Google Scholar] [CrossRef]
- Gilbert, D.; Rapley, E.; Shipley, J. Testicular germ cell tumours: Predisposition genes and the male germ cell niche. Nat. Rev. Cancer 2011, 11, 278–288. [Google Scholar] [CrossRef]
- Gurney, J.; Shaw, C.; Stanley, J.; Signal, V.; Sarfati, D. Cannabis exposure and risk of testicular cancer: A systematic review and meta-analysis. BMC Cancer 2015, 15, 897–906. [Google Scholar] [CrossRef]
- Hanna, N.H.; Einhorn, L.H. Testicular cancer—Discoveries and updates. N. Engl. J. Med. 2014, 371, 2005–2016. [Google Scholar] [CrossRef]
- Huyghe, E.; Matsuda, T.; Thonneau, P. Increasing incidence of testicular cancer worldwide: A review. J. Urol. 2003, 170, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Rajpert-De Meyts, E.; McGlynn, K.A.; Okamoto, K.; Jewett, M.A.; Bokemeyer, C. Testicular germ cell tumours. Lancet 2016, 387, 1762–1774. [Google Scholar] [CrossRef]
- Oosterhuis, J.W.; Looijenga, L.H.J. Germ Cell Tumors from a Developmental Perspective: Cells of Origin, Pathogenesis, and Molecular Biology (Emerging Patterns). In Pathology and Biology of Human Germ Cell Tumors; Nogales, F.F., Jimenez, R.E., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 23–129. [Google Scholar]
- McGlynn, K.A.; Trabert, B. Adolescent and adult risk factors for testicular cancer. Nat. Rev. Urol. 2012, 9, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Baler, R.D.; Compton, W.M.; Weiss, S.R. Adverse health effects of marijuana use. N. Engl. J. Med. 2014, 370, 2219–2227. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. Causal inference multiple imputation investigation of the impact of cannabinoids and other substances on ethnic differentials in US testicular cancer incidence. BMC Pharmacol. Toxicol. 2021, 22, 40–71. [Google Scholar] [CrossRef] [PubMed]
- Patsenker, E.; Stickel, F. Cannabinoids in liver diseases. Clin. Liver Dis. 2016, 7, 21–25. [Google Scholar] [CrossRef]
- Reichenbach, V.; Ros, J.; Fernandez-Varo, G.; Casals, G.; Melgar-Lesmes, P.; Campos, T.; Makriyannis, A.; Morales-Ruiz, M.; Jimenez, W. Prevention of fibrosis progression in CCl4-treated rats: Role of the hepatic endocannabinoid and apelin systems. J. Pharmacol. Exp. Ther. 2012, 340, 629–637. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Lin, H.C.; Huang, Y.T.; Lee, T.Y.; Hou, M.C.; Wang, Y.W.; Lee, F.Y.; Lee, S.D. Effect of chronic CB1 cannabinoid receptor an-tagonism on livers of rats with biliary cirrhosis. Clin. Sci. 2007, 112, 533–542. [Google Scholar] [CrossRef]
- Mukhopadhyay, B.; Schuebel, K.; Mukhopadhyay, P.; Cinar, R.; Godlewski, G.; Xiong, K.; Mackie, K.; Lizak, M.; Yuan, Q.; Goldman, D.; et al. Cannabinoid receptor 1 promotes hepatocellular carcinoma initiation and progression through multiple mechanisms. Hepatology 2015, 61, 1615–1626. [Google Scholar] [CrossRef]
- Petrick, J.L.; Florio, A.A.; Znaor, A.; Ruggieri, D.; Laversanne, M.; Alvarez, C.S.; Ferlay, J.; Valery, P.C.; Bray, F.; McGlynn, K.A. International trends in hepatocellular carcinoma incidence, 1978–2012. Int. J. Cancer 2020, 147, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Efird, J.T.; Friedman, G.D.; Sidney, S.; Klatsky, A.; Habel, L.A.; Udaltsova, N.V.; Van den Eeden, S.; Nelson, L.M. The risk for malignant primary adult-onset glioma in a large, multiethnic, managed-care cohort: Cigarette smoking and other lifestyle behaviors. J. Neuro-Oncol. 2004, 68, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.F.; Morgenstern, H.; Spitz, M.R.; Tashkin, D.P.; Yu, G.P.; Marshall, J.R.; Hsu, T.C.; Schantz, S.P. Marijuana use and increased risk of squamous cell carcinoma of the head and neck. Cancer. Epidemiol. Biomark. Prev. 1999, 8, 1071–1078. [Google Scholar]
- Hashibe, M.; Ford, D.E.; Zhang, Z.F. Marijuana smoking and head and neck cancer. J. Clin. Pharmacol. 2002, 42, 103S–107S. [Google Scholar] [CrossRef] [PubMed]
- Aldington, S.; Harwood, M.; Cox, B.; Weatherall, M.; Beckert, L.; Hansell, A.; Pritchard, A.; Robinson, G.; Beasley, R. Cannabis use and risk of lung cancer: A case-control study. Eur. Respir. J. 2008, 31, 280–286. [Google Scholar] [CrossRef]
- Voirin, N.; Berthiller, J.; Benhaim-Luzon, V.; Boniol, M.; Straif, K.; Ayoub, W.B.; Ayed, F.B.; Sasco, A.J. Risk of lung cancer and past use of cannabis in Tunisia. J. Thorac. Oncol. 2006, 1, 577–579. [Google Scholar] [CrossRef]
- Berthiller, J.; Straif, K.; Boniol, M.; Voirin, N.; Benhaim-Luzon, V.; Ayoub, W.B.; Dari, I.; Laouamri, S.; Hamdi-Cherif, M.; Bartal, M.; et al. Cannabis smoking and risk of lung cancer in men: A pooled analysis of three studies in Maghreb. J. Thorac. Oncol. 2008, 3, 1398–1403. [Google Scholar] [CrossRef]
- Moiche Bokobo, P.; Atxa de la Presa, M.A.; Cuesta Angulo, J. Transitional cell carcinoma in a young heavy marihuana smoker. Archiv. Esp. Urol. 2001, 54, 165–167. [Google Scholar]
- Chacko, J.A.; Heiner, J.G.; Siu, W.; Macy, M.; Terris, M.K. Association between marijuana use and transitional cell carcinoma. Urology 2006, 67, 100–104. [Google Scholar] [CrossRef]
- Nieder, A.M.; Lipke, M.C.; Madjar, S. Transitional cell carcinoma associated with marijuana: Case report and review of the liter-ature. Urology 2006, 67, 200. [Google Scholar] [CrossRef]
- Sidney, S.; Quesenberry, C.P., Jr.; Friedman, G.D.; Tekawa, I.S. Marijuana use and cancer incidence (California, United States). Cancer Causes Control 1997, 8, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Hashibe, M.; Morgenstern, H.; Cui, Y.; Tashkin, D.P.; Zhang, Z.F.; Cozen, W.; Mack, T.M.; Greenland, S. Marijuana use and the risk of lung and upper aerodigestive tract cancers: Results of a population-based case-control study. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Hashibe, M.; Straif, K.; Tashkin, D.P.; Morgenstern, H.; Greenland, S.; Zhang, Z.F. Epidemiologic review of marijuana use and cancer risk. Alcohol 2005, 35, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.Q.; Shu, X.O.; Steinbuch, M.; Severson, R.K.; Reaman, G.H.; Buckley, J.D.; Robison, L.L. Paternal military service and risk for childhood leukemia in offspring. Am. J. Epidemiol. 2000, 151, 231–240. [Google Scholar] [CrossRef]
- Robison, L.L.; Buckley, J.D.; Daigle, A.E.; Wells, R.; Benjamin, D.; Arthur, D.C.; Hammond, G.D. Maternal drug use and risk of childhood nonlymphoblastic leukemia among offspring. An epidemiologic investigation implicating marijuana (a report from the childrens cancer study group). Cancer 1989, 63, 1904–1911. [Google Scholar] [CrossRef]
- Bluhm, E.C.; Daniels, J.; Pollock, B.H.; Olshan, A.F. Maternal use of recreational drugs and neuroblastoma in offspring: A report from the Children’s Oncology Group (United States). Cancer Causes Control 2006, 17, 663–669. [Google Scholar] [CrossRef]
- Grufferman, S.; Schwartz, A.G.; Ruymann, F.B.; Maurer, H.M. Parents’ use of cocaine and marijuana and increased risk of rhabdomyosarcoma in their children. Cancer Causes Control 1993, 4, 217–224. [Google Scholar] [CrossRef]
- Grobner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. A geospatiotemporal and causal inference epidemiological exploration of substance and cannabinoid exposure as drivers of rising US pediatric cancer rates. BMC Cancer 2021, 21, 197–230. [Google Scholar] [CrossRef]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef] [PubMed]
- SEER Explorer. Available online: https://seer.cancer.gov/explorer/application.html (accessed on 10 February 2023).
- Reece, A.S.; Hulse, G.K. Epidemiological Overview of Multidimensional Chromosomal and Genome Toxicity of Cannabis Exposure in Congenital Anomalies and Cancer Development. Sci. Rep. 2021, 11, 13892–13912. [Google Scholar] [CrossRef] [PubMed]
- Forrester, M.B.; Merz, R.D. Risk of selected birth defects with prenatal illicit drug use, Hawaii, 1986-2002. J. Toxicol. Environ. Health 2007, 70, 7–18. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Cannabis Teratology Explains Current Patterns of Coloradan Congenital Defects: The Contribution of Increased Cannabinoid Exposure to Rising Teratological Trends. Clin. Pediatr. 2019, 58, 1085–1123. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Cannabis Consumption Patterns Explain the East-West Gradient in Canadian Neural Tube Defect Incidence: An Ecological Study. Glob Pediatr Health 2019, 6, 1–12. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Cannabis in Pregnancy—Rejoinder, Exposition and Cautionary Tales. Psychiatr. Times 2020, 37. Available online: https://www.psychiatrictimes.com/view/cannabis-pregnancy-rejoinder-exposition-cautionary-tales. (accessed on 10 January 2023).
- Reece, A.S.; Hulse, G.K. Broad Spectrum epidemiological contribution of cannabis and other substances to the teratological profile of northern New South Wales: Geospatial and causal inference analysis. BMC Pharmacol. Toxicol. 2020, 21, 75–103. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Geotemporospatial and causal inference epidemiological analysis of US survey and overview of cannabis, cannabidiol and cannabinoid genotoxicity in relation to congenital anomalies 2001–2015. BMC Pediatr. 2022, 22, 47–124. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Cannabinoid Genotoxicity and Congenital Anomalies: A Convergent Synthesis of European and USA Datasets. In Cannabis, Cannabinoids and Endocannabinoids; Preedy, V., Patel, V., Eds.; Elsevier: London, UK, 2022; Volume 1, pp. 71–92, Chapter 5. [Google Scholar]
- Reece, A.S.; Hulse, G.K. Cannabinoid and substance relationships of European congenital anomaly patterns: A space-time panel regression and causal inferential study. Environ. Epigenet. 2022, 8, dvab015. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Congenital anomaly epidemiological correlates of Δ8THC across USA 2003–16: Panel regression and causal inferential study. Environ. Epigenet. 2022, 8, dvac012. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. European epidemiological patterns of cannabis- and substance-related congenital cardiovascular anomalies: Geospatiotemporal and causal inferential study. Environ. Epigenet. 2022, 8, dvac015. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. Effects of cannabis on congenital limb anomalies in 14 European nations: A geospatiotemporal and causal inferential study. Environ. Epigenet. 2022, 8, dvac016. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. Chapter 3: Geospatiotemporal and Causal Inferential Analysis of United States Congenital Anomalies as a Function of Multiple Cannabinoid- and Substance- Exposures: Phenocopying Thalidomide and Hundred Megabase-Scale Genotoxicity. In Epidemiology of Cannabis: Genotoxicity and Neurotoxicity, Epigenomics and Aging; Elsevier: New York, NY, USA, 2023; Volume 1, In Press: 2500. [Google Scholar]
- Reece, A.S.; Hulse, G.K. Congenital Gastrointestinal Anomalies in Europe 2010–2019: A Geo-Spatiotemporal and Causal Inferential Study of Epidemiological Patterns in Relationship to Cannabis- and Substance Exposure. Gastrointest. Insights 2023, 14, 64–109. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. European Epidemiological Patterns of Cannabis- and Substance-Related Body Wall Congenital Anomalies: Geospatiotemporal and Causal Inferential Study. Int. J. Environ. Res. Public Health 2022, 19, 9027. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. European Epidemiological Patterns of Cannabis- and Substance- Related Congenital Chromosomal Anomalies: Geospatiotemporal and Causal Inferential Study. Int. J. Environ. Res. Public Health 2022, 19, 13769. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. European Epidemiological Patterns of Cannabis- and Substance-Related Congenital Neurological Anomalies: Geospatiotemporal and Causal Inferential Study. Int. J. Environ. Res. Public Health 2022, 20, 441. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. European Epidemiological Patterns of Cannabis- and Substance- Related Congenital Uronephrological Anomalies: Geospatiotemporal and Causal Inferential Study. Int. J. Environ. Res. Public Health 2022, 19, 13769. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Cannabis- and Substance-Related Epidemiological Patterns of Chromosomal Congenital Anomalies in Europe: Geospatiotemporal and Causal Inferential Study. Int. J. Environ. Res. Public Health 2022, 19, 11208. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. Canadian Cannabis Consumption and Patterns of Congenital Anomalies: An Ecological Geospatial Analysis. J. Addict. Med. 2020, 14, e195–e210. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Geospatiotemporal and Causal Inferential Study of European Epidemiological Patterns of Cannabis- and Substance-Related Congenital Orofacial Anomalies. J. Xenobiot. 2023, 13, 42–74. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Chromothripsis and epigenomics complete causality criteria for cannabis- and addiction-connected carcinogenicity, congenital toxicity and heritable genotoxicity. Mutat. Res. 2016, 789, 15–25. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Patterns of Cannabis- and Substance- Related Congenital General Anomalies in Europe: A Geospatiotemporal and Causal Inferential Study. Pediatr. Rep. 2023, 15, 69–118. [Google Scholar] [CrossRef]
- Phillips, K.T.; Pedula, K.L.; Choi, N.G.; Tawara, K.K.; Simiola, V.; Satre, D.D.; Owen-Smith, A.; Lynch, F.F.; Dickerson, J. Chronic health conditions, acute health events, and healthcare utilization among adults over age 50 in Hawai’i who use cannabis: A matched cohort study. Drug Alcohol Depend. 2022, 234, 109387. [Google Scholar] [CrossRef]
- Reece, A.S.; Norman, A.; Hulse, G.K. Cannabis exposure as an interactive cardiovascular risk factor and accelerant of organismal ageing: A longitudinal study. BMJ Open 2016, 6, e011891–e011901. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.P.; Danoff, J.S.; Costello, M.A.; Hunt, G.L.; Hellwig, A.F.; Krol, K.M.; Gregory, S.G.; Giamberardino, S.N.; Sugden, K.; Connelly, J.J. Lifetime marijuana use and epigenetic age acceleration: A 17-year prospective examination. Drug Alcohol Depend. 2022, 233, 109363. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Y.; Zhang, B.; Lin, X.; Fu, X.; An, Y.; Zou, Y.; Wang, J.X.; Wang, Z.; Yu, T. Lactate metabolism in human health and disease. Signal Transduct. Target Ther. 2022, 7, 305. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J. How lactate links cannabis to social behaviour. Nature 2020, 583, 526–527. [Google Scholar] [CrossRef]
- Martin, L.J.; Cairns, E.A.; Heblinski, M.; Fletcher, C.; Krycer, J.R.; Arnold, J.C.; McGregor, I.S.; Bowen, M.T.; Anderson, L.L. Cannabichromene and Δ-Tetrahydrocannabinolic Acid Identified as Lactate Dehydrogenase-A Inhibitors by in Silico and in Vitro Screening. J. Nat. Prod. 2021, 84, 1469–1477. [Google Scholar] [CrossRef]
- Papadakis, D.P.; Michael, C.M.; Kephalas, T.A.; Miras, C.J. Effects of cannabis smoking in blood lactic acid and glucose in humans. Experientia 1974, 30, 1183–1184. [Google Scholar] [CrossRef]
- Yang, Z.; Yan, C.; Ma, J.; Peng, P.; Ren, X.; Cai, S.; Shen, X.; Wu, Y.; Zhang, S.; Wang, X.; et al. Lactylome analysis suggests lactylation-dependent mechanisms of metabolic adaptation in hepatocellular carcinoma. Nat. Metab. 2023, 5, 61–79. [Google Scholar] [CrossRef]
- Sarafian, T.A.; Habib, N.; Oldham, M.; Seeram, N.; Lee, R.P.; Lin, L.; Tashkin, D.P.; Roth, M.D. Inhaled marijuana smoke disrupts mitochondrial energetics in pulmonary epithelial cells in vivo. Am. J. Physiol. 2006, 290, L1202–L1209. [Google Scholar] [CrossRef]
- Badawy, Z.S.; Chohan, K.R.; Whyte, D.A.; Penefsky, H.S.; Brown, O.M.; Souid, A.K. Cannabinoids inhibit the respiration of human sperm. Fertil. Steril. 2009, 91, 2471–2476. [Google Scholar] [CrossRef]
- Wolff, V.; Schlagowski, A.I.; Rouyer, O.; Charles, A.L.; Singh, F.; Auger, C.; Schini-Kerth, V.; Marescaux, C.; Raul, J.S.; Zoll, J.; et al. Tetra-hydrocannabinol induces brain mitochondrial respiratory chain dysfunction and increases oxidative stress: A potential mechanism involved in cannabis-related stroke. BioMed Res. Int. 2015, 2015, 323706. [Google Scholar] [CrossRef]
- Rupprecht, A.; Theisen, U.; Wendt, F.; Frank, M.; Hinz, B. The Combination of Δ-Tetrahydrocannabinol and Cannabidiol Suppresses Mitochondrial Respiration of Human Glioblastoma Cells via Downregulation of Specific Respiratory Chain Proteins. Cancers 2022, 14, 3129. [Google Scholar] [CrossRef]
- Chiu, P.; Karler, R.; Craven, C.; Olsen, D.M.; Turkanis, S.A. The influence of delta9-tetrahydrocannabinol, cannabinol and cannabidiol on tissue oxygen consumption. Res. Commun. Chem. Pathol. Pharmacol. 1975, 12, 267–286. [Google Scholar] [PubMed]
- Koch, M.; Varela, L.; Kim, J.G.; Kim, J.D.; Hernandez-Nuno, F.; Simonds, S.E.; Castorena, C.M.; Vianna, C.R.; Elmquist, J.K.; Morozov, Y.M.; et al. Hypothalamic POMC neurons promote cannabinoid-induced feeding. Nature 2015, 519, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Hebert-Chatelain, E.; Desprez, T.; Serrat, R.; Bellocchio, L.; Soria-Gomez, E.; Busquets-Garcia, A.; Pagano Zottola, A.C.; Delamarre, A.; Cannich, A.; Vincent, P.; et al. A cannabinoid link between mitochondria and memory. Nature 2016, 539, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Harkany, T.; Horvath, T.L. (S)Pot on Mitochondria: Cannabinoids Disrupt Cellular Respiration to Limit Neuronal Activity. Cell Metab. 2017, 25, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.W.; Newton, C.; Larsen, K.; Lu, L.; Perkins, I.; Nong, L.; Friedman, H. The cannabinoid system and immune modulation. J. Leukocyte Biol. 2003, 74, 486–496. [Google Scholar] [CrossRef]
- Bisogno, T.; Di Marzo, V. Short- and long-term plasticity of the endocannabinoid system in neuropsychiatric and neurological disorders. Pharmacol. Res. 2007, 56, 428–442. [Google Scholar] [CrossRef]
- Benamar, K.; Geller, E.B.; Adler, M.W. First in vivo evidence for a functional interaction between chemokine and cannabinoid systems in the brain. J. Pharmacol. Exp. Ther. 2008, 325, 641–645. [Google Scholar] [CrossRef]
- Chandra, L.C.; Kumar, V.; Torben, W.; Vande Stouwe, C.; Winsauer, P.; Amedee, A.; Molina, P.E.; Mohan, M. Chronic administration of Delta9-tetrahydrocannabinol induces intestinal anti-inflammatory microRNA expression during acute simian immunodeficiency virus infection of rhesus macaques. J. Virol. 2015, 89, 1168–1181. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, T.K.; Meissler, J.J. Effects of Cannabinoids on T-cell Function and Resistance to Infection. J. Neuroimmune Pharmacol. 2015, 10, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Zumbrun, E.E.; Sido, J.M.; Nagarkatti, P.S.; Nagarkatti, M. Epigenetic Regulation of Immunological Alterations Following Prenatal Exposure to Marijuana Cannabinoids and its Long Term Consequences in Offspring. J. Neuroimmune Pharmacol. 2015, 10, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Chiurchiu, V. Endocannabinoids and Immunity. Cannabis Cannabinoid Res. 2016, 1, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Gallily, R.; Yekhtin, Z. Avidekel Cannabis extracts and cannabidiol are as efficient as Copaxone in suppressing EAE in SJL/J mice. Inflammopharmacology 2019, 27, 167–173. [Google Scholar] [CrossRef]
- Kaplan, B.L.F. Evaluation of Marijuana Compounds on Neuroimmune Endpoints in Experimental Autoimmune Encephalomyelitis. Curr. Protoc. Toxicol. 2018, 75, 11.25.1–11.25.22. [Google Scholar] [CrossRef]
- Pavarin, R.M.; Berardi, D. Mortality risk in a cohort of subjects reported by authorities for cannabis possession for personal use. Results of a longitudinal study. Epidemiol. Prev. 2011, 35, 89–93. [Google Scholar]
- von Greiff, N.; Skogens, L.; Berlin, M.; Bergmark, A. Mortality and Cause of Death-A 30-Year Follow-Up of Substance Misusers in Sweden. Subst. Use Misuse 2018, 53, 2043–2051. [Google Scholar] [CrossRef]
- Calabria, B.; Degenhardt, L.; Hall, W.; Lynskey, M. Does cannabis use increase the risk of death? Systematic review of epidemi-ological evidence on adverse effects of cannabis use. Drug Alcohol Rev. 2010, 29, 318–330. [Google Scholar] [CrossRef]
- Arendt, M.; Munk-Jørgensen, P.; Sher, L.; Jensen, S.O. Mortality among individuals with cannabis, cocaine, amphetamine, MDMA, and opioid use disorders: A nationwide follow-up study of Danish substance users in treatment. Drug Alcohol Depend. 2011, 114, 134–139. [Google Scholar] [CrossRef]
- Davstad, I.; Allebeck, P.; Leifman, A.; Stenbacka, M.; Romelsjo, A. Self-reported drug use and mortality among a nationwide sample of Swedish conscripts—A 35-year follow-up. Drug Alcohol Depend 2011, 118, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Muhuri, P.K.; Gfroerer, J.C. Mortality associated with illegal drug use among adults in the United States. Am. J. Drug Alcohol Abuse 2011, 37, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, R.C.; Cunningham, J.K.; Verdichevski, M.; Sykes, J.; Jaffer, S.R.; Kish, S.J. All-cause mortality among individuals with disorders related to the use of methamphetamine: A comparative cohort study. Drug Alcohol Depend. 2012, 125, 290–294. [Google Scholar] [CrossRef]
- Hser, Y.I.; Kagihara, J.; Huang, D.; Evans, E.; Messina, N. Mortality among substance-using mothers in California: A 10-year prospective study. Addiction 2012, 107, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Frost, L.; Mostofsky, E.; Rosenbloom, J.I.; Mukamal, K.J.; Mittleman, M.A. Marijuana use and long-term mortality among survivors of acute myocardial infarction. Am. Heart J. 2013, 165, 170–175. [Google Scholar] [CrossRef]
- Desai, R.; Patel, U.; Sharma, S.; Amin, P.; Bhuva, R.; Patel, M.S.; Sharma, N.; Shah, M.; Patel, S.; Savani, S.; et al. Recreational Marijuana Use and Acute Myocardial Infarction: Insights from Nationwide Inpatient Sample in the United States. Cureus 2017, 9, e1816. [Google Scholar] [PubMed]
- DeFilippis, E.M.; Singh, A.; Divakaran, S.; Gupta, A.; Collins, B.L.; Biery, D.; Qamar, A.; Fatima, A.; Ramsis, M.; Piplas, D.; et al. Cocaine and Marijuana Use among Young Adults Presenting with Myocardial Infarction: The Partners YOUNG-MI Registry. J. Am. Coll. Cardiol. 2018, 71, 2540–2551. [Google Scholar] [CrossRef]
- Fridell, M.; Bäckström, M.; Hesse, M.; Krantz, P.; Perrin, S.; Nyhlén, A. Prediction of psychiatric comorbidity on premature death in a cohort of patients with substance use disorders: A 42-year follow-up. BMC Psychiatr 2019, 19, 150. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Epidemiology of Cannabis: Genotoxicity, Neurotoxicity, Epigenomics and Aging; Elsevier: New York, NY, USA, 2023; Volume 1. [Google Scholar]
- Yang, J.H.; Hayano, M.; Griffin, P.T.; Amorim, J.A.; Bonkowski, M.S.; Apostolides, J.K.; Salfati, E.L.; Blanchette, M.; Munding, E.M.; Bhakta, M.; et al. Loss of epigenetic information as a cause of mammalian aging. Cell 2023, 186, 305–326.e27. [Google Scholar] [CrossRef]
- Schultz, M.B.; Sinclair, D.A. When stem cells grow old: Phenotypes and mechanisms of stem cell aging. Development 2016, 143, 3–14. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Chapter 5: Multivalent Cannabinoid Epigenotoxicities and Multigenerational Aging. In Epidemiology of Cannabis: Genotoxicity, Neurotoxicity, Epigenomics and Aging; Elsevier: New York, NY, USA, 2023; Volume 1, In Press: 2500. [Google Scholar]
- Reece, A.S.; Hulse, G.K. Epigenomic and Other Evidence for Cannabis-Induced Aging Contextualized in a Synthetic Epidemiologic Overview of Cannabinoid-Related Teratogenesis and Cannabinoid-Related Carcinogenesis. Int. J. Environ. Res. Public Health 2022, 19, 16721. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. Clinical Epigenomic Explanation of the Epidemiology of Cannabinoid Genotoxicity Manifesting as Transgenerational Teratogenesis, Cancerogenesis and Aging Acceleration. Int. J. Environ. Res. Public Health 2023, 20, 3360. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. Quadruple Convergence—Rising Cannabis Prevalence, Intensity, Concentration and Use Disorder Treatment. Lancet Reg. Health Eur. 2021, 10, 100245–100246. [Google Scholar] [CrossRef] [PubMed]
- Manthey, J.; Freeman, T.P.; Kilian, C.; Lopez-Pelayo, H.; Rehm, J. Public health monitoring of cannabis use in Europe: Prevalence of use, cannabis potency, and treatment rates. Lancet Reg. Health Eur. 2021, 10, 100227. [Google Scholar] [CrossRef] [PubMed]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef]
- Wala, J.; Beroukhim, R. The oncogene makes its escape. Science 2016, 351, 1398–1399. [Google Scholar] [CrossRef]
- Weintraub, A.S.; Li, C.H.; Zamudio, A.V.; Sigova, A.A.; Hannett, N.M.; Day, D.S.; Abraham, B.J.; Cohen, M.A.; Nabet, B.; Buckley, D.L.; et al. YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 2017, 171, 1573–1588.e28. [Google Scholar] [CrossRef]
- Isoda, T.; Morio, T.; Takagi, M. Noncoding RNA transcription at enhancers and genome folding in cancer. Cancer Sci. 2019, 110, 2328–2336. [Google Scholar] [CrossRef]
- Petrovic, J.; Zhou, Y.; Fasolino, M.; Goldman, N.; Schwartz, G.W.; Mumbach, M.R.; Nguyen, S.C.; Rome, K.S.; Sela, Y.; Zapataro, Z.; et al. Oncogenic Notch Promotes Long-Range Regulatory Interactions within Hyperconnected 3D Cliques. Mol. Cell 2019, 73, 1174–1190.e12. [Google Scholar] [CrossRef]
- Yang, M.; Vesterlund, M.; Siavelis, I.; Moura-Castro, L.H.; Castor, A.; Fioretos, T.; Jafari, R.; Lilljebjörn, H.; Odom, D.T.; Olsson, L.; et al. Proteogenomics and Hi-C reveal transcriptional dysregulation in high hyperdiploid childhood acute lymphoblastic leukemia. Nat. Commun. 2019, 10, 1519. [Google Scholar] [CrossRef]
- Kloetgen, A.; Thandapani, P.; Ntziachristos, P.; Ghebrechristos, Y.; Nomikou, S.; Lazaris, C.; Chen, X.; Hu, H.; Bakogianni, S.; Wang, J.; et al. Three-dimensional chromatin landscapes in T cell acute lymphoblastic leukemia. Nat. Genet. 2020, 52, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Diedrich, J.D.; Dong, Q.; Ferguson, D.C.; Bergeron, B.P.; Autry, R.J.; Qian, M.; Yang, W.; Smith, C.; Papizan, J.B.; Connelly, J.P.; et al. Profiling chromatin accessibility in pediatric acute lymphoblastic leukemia identifies subtype-specific chromatin landscapes and gene regulatory networks. Leukemia 2021, 35, 3078–3091. [Google Scholar] [CrossRef] [PubMed]
- Heide, T.; Househam, J.; Cresswell, G.D.; Spiteri, I.; Lynn, C.; Mossner, M.; Kimberley, C.; Fernandez-Mateos, J.; Chen, B.; Zapata, L.; et al. The co-evolution of the genome and epigenome in colorectal cancer. Nature 2022, 611, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Househam, J.; Heide, T.; Cresswell, G.D.; Spiteri, I.; Kimberley, C.; Zapata, L.; Lynn, C.; James, C.; Mossner, M.; Fernandez-Mateos, J.; et al. Phenotypic plasticity and genetic control in colorectal cancer evolution. Nature 2022, 611, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Lomakin, A.; Svedlund, J.; Strell, C.; Gataric, M.; Shmatko, A.; Rukhovich, G.; Park, J.S.; Ju, Y.S.; Dentro, S.; Kleshchevnikov, V.; et al. Spatial genomics maps the structure, nature and evolution of cancer clones. Nature 2022, 611, 594–602. [Google Scholar] [CrossRef]
- Luquette, L.J.; Miller, M.B.; Zhou, Z.; Bohrson, C.L.; Zhao, Y.; Jin, H.; Gulhan, D.; Ganz, J.; Bizzotto, S.; Kirkham, S.; et al. Single-cell genome sequencing of human neurons identifies somatic point mutation and indel enrichment in regulatory elements. Nat. Genet. 2022, 54, 1564–1571. [Google Scholar] [CrossRef]
- Wang, D.; Wu, W.; Callen, E.; Pavani, R.; Zolnerowich, N.; Kodali, S.; Zong, D.; Wong, N.; Noriega, S.; Nathan, W.J.; et al. Active DNA demethylation promotes cell fate specification and the DNA damage response. Science 2022, 378, 983–989. [Google Scholar] [CrossRef]
- Xu, J.; Song, F.; Lyu, H.; Kobayashi, M.; Zhang, B.; Zhao, Z.; Hou, Y.; Wang, X.; Luan, Y.; Jia, B.; et al. Subtype-specific 3D genome alteration in acute myeloid leukaemia. Nature 2022, 611, 387–398. [Google Scholar] [CrossRef]
- Xu, Z.; Lee, D.S.; Chandran, S.; Le, V.T.; Bump, R.; Yasis, J.; Dallarda, S.; Marcotte, S.; Clock, B.; Haghani, N.; et al. Structural variants drive context-dependent oncogene activation in cancer. Nature 2022, 612, 564–572. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, H.; Luan, Y.; Liu, T.; Yang, W.; Roberts, K.G.; Qian, M.X.; Zhang, B.; Yang, W.; Perez-Andreu, V.; et al. Noncoding genetic variation in GATA3 increases acute lymphoblastic leukemia risk through local and global changes in chromatin conformation. Nat. Genet. 2022, 54, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Isoda, T.; Moore, A.J.; He, Z.; Chandra, V.; Aida, M.; Denholtz, M.; Piet van Hamburg, J.; Fisch, K.M.; Chang, A.N.; Fahl, S.P.; et al. Non-coding Transcription Instructs Chromatin Folding and Compartmentalization to Dictate Enhancer-Promoter Communication and T Cell Fate. Cell 2017, 171, 103–119.e118. [Google Scholar] [CrossRef] [PubMed]
- Blevins, R.D.; Regan, J.D. Delta-9-Tetrahydrocannabinol: Effect on macromolecular synthesis in human and other mammalian cells. Arch. Toxicol. 1976, 35, 127–135. [Google Scholar] [CrossRef] [PubMed]
- McClean, D.K.; Zimmerman, A.M. Action of delta 9-tetrahydrocannabinol on cell division and macromolecular synthesis in division-synchronized protozoa. Pharmacology 1976, 14, 307–321. [Google Scholar] [CrossRef]
- Nahas, G.G.; Morishima, A.; Desoize, B. Effects of cannabinoids on macromolecular synthesis and replication of cultured lymphocytes. Fed. Proc. 1977, 36, 1748–1752. [Google Scholar]
- Mon, M.J.; Jansing, R.L.; Doggett, S.; Stein, J.L.; Stein, G.S. Influence of delta9-tetrahydrocannabinol on cell proliferation and macromolecular biosynthesis in human cells. Biochem. Pharmacol. 1978, 27, 1759–1765. [Google Scholar] [CrossRef]
- Mon, M.J.; Haas, A.E.; Stein, J.L.; Stein, G.S. Influence of psychoactive and nonpsychoactive cannabinoids on cell proliferation and macromolecular biosynthesis in human cells. Biochem. Pharmacol. 1981, 30, 31–43. [Google Scholar] [CrossRef]
- Schrott, R.; Modliszewski, J.L.; Hawkey, A.B.; Grenier, C.; Holloway, Z.; Evans, J.; Pippen, E.; Corcoran, D.L.; Levin, E.D.; Murphy, S.K. Sperm DNA methylation alterations from cannabis extract exposure are evident in offspring. Epigenet. Chromatin 2022, 15, 33. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Novel Insights into Potential Cannabis-Related Cancerogenesis from Recent Key Whole Epigenome Screen of Cannabis Dependence and Withdrawal: Epidemiological Comment and Explication of Schrott et.al. Genes 2022, 14, 32. [Google Scholar] [CrossRef]
- Schrott, R.; Murphy, S.K.; Modliszewski, J.L.; King, D.E.; Hill, B.; Itchon-Ramos, N.; Raburn, D.; Price, T.; Levin, E.D.; Vandrey, R.; et al. Refraining from use diminishes cannabis-associated epigenetic changes in human sperm. Environ. Epigenet. 2021, 7, dvab009. [Google Scholar] [CrossRef]
- Gabriele, M.; Brandão, H.B.; Grosse-Holz, S.; Jha, A.; Dailey, G.M.; Cattoglio, C.; Hsieh, T.S.; Mirny, L.; Zechner, C.; Hansen, A.S. Dynamics of CTCF- and cohesin-mediated chromatin looping revealed by live-cell imaging. Science 2022, 376, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Davis, E.S.; Daugird, T.A.; Zhao, S.; Quiroga, I.Y.; Uryu, H.; Li, J.; Storey, A.J.; Tsai, Y.H.; Keeley, D.P.; et al. Phase separation drives aberrant chromatin looping and cancer development. Nature 2021, 595, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Cheloshkina, K.; Poptsova, M. Comprehensive analysis of cancer breakpoints reveals signatures of genetic and epigenetic contribution to cancer genome rearrangements. PLoS Comput. Biol. 2021, 17, e1008749. [Google Scholar] [CrossRef] [PubMed]
- Young, N.L.; Dere, R. Mechanistic insights into KDM4A driven genomic instability. Biochem. Soc. Trans. 2021, 49, 93–105. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, Y.; Ramesh, K.H.; Naeem, R.; Wang, Y. Karyotypic complexity, TP53 pathogenic variants, and increased number of variants on Next-Generation Sequencing are associated with disease progression in a North American Adult T-Cell Leukemia/Lymphoma cohort. Int. J. Lab. Hematol. 2021, 43, 651–657. [Google Scholar] [CrossRef]
- Kelso, A.A.; Lopezcolorado, F.W.; Bhargava, R.; Stark, J.M. Distinct roles of RAD52 and POLQ in chromosomal break repair and replication stress response. PLoS Genet. 2019, 15, e1008319. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, Y.; Takata, K.I.; Nowak, B.; Liu, C.; Wood, R.D.; Hittelman, W.N.; Plunkett, W. CNDAC-Induced DNA Double-Strand Breaks Cause Aberrant Mitosis Prior to Cell Death. Mol. Cancer Ther. 2019, 18, 2283–2295. [Google Scholar] [CrossRef]
- Zhao, S.; Klattenhoff, A.W.; Thakur, M.; Sebastian, M.; Kidane, D. Mutation in DNA Polymerase Beta Causes Spontaneous Chromosomal Instability and Inflammation-Associated Carcinogenesis in Mice. Cancers 2019, 11, 1160. [Google Scholar] [CrossRef]
- Harrod, A.; Lane, K.A.; Downs, J.A. The role of the SWI/SNF chromatin remodelling complex in the response to DNA double strand breaks. DNA Repair 2020, 93, 102919. [Google Scholar] [CrossRef]
- Thompson, R.F.; Atzmon, G.; Gheorghe, C.; Liang, H.Q.; Lowes, C.; Greally, J.M.; Barzilai, N. Tissue-specific dysregulation of DNA methylation in aging. Aging Cell 2010, 9, 506–518. [Google Scholar] [CrossRef]
- Zykovich, A.; Hubbard, A.; Flynn, J.M.; Tarnopolsky, M.; Fraga, M.F.; Kerksick, C.; Ogborn, D.; MacNeil, L.; Mooney, S.D.; Melov, S. Genome-wide DNA methylation changes with age in disease-free human skeletal muscle. Aging Cell 2014, 13, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Ashapkin, V.V.; Kutueva, L.I.; Vanyushin, B.F. Aging as an Epigenetic Phenomenon. Curr. Genom. 2017, 18, 385–407. [Google Scholar] [CrossRef]
- Aavik, E.; Babu, M.; Ylä-Herttuala, S. DNA methylation processes in atheosclerotic plaque. Atherosclerosis 2019, 281, 168–179. [Google Scholar] [CrossRef]
- Kane, A.E.; Sinclair, D.A. Epigenetic changes during aging and their reprogramming potential. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 61–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, K.; Zhu, J.K. A model for the aberrant DNA methylomes in aging cells and cancer cells. Biochem. Soc. Trans. 2019, 47, 997–1003. [Google Scholar] [CrossRef]
- DiNieri, J.A.; Wang, X.; Szutorisz, H.; Spano, S.M.; Kaur, J.; Casaccia, P.; Dow-Edwards, D.; Hurd, Y.L. Maternal cannabis use alters ventral striatal dopamine D2 gene regulation in the offspring. Biol. Psychiatr. 2011, 70, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Szutorisz, H.; DiNieri, J.A.; Sweet, E.; Egervari, G.; Michaelides, M.; Carter, J.M.; Ren, Y.; Miller, M.L.; Blitzer, R.D.; Hurd, Y.L. Parental THC exposure leads to compulsive heroin-seeking and altered striatal synaptic plasticity in the subsequent generation. Neuropsychopharmacology 2014, 39, 1315–1323. [Google Scholar] [CrossRef]
- Watson, C.T.; Szutorisz, H.; Garg, P.; Martin, Q.; Landry, J.A.; Sharp, A.J.; Hurd, Y.L. Genome-Wide DNA Methylation Profiling Reveals Epigenetic Changes in the Rat Nucleus Accumbens Associated with Cross-Generational Effects of Adolescent THC Exposure. Neuropsychopharmacology 2015, 40, 2993–3005. [Google Scholar] [CrossRef]
- Szutorisz, H.; Hurd, Y.L. Epigenetic Effects of Cannabis Exposure. Biol. Psychiatr. 2016, 79, 586–594. [Google Scholar] [CrossRef]
- Murphy, S.K.; Itchon-Ramos, N.; Visco, Z.; Huang, Z.; Grenier, C.; Schrott, R.; Acharya, K.; Boudreau, M.H.; Price, T.M.; Raburn, D.J.; et al. Cannabinoid exposure and altered DNA methylation in rat and human sperm. Epigenetics 2018, 13, 1208–1221. [Google Scholar] [CrossRef]
- Szutorisz, H.; Hurd, Y.L. High times for cannabis: Epigenetic imprint and its legacy on brain and behavior. Neurosci. Biobehav. Rev. 2018, 85, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Schrott, R.; Acharya, K.; Itchon-Ramos, N.; Hawkey, A.B.; Pippen, E.; Mitchell, J.T.; Kollins, S.H.; Levin, E.D.; Murphy, S.K. Cannabis use is associated with potentially heritable widespread changes in autism candidate gene DLGAP2 DNA methylation in sperm. Epigenetics 2020, 15, 161–173. [Google Scholar] [CrossRef]
- Ellis, R.J.; Bara, A.; Vargas, C.A.; Frick, A.L.; Loh, E.; Landry, J.; Uzamere, T.O.; Callens, J.E.; Martin, Q.; Rajarajan, P.; et al. Prenatal Δ-Tetrahydrocannabinol Exposure in Males Leads to Motivational Disturbances Related to Striatal Epigenetic Dysregulation. Biol. Psychiatr. 2021, 92, 127–138. [Google Scholar] [CrossRef]
- Schrott, R.; Greeson, K.W.; King, D.; Symosko Crow, K.M.; Easley, C.A., 4th; Murphy, S.K. Cannabis alters DNA methylation at maternally imprinted and autism candidate genes in spermatogenic cells. Syst. Biol. Reprod. Med. 2022, 68, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K.; Wang, W. Cannabis, Cannabidiol, Cannabinoids and Multigenerational Policy. Engineering 2023, 23, 29–32. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Response to Chen et.al. Arch. Public Health 2022, 80, 235. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S. Rapid Response: Known Cannabis Teratogenicity Needs to be Carefully Considered. BMJ 2018, 362, k3357. [Google Scholar]
- Reece, A.S. Rapid Response: Cannabinoid Genotoxic Trifecta—Cancerogenesis, Clinical Teratogenesis and Cellular Ageing. Br. Med. J. 2022, 376, n3114. [Google Scholar]
- Reece, A.S. hGK: Reviewing Medical Cannabis, Drug Development in Reverse—And the Great Genotoxic Trifecta. J. Clin. Med. 2023; in press. [Google Scholar]
- Reece, A.S.; Hulse, G.K. Geotemporospatial and Causal Inferential Epidemiological Overview and Survey of USA Cannabis, Cannabidiol and Cannabinoid Genotoxicity Expressed in Cancer Incidence 2003–2017: Part 1—Continuous Bivariate Analysis. Arch. Public Health 2022, 80, 99–133. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Geotemporospatial and Causal Inferential Epidemiological Overview and Survey of USA Cannabis, Cannabidiol and Cannabinoid Genotoxicity Expressed in Cancer Incidence 2003–2017: Part 2—Categorical Bivariate Analysis and Attributable Fractions. Arch. Public Health 2022, 80, 100–135. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. Geotemporospatial and Causal Inferential Epidemiological Overview and Survey of USA Cannabis, Cannabidiol and Cannabinoid Genotoxicity Expressed in Cancer Incidence 2003–2017: Part 3—Spatiotemporal, Multivariable and Causal Inferential Pathfinding and Exploratory Analyses of Prostate and Ovarian Cancers. Arch. Public Health 2022, 80, 100–136. [Google Scholar] [PubMed]
- Chioccarelli, T.; Cacciola, G.; Altucci, L.; Lewis, S.E.; Simon, L.; Ricci, G.; Ledent, C.; Meccariello, R.; Fasano, S.; Pierantoni, R.; et al. Cannabinoid receptor 1 influences chromatin remodeling in mouse spermatids by affecting content of transition protein 2 mRNA and histone displacement. Endocrinology 2010, 151, 5017–5029. [Google Scholar] [CrossRef]
- Rossato, M.; Ion Popa, F.; Ferigo, M.; Clari, G.; Foresta, C. Human sperm express cannabinoid receptor Cb1, the activation of which inhibits motility, acrosome reaction, and mitochondrial function. J. Clin. Endocrinol. Metab. 2005, 90, 984–991. [Google Scholar] [CrossRef]
- European Cancer Information System Block Data. Available online: https://ecis.jrc.ec.europa.eu (accessed on 20 December 2022).
- CI5Plus: Cancer Incidence in Five Continents Time Trends, Volume XI: Detailed Data. Available online: https://ci5.iarc.fr/CI5plus/Pages/download.aspx. (accessed on 10 February 2023).
- Doll, R.; Payne, P.; Waterhouse, J. Cancer Incidence in Five Continents: A Technical Report; Springer: Berlin, Germany, 1966; Volume 1. [Google Scholar]
- Dyba, T.; Hakulinen, T. Comparison of different approaches to incidence prediction based on simple interpolation techniques. Stat. Med. 2000, 19, 1741–1752. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J. Age Standardization. In CI5XI Cancer Incidence in Five Continents Volume XI; Bray, F., Colombet, M., Mery, L., Piñeros, M., Znaor, A., Zanetti, R., Ferlay, J., Eds.; International Agency for Research on Cancer: Lyon, France, 2017; Volume 1. [Google Scholar]
- Global Health Observatory. Available online: https://www.who.int/data/gho/data/indicators/indicator-details/GHO/total-(recorded-unrecorded)-alcohol-per-capita-(15-)-consumption (accessed on 10 February 2023).
- European Monitoring Centre for Drugs and Drug Addiction (EMCDDA): Statistical Bulletin 2021—Prevalence of Drug Use. Available online: https://www.emcdda.europa.eu/data/stats2021/gps_en (accessed on 20 December 2022).
- Adjusted Net National Income per Capita (Current US$). Available online: https://data.worldbank.org/indicator/NY.ADJ.NNTY.PC.CD (accessed on 10 February 2023).
- R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 10 February 2023).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; Francios, R.; Groelmund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686–1691. [Google Scholar] [CrossRef]
- ggpubr: ‘ggplot2’ Based Publication Ready Plots. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 10 February 2023).
- cowplot: Streamlined Plot Theme and Plot Annotations for ‘ggplot2’. Available online: https://CRAN.R-project.org/package=cowplot (accessed on 10 February 2023).
- Patchwork: The Composer of Plots_. R Package Version 1.1.2. Available online: https://CRAN.R-project.org/package=patchwork (accessed on 10 February 2023).
- Pebesma, E. Simple Features for R: Standardized Support for Spatial Vector Data. R J. 2018, 10, 439–446. [Google Scholar] [CrossRef]
- RNaturalEarth: World Map Data from Natural Earth. Available online: https://CRAN.R-project.org/package=rnaturalearth (accessed on 10 February 2023).
- Viridis: Default Color Maps from ‘Matplotlib’. Available online: https://CRAN.R-project.org/package=viridis (accessed on 10 February 2023).
- RColorBrewer: ColorBrewer Palettes. Available online: https://CRAN.R-project.org/package=RColorBrewer (accessed on 10 February 2023).
- Colorplaner: Ggplot2 Extension to Visualize Two Variables Per Color Aesthetic Through Colorspace Projection. Available online: https://github.com/wmurphyrd/colorplaner (accessed on 10 February 2023).
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Holm, S. A Simple Sequentially Rejective Multiple Test Procedure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinf. 2008, 9, 559. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. Fast R Functions for Robust Correlations and Hierarchical Clustering. J. Stat. Softw. 2012, 46, i11. [Google Scholar] [CrossRef]
- R Package “Corrplot”: Visualization of a Correlation Matrix. Available online: https://github.com/taiyun/corrplot (accessed on 10 February 2023).
- epiR: Tools for the Analysis of Epidemiological Data. Available online: https://fvas.unimelb.edu.au/research/groups/veterinary-epidemiology-melbourne; (accessed on 28 December 2022).
- Collapse: Advanced and Fast Data Transformation. Available online: https://CRAN.R-project.org/package=collapse (accessed on 10 February 2023).
- Pinheiro, J.; Bates, D.; DebRoy, S.; Sarkar, D.; R Core Team. Nlme: Linear and Nonlinear Mixed Effects Models, vol. 1: R: Comprehensive R Archive Network. 2020. Available online: https://cran.r-project.org/web/packages/nlme//nlme.pdf (accessed on 15 July 2023).
- Package ‘plm’. Available online: https://cran.r-project.org/web/packages/plm/plm.pdf (accessed on 10 February 2023).
- Broom.mixed: Tidying Methods for Mixed Models. Available online: http://github.com/bbolker/broom.mixed (accessed on 10 February 2023).
- Broom: Convert Statistical Objects into Tidy Tibbles. Available online: https://CRAN.R-project.org/package=broom (accessed on 10 February 2023).
- Health Effects of Cigarette Smoking. Available online: https://www.cdc.gov/tobacco/data_statistics/fact_sheets/health_effects/effects_cig_smoking/index.htm#:~:text=Smoking%20can%20cause%20lung%20disease%20by%20damaging%20your,smoking%20causes%20most%20cases%20of%20lung%20cancer.%201%2C2 (accessed on 10 February 2023).
- Wal, W.; Geskus, R. Ipw: An R Package for Inverse Probability Weighting. J. Stat. Softw. 2011, 43, 1–23. [Google Scholar] [CrossRef]
- Mathur, M.B.; Ding, P.; Riddell, C.A.; VanderWeele, T.J. Web Site and R Package for Computing E-values. Epidemiology 2018, 29, e45–e47. [Google Scholar] [CrossRef]
- VanderWeele, T.J.; Ding, P.; Mathur, M. Technical Considerations in the Use of the E-Value. J. Causal Inference 2019, 7, 1–11. [Google Scholar] [CrossRef]
- VanderWeele, T.J.; Ding, P. Sensitivity Analysis in Observational Research: Introducing the E-Value. Ann. Intern. Med. 2017, 167, 268–274. [Google Scholar] [CrossRef] [PubMed]
- VanderWeele, T.J.; Martin, J.N.; Mathur, M.B. E-values and incidence density sampling. Epidemiology 2020, 31, e51–e52. [Google Scholar] [CrossRef] [PubMed]
- VanderWeele, T.J.; Mathur, M.B. Commentary: Developing best-practice guidelines for the reporting of E-values. Int. J. Epidemiol. 2020, 49, 1495–1497. [Google Scholar] [CrossRef] [PubMed]
- Pearl, J.; Mackaenzie, D. The Book of Why. The New Science of Cause and Effect; Basic Books: New York, NY, USA, 2019; Volume 1. [Google Scholar]
- Package ‘EValue’. Available online: https://cran.r-project.org/web/packages/EValue/EValue.pdf (accessed on 10 February 2023).
- Health Effects of Cigarette Smoking: Smoking and Cancer. Available online: https://www.cdc.gov/tobacco/data_statistics/fact_sheets/health_effects/effects_cig_smoking/index.htm#cancer (accessed on 10 February 2023).
- Reece, A.S.; Hulse, G.K. Geospatiotemporal and causal inference study of cannabis and other drugs as risk factors for female breast cancer USA 2003–2017. Environ. Epigenet. 2022, 8, dvac006. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. Cannabinoid exposure as a major driver of pediatric acute lymphoid Leukaemia rates across the USA: Combined geospatial, multiple imputation and causal inference study. BMC Cancer 2021, 21, 984–1017. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Chapter 4: Geospatiotemporal and Causal Inferential Epidemiological Survey and Exploration of Cannabinoid- and Substance- Related Carcinogenesis in USA 2003–2017. In Epidemiology of Cannabis: Genotoxicity and Neurotoxicity, Epigenomics and Aging; Elsevier: New York, NY, USA, 2023; Volume 1, In Press: 2500. [Google Scholar]
- Reece, A.S.; Hulse, G.K. State Trends of Cannabis Liberalization as a Causal Driver of Increasing Testicular Cancer Rates across the USA. Int. J. Environ. Res. Public Health 2022, 19, 12759. [Google Scholar] [CrossRef]
- Patsenker, E.; Stoll, M.; Millonig, G.; Agaimy, A.; Wissniowski, T.; Schneider, V.; Mueller, S.; Brenneisen, R.; Seitz, H.K.; Ocker, M.; et al. Cannabinoid receptor type I modulates alcohol-induced liver fibrosis. Mol. Med. 2011, 17, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. Cannabis- and Substance-Related Carcinogenesis in Europe: A Lagged Causal Inferential Panel Re-gression Modelling Study. Int. J. Environ. Res. Public Health, 2023; in press. [Google Scholar]
- Colucci-D’amato, L.; Speranza, L.; Volpicelli, F. Neurotrophic Factor BDNF, Physiological Functions and Therapeutic Potential in Depression, Neurodegeneration and Brain Cancer. Int. J. Mol. Sci. 2020, 21, 7777. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Michael, I.P.; Zhang, P.; Saghafinia, S.; Knott, G.; Jiao, W.; McCabe, B.D.; Galván, J.A.; Robinson, H.P.C.; Zlobec, I.; et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 2019, 573, 526–531. [Google Scholar] [CrossRef]
- Kuijten, R.R.; Bunin, G.R.; Nass, C.C.; Meadows, A.T. Gestational and familial risk factors for childhood astrocytoma: Results of a case-control study. Cancer Res. 1990, 50, 2608–2612. [Google Scholar]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Liu, Z.; Jiang, Y.; Yuan, H.; Fang, Q.; Cai, N.; Suo, C.; Jin, L.; Zhang, T.; Chen, X. The trends in incidence of primary liver cancer caused by specific etiologies: Results from the Global Burden of Disease Study 2016 and implications for liver cancer prevention. J. Hepatol. 2019, 70, 674–683. [Google Scholar] [CrossRef]
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73 (Suppl. S1), 4–13. [Google Scholar] [CrossRef]
- Yu, L.X.; Schwabe, R.F. The gut microbiome and liver cancer: Mechanisms and clinical translation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 527–539. [Google Scholar] [CrossRef]
- Mukhopadhyay, B.; Cinar, R.; Yin, S.; Liu, J.; Tam, J.; Godlewski, G.; Harvey-White, J.; Mordi, I.; Cravatt, B.F.; Lotersztajn, S.; et al. Hyperactivation of anandamide synthesis and regulation of cell-cycle progression via cannabinoid type 1 (CB1) receptors in the regenerating liver. Proc. Natl. Acad. Sci. USA 2011, 108, 6323–6328. [Google Scholar] [CrossRef] [PubMed]
- Abboud, Y.; Samaan, J.S.; Oh, J.; Jiang, Y.; Randhawa, N.; Lew, D.; Ghaith, J.; Pala, P.; Leyson, C.; Watson, R.; et al. Increasing Pancreatic Cancer Incidence in Young Women in the US: A Population-Based Time-Trend Analysis, 2001–2018. Gastroenterology 2023, 164, 978–989.e6. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. Cannabis Could be the Missing Environmental Pancreatic Carcinogen Hiding in Plain View. Gastroenterology 2023, 23. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Sociodemographically Stratified Exploration of Pancreatic Cancer Incidence in Younger US Patients: Implication of Cannabis Exposure as a Risk Factor. Gastroenterol. Insights 2023, 14, 204–235. [Google Scholar] [CrossRef]
- VanverWeele, T.J.; Mathur, M.; Chen, Y. Outcome-Wide Longitudinal Designs for Causal Inference: A New Template for Empirical Studies. Stat. Sci. 2020, 35, 437–466. [Google Scholar]
- Raad, H.; Cornelius, V.; Chan, S.; Williamson, E.; Cro, S. An evaluation of inverse probability weighting using the propensity score for baseline covariate adjustment in smaller population randomised controlled trials with a continuous outcome. BMC Med. Res. Methodol. 2020, 20, 70. [Google Scholar] [CrossRef]
- Seaman, S.R.; White, I.R. Review of inverse probability weighting for dealing with missing data. Stat. Methods Med. Res. 2013, 22, 278–295. [Google Scholar] [CrossRef]
- Hill, A.B. The Environment and Disease: Association or Causation? Proc. R. Soc. Med. 1965, 58, 295–300. [Google Scholar] [CrossRef]
- Nahas, G.G. Keep Off the Grass; P. S. Eriksson: Middlebury, VT, USA, 1990; Volume 1. [Google Scholar]
- Cozens, D.D.; Clark, R.; Palmer, A.K.; Hardy, N.; Nahas, G.G.; Harvey, D.J. The effect of a crude marihuana extract on embryonic and foetal development of the rabbit. Adv. Biosci. 1978, 22–23, 469–477. [Google Scholar]
- Cozens, D.D.; Nahas, G.G.; Harvey, D. Prenatal Exposure to Cannabis and Fetal Development. In Marijuana in Medicine; Nahas, G.G., Sutin, K.M., Harvey, D.J., Agurell, S., Eds.; Humana Press: Totowa, NJ, USA; New York, NY, USA, 1999; Volume 1, pp. 431–440. [Google Scholar]
- Huang, H.F.S.; Nahas, G.G.; Hembree, W.C. Effects of Marijuana Inhalation on Spermatogenesis of the Rat. In Marijuana in Medicine; Nahas, G.G., Sutin, K.M., Harvey, D.J., Agurell, S., Eds.; Humana Press: Totowa, NJ, USA; New York, NY, USA, 1999; Volume 1, pp. 359–366. [Google Scholar]
- Reece, A.S.; Hulse, G.K. Impacts of cannabinoid epigenetics on human development: Reflections on Murphy et. al. ‘cannabinoid exposure and altered DNA methylation in rat and human sperm’ epigenetics 2018; 13: 1208-1221. Epigenetics 2019, 14, 1041–1056. [Google Scholar] [CrossRef]
- Reece, A.S.; Wang, W.; Hulse, G.K. Pathways from epigenomics and glycobiology towards novel biomarkers of addiction and its radical cure. Med. hypotheses 2018, 116, 10–21. [Google Scholar] [CrossRef]
- Wilson, R.G., Jr.; Tahir, S.K.; Mechoulam, R.; Zimmerman, S.; Zimmerman, A.M. Cannabinoid enantiomer action on the cytoarchitecture. Cell Biol. Int. 1996, 20, 147–157. [Google Scholar] [CrossRef]
- Tahir, S.K.; Trogadis, J.E.; Stevens, J.K.; Zimmerman, A.M. Cytoskeletal organization following cannabinoid treatment in undifferentiated and differentiated PC12 cells. Biochem. Cell Biol. 1992, 70, 1159–1173. [Google Scholar] [CrossRef]
- Zimmerman, S.; Zimmerman, A.M. Genetic effects of marijuana. Int. J. Addict. 1990, 25, 19–33. [Google Scholar] [CrossRef]
- Tahir, S.K.; Zimmerman, A.M. Influence of marihuana on cellular structures and biochemical activities. Pharmacol. Biochem. Behav. 1991, 40, 617–623. [Google Scholar] [CrossRef]
- Busch, F.W.; Seid, D.A.; Wei, E.T. Mutagenic activity of marihuana smoke condensates. Cancer Lett. 1979, 6, 319–324. [Google Scholar] [CrossRef]
- Cannabis: A Smoking Gun; British Lung Foundation: London, UK, 2005.
- Sarafian, T.A.; Magallanes, J.A.; Shau, H.; Tashkin, D.; Roth, M.D. Oxidative stress produced by marijuana smoke. An adverse effect enhanced by cannabinoids. Am. J. Respir. Cell Mol. Biol. 1999, 20, 1286–1293. [Google Scholar] [CrossRef]
- Zimmerman, A.M.; Zimmerman, S.; Raj, A.Y. Effects of Cannabinoids on Spermatogensis in Mice. In Marijuana and Medicine; Nahas, G.G., Sutin, K.M., Harvey, D.J., Agurell, S., Eds.; Humana Press: Totowa, NJ, USA; New York, NY, USA, 1999; Volume 1, pp. 347–358. [Google Scholar]
- Morishima, A: Effects of cannabis and natural cannabinoids on chromosomes and ova. NIDA Res. Monogr. 1984, 44, 25–45.
- Russo, C.; Ferk, F.; Mišík, M.; Ropek, N.; Nersesyan, A.; Mejri, D.; Holzmann, K.; Lavorgna, M.; Isidori, M.; Knasmüller, S. Low doses of widely consumed cannabinoids (cannabidiol and cannabidivarin) cause DNA damage and chromosomal aberrations in human-derived cells. Arch. Toxicol. 2019, 93, 179–188. [Google Scholar] [CrossRef]
- Stenchever, M.A.; Kunysz, T.J.; Allen, M.A. Chromosome breakage in users of marihuana. Am. J. Obstet. Gynecol. 1974, 118, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Leuchtenberger, C.; Leuchtenberger, R. Morphological and cytochemical effects of marijuana cigarette smoke on epithelioid cells of lung explants from mice. Nature 1971, 234, 227–229. [Google Scholar] [CrossRef]
- Leuchtenberger, C.; Leuchtenberger, R.; Schneider, A. Effects of marijuana and tobacco smoke on human lung physiology. Nature 1973, 241, 137–139. [Google Scholar] [CrossRef]
- Kloosterman, W.P. Making heads or tails of shattered chromosomes. Science 2015, 348, 1205–1206. [Google Scholar] [CrossRef] [PubMed]
- de Pagter, M.S.; van Roosmalen, M.J.; Baas, A.F.; Renkens, I.; Duran, K.J.; van Binsbergen, E.; Tavakoli-Yaraki, M.; Hochstenbach, R.; van der Veken, L.T.; Cuppen, E.; et al. Chromothripsis in healthy individuals affects multiple protein-coding genes and can result in severe congenital abnormalities in offspring. Am. J. Hum. Genet. 2015, 96, 651–656. [Google Scholar] [CrossRef]
- Kloosterman, W.P.; Guryev, V.; van Roosmalen, M.; Duran, K.J.; de Bruijn, E.; Bakker, S.C.; Letteboer, T.; van Nesselrooij, B.; Hochstenbach, R.; Poot, M.; et al. Chromothripsis as a mechanism driving complex de novo structural rearrangements in the germline. Hum. Mol. Genet. 2011, 20, 1916–1924. [Google Scholar] [CrossRef]
- Kuznetsova, A.Y.; Seget, K.; Moeller, G.K.; De Pagter, M.S.; de Roos, J.A.D.M.; Dürrbaum, M.; Kuffer, C.; Müller, S.; Zaman, G.J.R.; Kloosterman, W.P.; et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle 2015, 14, 2810–2820. [Google Scholar] [CrossRef]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef]
- Hatch, E.M.; Hetzer, M.W. Linking Micronuclei to Chromosome Fragmentation. Cell 2015, 161, 1502–1504. [Google Scholar] [CrossRef]
- Lusk, C.P.; King, M.C. Rotten to the Core: Why Micronuclei Rupture. Dev. Cell 2018, 47, 265–266. [Google Scholar] [CrossRef]
- Terzoudi, G.I.; Karakosta, M.; Pantelias, A.; Hatzi, V.I.; Karachristou, I.; Pantelias, G. Stress induced by premature chromatin condensation triggers chromosome shattering and chromothripsis at DNA sites still replicating in micronuclei or multinucleate cells when primary nuclei enter mitosis. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 793, 185–198. [Google Scholar] [CrossRef]
- Norppa, H.; Falck, G.C. What do human micronuclei contain? Mutagenesis 2003, 18, 221–233. [Google Scholar] [CrossRef]
- Knouse, K.A.; Amon, A. Cell biology: The micronucleus gets its big break. Nature 2015, 522, 162–163. [Google Scholar] [CrossRef] [PubMed]
- Waldron, D. Genome stability: Chromothripsis and micronucleus formation. Nat. Rev. Genet. 2015, 16, 376–377. [Google Scholar] [CrossRef]
- Fenech, M.; Knasmueller, S.; Bolognesi, C.; Holland, N.; Bonassi, S.; Kirsch-Volders, M. Micronuclei as biomarkers of DNA damage, aneuploidy, inducers of chromosomal hypermutation and as sources of pro-inflammatory DNA in humans. Mutat. Res. Mol. Mech. Mutagen. 2020, 786, 108342. [Google Scholar] [CrossRef]
- Shoshani, O.; Brunner, S.F.; Yaeger, R.; Ly, P.; Nechemia-Arbely, Y.; Kim, D.H.; Fang, R.; Castillon, G.A.; Yu, M.; Li, J.S.Z.; et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature 2021, 591, 137–141. [Google Scholar] [CrossRef]
- Martire, S.; Nguyen, J.; Sundaresan, A.; Banaszynski, L.A. Differential contribution of p300 and CBP to regulatory element acetylation in mESCs. BMC Mol. Cell Biol. 2020, 21, 55. [Google Scholar] [CrossRef]
- Rampersaud, A.; Lodato, N.J.; Shin, A.; Waxman, D.J. Widespread epigenetic changes to the enhancer landscape of mouse liver induced by a specific xenobiotic agonist ligand of the nuclear receptor CAR. Toxicol. Sci. 2019, 171, 315–338. [Google Scholar] [CrossRef]
- Tchurikov, N.A.; Uroshlev, L.A.; Klushevskaya, E.S.; Alembekov, I.R.; Lagarkova, M.A.; Kravatskaya, G.I.; Makeev, V.Y.; Kravatsky, Y.V. Chromosomal Translocations in NK-Cell Lymphomas Originate from Inter-Chromosomal Contacts of Active rDNA Clusters Possessing Hot Spots of DSBs. Cancers 2021, 13, 3889. [Google Scholar] [CrossRef]
- Sarafian, T.A.; Kouyoumjian, S.; Khoshaghideh, F.; Tashkin, D.P.; Roth, M.D. Delta 9-tetrahydrocannabinol disrupts mitochondrial function and cell energetics. Am. J. Physiol. 2003, 284, L298–L306. [Google Scholar]
- Benard, G.; Massa, F.; Puente, N.; Lourenco, J.; Bellocchio, L.; Soria-Gomez, E.; Matias, I.; Delamarre, A.; Metna-Laurent, M.; Cannich, A.; et al. Mitochondrial CB receptors regulate neuronal energy metabolism. Nat. Neurosci. 2012, 15, 558–564. [Google Scholar] [CrossRef]
- Wang, J.; Yuan, W.; Li, M.D. Genes and pathways co-associated with the exposure to multiple drugs of abuse, including alcohol, amphetamine/methamphetamine, cocaine, marijuana, morphine, and/or nicotine: A review of proteomics analyses. Mol. Neurobiol. 2011, 44, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Henrich, R.T.; Nogawa, T.; Morishima, A. In vitro induction of segregational errors of chromosomes by natural cannabinoids in normal human lymphocytes. Environ. Mutagen. 1980, 2, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Gadadhar, S.; Alvarez Viar, G.; Hansen, J.N.; Gong, A.; Kostarev, A.; Ialy-Radio, C.; Leboucher, S.; Whitfield, M.; Ziyyat, A.; Touré, A.; et al. Tubulin glycylation controls axonemal dynein activity, flagellar beat, and male fertility. Science 2021, 371, eabd4914. [Google Scholar] [CrossRef]
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated Molecular Characterization of Testicular Germ Cell Tumors. Cell Rep. 2018, 23, 3392–3406. [Google Scholar] [CrossRef]
- Human Genome Project Information Archive 1990–2003. Available online: https://web.ornl.gov/sci/techresources/Human_Genome/posters/chromosome/chromo18.shtml (accessed on 10 February 2023).
- Hall, W.; Degenhardt, L. Adverse health effects of non-medical cannabis use. Lancet 2009, 374, 1383–1391. [Google Scholar] [CrossRef]
- Leuchtenberger, C.; Leuchtenberger, R.; Ritter, U.; Inui, N. Effects of marijuana and tobacco smoke on DNA and chromosomal complement in human lung explants. Nature 1973, 242, 403–404. [Google Scholar] [CrossRef]
- Zimmerman, A.M.; Raj, A.Y. Influence of cannabinoids on somatic cells in vivo. Pharmacology 1980, 21, 277–287. [Google Scholar] [CrossRef]
- Kuhn, J.; Dumont, S. Mammalian kinetochores count attached microtubules in a sensitive and switch-like manner. J. Cell Biol. 2019, 218, 3583–3596. [Google Scholar] [CrossRef]
- Fukagawa, T. Critical histone post-translational modifications for centromere function and propagation. Cell Cycle 2017, 16, 1259–1265. [Google Scholar] [CrossRef]
- Ryu, H.Y.; Hochstrasser, M. Histone sumoylation and chromatin dynamics. Nucleic Acids Res 2021, 49, 6043–6052. [Google Scholar] [CrossRef] [PubMed]
- Gowran, A.; Murphy, C.E.; Campbell, V.A. Delta-tetrahydrocannabinol regulates the p53 post-translational modifiers Murine double minute 2 and the Small Ubiquitin MOdifier protein in the rat brain. FEBS Lett. 2009, 583, 3412–3418. [Google Scholar] [CrossRef]
- Wartosch, L.; Schindler, K.; Schuh, M.; Gruhn, J.R.; Hoffmann, E.R.; McCoy, R.C.; Xing, J. Origins and mechanisms leading to aneuploidy in human eggs. Prenat. Diagn. 2021, 41, 620–630. [Google Scholar] [CrossRef]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef]
- Brieghel, C.; Aarup, K.; Torp, M.H.; Andersen, M.A.; Yde, C.W.; Tian, X.; Wiestner, A.; Ahn, I.E.; Niemann, C.U. Clinical Outcomes in Patients with Multi-Hit TP53 Chronic Lymphocytic Leukemia Treated with Ibrutinib. Clin. Cancer Res. 2021, 27, 4531–4538. [Google Scholar] [CrossRef]
- Fornalski, K.W.; Dobrzyński, L. Modeling of single cell cancer transformation using phase transition theory: Application of the Avrami equation. Radiat. Environ. Biophys. 2022, 61, 169–175. [Google Scholar] [CrossRef]
- Mitchell, S.R.; Gopakumar, J.; Jaiswal, S. Insights into clonal hematopoiesis and its relation to cancer risk. Curr. Opin. Genet. Dev. 2021, 66, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Lu, Y.; Zhang, P.; Chen, D.; Liu, X.; Du, X.; Cao, J.; Ye, P.; Chen, L.; Li, S.; et al. Lenalidomide, bortezomib and dexamethasone followed by tandem- autologous stem cell transplantation is an effective treatment modality for multi-hit multiple myeloma. Leuk. Res. 2021, 110, 106710. [Google Scholar] [CrossRef]
- Morishima, A.; Henrich, R.T.; Jayaraman, J.; Nahas, G.G. Hypoploid metaphases in cultured lymphocytes of marihuana smokers. Adv. Biosci. 1978, 22–23, 371–376. [Google Scholar]
- Malouf, C.; Ottersbach, K. Molecular processes involved in B cell acute lymphoblastic leukaemia. Cell Mol. Life Sci. 2018, 75, 417–446. [Google Scholar] [CrossRef]
- Disruption of Interlocking Synchrony Between Metabolome and Epigenome Key to Understanding Widespread Embyrotoxicity and Carcinogenicity of Diverse Cannabinoids. Available online: https://www.bmj.com/content/377/bmj.o1567/rr-0 (accessed on 10 February 2023).















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer | β-Estimate | Std Error | p-Value | P. Adj. Holm | E-Value Estimate | 95% Lower Bound of E-Value |
|---|---|---|---|---|---|---|
| Non-Seminoma | 0.2348 | 0.0233 | 5.36 × 10−12 | 1.34 × 10−10 | 2.10 | 1.92 |
| Cervix | 0.0533 | 0.0022 | 1.01 × 10−112 | 4.15 × 10−111 | 1.40 | 1.37 |
| Lung | 0.0259 | 0.0013 | 3.51 × 10−75 | 1.33 × 10−73 | 1.34 | 1.32 |
| Stomach | 0.0437 | 0.0022 | 8.16 × 10−77 | 3.26 × 10−75 | 1.34 | 1.32 |
| Ovary | 0.0339 | 0.0017 | 3.42 × 10−76 | 1.33 × 10−74 | 1.34 | 1.32 |
| Kidney | 0.0288 | 0.0020 | 3.90 × 10−45 | 1.44 × 10−43 | 1.28 | 1.26 |
| All Cancers nNMSC | 0.0150 | 0.0012 | 1.67 × 10−33 | 5.50 × 10−32 | 1.27 | 1.24 |
| Pancreas | 0.0247 | 0.0019 | 1.19 × 10−38 | 4.16 × 10−37 | 1.27 | 1.24 |
| Corpus Uteri | 0.0350 | 0.0028 | 1.34 × 10−34 | 4.56 × 10−33 | 1.25 | 1.23 |
| Larynx | 0.0202 | 0.0020 | 2.26 × 10−23 | 6.78 × 10−22 | 1.23 | 1.20 |
| Prostate | 0.0188 | 0.0019 | 4.13 × 10−22 | 1.20 × 10−20 | 1.22 | 1.20 |
| Leukaemia | 0.0264 | 0.0030 | 9.97 × 10−18 | 2.79 × 10−16 | 1.21 | 1.18 |
| All Cancers | 0.0132 | 0.0019 | 1.95 × 10−11 | 4.69 × 10−10 | 1.22 | 1.18 |
| Oesophagus | 0.0205 | 0.0024 | 1.24 × 10−16 | 3.34 × 10−15 | 1.20 | 1.17 |
| Seminoma | 0.0128 | 0.0050 | 0.0160 | 0.1437 | 1.38 | 1.15 |
| Breast | 0.0178 | 0.0039 | 4.39 × 10−06 | 7.90 × 10−5 | 1.14 | 1.10 |
| Oropharynx | 0.0213 | 0.0096 | 0.0280 | 0.1894 | 1.16 | 1.05 |
| Leukaemia—Lymphoid | −0.0008 | 0.0028 | 0.7663 | 1.0000 | 1.05 | - |
| Colorectum | −0.0018 | 0.0013 | 0.1638 | 0.6553 | 1.07 | - |
| Brain | −0.0063 | 0.0018 | 5.34 × 10−4 | 0.0080 | 1.12 | - |
| Vulva and Vagina | −0.0059 | 0.0019 | 0.0021 | 0.0256 | 1.14 | - |
| Anus | −0.0055 | 0.0016 | 8.48 × 10−4 | 0.0115 | 1.14 | - |
| Leukaemia—Myeloid | −0.0103 | 0.0046 | 0.0269 | 0.1894 | 1.14 | - |
| Penis | −0.0056 | 0.0017 | 8.22 × 10−4 | 0.0115 | 1.15 | - |
| Non-Hodgkin’s Lymphoma | −0.0083 | 0.0017 | 1.05 × 10−6 | 1.99 × 10−5 | 1.15 | - |
| Bladder | −0.0104 | 0.0018 | 7.02 × 10−9 | 1.40 × 10−7 | 1.16 | - |
| Gallbladder and Biliary | −0.0108 | 0.0027 | 7.95 × 10−5 | 0.0013 | 1.17 | - |
| Ovarian Dysgerminoma | −0.0014 | 0.0030 | 0.6599 | 1.0000 | 1.17 | - |
| Melanoma | −0.0145 | 0.0022 | 3.22 × 10−11 | 7.40 × 10−10 | 1.18 | - |
| Medulloblastoma | −0.0023 | 0.0054 | 0.6807 | 1.0000 | 1.18 | - |
| Testis | −0.0117 | 0.0018 | 1.98 × 10−10 | 4.35 × 10−9 | 1.21 | - |
| Kaposi | −0.0081 | 0.0044 | 0.0643 | 0.3213 | 1.21 | - |
| Vagina | −0.0064 | 0.0025 | 0.0124 | 0.1243 | 1.22 | - |
| Liver | −0.0192 | 0.0027 | 3.63 × 10−12 | 9.43 × 10−11 | 1.22 | - |
| Oropharynx_Broad | −0.0172 | 0.0038 | 8.05 × 10−6 | 1.37E-04 | 1.22 | - |
| Hepatocellular | −0.0098 | 0.0042 | 0.0237 | 0.1894 | 1.24 | - |
| Mesothelioma | −0.0291 | 0.0097 | 0.0032 | 0.0351 | 1.28 | - |
| Hodgkin’s | −0.0160 | 0.0014 | 2.24 × 10−28 | 6.93 × 10−27 | 1.29 | - |
| Myeloma | −0.0175 | 0.0015 | 1.06 × 10−31 | 3.40 × 10−30 | 1.30 | - |
| Thyroid | −0.0390 | 0.0028 | 2.04 × 10−40 | 7.36 × 10−39 | 1.33 | - |
| Vulva | −0.0309 | 0.0047 | 3.98 × 10−10 | 8.37 × 10−9 | 1.39 | - |
| Cancer | β-Estimate | Std. Error | p-Value | P. Adj. Holm | E-Value Estimate | Lower Bound E-Value |
|---|---|---|---|---|---|---|
| Oesophagus | 0.1388 | 0.0059 | 5.92 × 10−108 | 2.43 × 10−106 | 1.80 | 1.75 |
| All Cancers | 0.0888 | 0.0069 | 3.72 × 10−34 | 1.12 × 10−32 | 1.85 | 1.74 |
| Cervix | 0.1395 | 0.0060 | 1.39 × 10−105 | 5.56 × 10−104 | 1.79 | 1.74 |
| Prostate | 0.0950 | 0.0048 | 1.12 × 10−78 | 4.36 × 10−77 | 1.70 | 1.64 |
| Lung | 0.0663 | 0.0036 | 3.94 × 10−69 | 1.50 × 10−67 | 1.66 | 1.60 |
| All Cancers nNMSC | 0.0456 | 0.0032 | 4.20 × 10−44 | 1.47 × 10−42 | 1.58 | 1.52 |
| Kidney | 0.0750 | 0.0054 | 3.29 × 10−42 | 1.12 × 10−40 | 1.54 | 1.48 |
| Leukaemia—Lymphoid | 0.0547 | 0.0091 | 4.19 × 10−09 | 9.63 × 10−08 | 1.61 | 1.46 |
| Ovary | 0.0652 | 0.0049 | 6.49 × 10−38 | 2.08 × 10−36 | 1.52 | 1.46 |
| Stomach | 0.0711 | 0.0064 | 1.65 × 10−27 | 4.62 × 10−26 | 1.45 | 1.40 |
| Pancreas | 0.0547 | 0.0051 | 2.34 × 10−26 | 6.31 × 10−25 | 1.45 | 1.39 |
| Breast | 0.0771 | 0.0104 | 1.82 × 10−13 | 4.72 × 10−12 | 1.34 | 1.28 |
| Larynx | 0.0332 | 0.0054 | 1.05 × 10−9 | 2.52 × 10−8 | 1.31 | 1.24 |
| Leukaemia—Myeloid | 0.0517 | 0.0163 | 0.0017 | 0.0284 | 1.38 | 1.21 |
| Anus | 0.0143 | 0.0038 | 0.0002 | 0.0036 | 1.25 | 1.16 |
| Leukaemia | 0.0305 | 0.0083 | 0.0003 | 0.0051 | 1.23 | 1.15 |
| Corpus Uteri | 0.0290 | 0.0080 | 0.0003 | 0.0061 | 1.22 | 1.14 |
| Penis | 0.0109 | 0.0038 | 0.0036 | 0.0542 | 1.21 | 1.11 |
| Melanoma | 0.0138 | 0.0059 | 0.0200 | 0.2398 | 1.17 | 1.06 |
| Vulva and Vagina | 0.0048 | 0.0044 | 0.2774 | 1.0000 | 1.12 | 1.00 |
| Non-Hodgkin’s Lymphoma | 0.0067 | 0.0046 | 0.1458 | 1.0000 | 1.13 | 1.00 |
| Vulva | 0.0104 | 0.0190 | 0.5831 | 1.0000 | 1.19 | 1.00 |
| Oropharynx_Broad | 0.0152 | 0.0166 | 0.3621 | 1.0000 | 1.20 | 1.00 |
| Vagina | 0.0151 | 0.0097 | 0.1221 | 1.0000 | 1.37 | 1.00 |
| Colorectum | −0.0009 | 0.0036 | 0.7948 | 1.0000 | 1.05 | - |
| Brain | −0.0113 | 0.0049 | 0.0216 | 0.2398 | 1.17 | - |
| Non-Seminoma | −0.0316 | 0.4242 | 0.9411 | 1.0000 | 1.17 | - |
| Testis | −0.0106 | 0.0043 | 0.0130 | 0.1825 | 1.19 | - |
| Myeloma | −0.0172 | 0.0035 | 9.02 × 10−7 | 1.98 × 10−5 | 1.29 | - |
| Hepatocellular | −0.0232 | 0.0170 | 0.1779 | 1.0000 | 1.41 | - |
| Liver | −0.0749 | 0.0061 | 5.30 × 10−33 | 1.54 × 10−31 | 1.54 | - |
| Bladder | −0.0678 | 0.0046 | 5.27 × 10−47 | 1.90 × 10−45 | 1.56 | - |
| Hodgkin’s | −0.0425 | 0.0032 | 8.56 × 10−38 | 2.65 × 10−36 | 1.56 | - |
| Gallbladder and Biliary | −0.0721 | 0.0053 | 8.46 × 10−39 | 2.79 × 10−37 | 1.60 | - |
| Ovarian Dysgerminoma | −0.0102 | 0.0143 | 0.4840 | 1.0000 | 1.64 | - |
| Kaposi | −0.0430 | 0.0179 | 0.0174 | 0.2260 | 1.65 | - |
| Mesothelioma | −0.1087 | 0.0353 | 0.0024 | 0.0382 | 1.69 | - |
| Oropharynx | −0.2054 | 0.0290 | 6.49 × 10−12 | 1.62 × 10−10 | 1.70 | - |
| Thyroid | −0.1136 | 0.0063 | 3.01 × 10−66 | 1.11 × 10−64 | 1.72 | - |
| Medulloblastoma | −0.0374 | 0.0272 | 0.1908 | 1.0000 | 2.43 | - |
| Seminoma | −0.0882 | 0.0250 | 0.0011 | 0.0203 | 3.01 | - |
| Cancer | β-Estimate | Std. Error | p-Value | P. Adj. Holm | E-Value Estimate | E-Value Lower Bound |
|---|---|---|---|---|---|---|
| All Cancers nNMSC | 2.6457 | 0.0778 | 4.69 × 10−180 | 1.41 × 10−178 | 4.88 × 1013 | 8.28 × 1012 |
| All Cancers | 2.8076 | 0.1071 | 6.70 × 10−102 | 1.94 × 10−100 | 3.15 × 1010 | 5.46 × 109 |
| Prostate | 5.2624 | 0.2489 | 1.55 × 10−86 | 4.35 × 10−85 | 4.59 × 107 | 9.56 × 106 |
| Breast | 2.4686 | 0.1369 | 1.36 × 10−65 | 3.67 × 10−64 | 3.51 × 106 | 7.37 × 105 |
| Melanoma | 5.9745 | 0.3598 | 8.89 × 10−57 | 2.31 × 10−55 | 5.59 × 105 | 1.28 × 105 |
| Kidney | 3.5398 | 0.2357 | 1.64 × 10−47 | 4.11 × 10−46 | 3.32 × 105 | 6.95 × 104 |
| Colorectum | 2.5415 | 0.1742 | 4.61 × 10−45 | 1.11 × 10−43 | 2.03 × 105 | 4.33 × 104 |
| Pancreas | 3.6625 | 0.2554 | 1.04 × 10−43 | 2.40 × 10−42 | 1.67 × 105 | 3.55 × 104 |
| Testis | 4.5855 | 0.3263 | 2.84 × 10−41 | 6.25 × 10−40 | 5.74 × 107 | 5.26 × 106 |
| Thyroid | 4.9267 | 0.3666 | 3.26 × 10−38 | 6.85 × 10−37 | 8.56 × 107 | 6.63 × 106 |
| Non-Hodgkin’s Lymphoma | 3.7844 | 0.2854 | 7.36 × 10−38 | 1.47 × 10−36 | 6.82 × 104 | 1.46 × 104 |
| Lung | 2.0357 | 0.1687 | 5.35 × 10−32 | 1.02 × 10−30 | 3.40 × 104 | 7.01 × 103 |
| Anus | 3.6672 | 0.3097 | 1.61 × 10−30 | 2.89 × 10−29 | 2.77 × 106 | 2.68 × 105 |
| Oesophagus | 3.5244 | 0.3847 | 1.71 × 10−19 | 2.90 × 10−18 | 2.58 × 103 | 557.87 |
| Leukaemia—Myeloid | 4.2458 | 0.6638 | 8.92 × 10−10 | 1.43 × 10−8 | 2.22 × 105 | 6.35 × 103 |
| Oropharynx_Broad | 2.5577 | 0.4523 | 2.82 × 10−8 | 4.23 × 10−7 | 5.05 × 102 | 74.07 |
| Leukaemia—Lymphoid | 1.6165 | 0.4299 | 2.16 × 10−4 | 0.0026 | 1784.06 | 51.56 |
| Brain | 1.1480 | 0.3174 | 3.09 × 10−4 | 0.0034 | 19.93 | 5.26 |
| Myeloma | 0.8656 | 0.2619 | 9.80 × 10−4 | 0.0098 | 127.86 | 10.41 |
| Corpus Uteri | 1.2936 | 0.4601 | 0.0050 | 0.0400 | 16.44 | 3.24 |
| Liver | 0.7809 | 0.3581 | 0.0294 | 0.2059 | 37.76 | 2.05 |
| Hodgkin’s | 0.4718 | 0.3032 | 0.1200 | 0.5999 | 12.08 | 1.00 |
| Cervix | 0.1742 | 0.3411 | 0.6097 | 1.0000 | 2.38 | 1.00 |
| Bladder | 0.0250 | 0.2849 | 0.9302 | 1.0000 | 1.35 | 1.00 |
| Kaposi | 0.0346 | 0.4840 | 0.9432 | 1.0000 | 2.09 | 1.00 |
| Ovary | −0.0127 | 0.3205 | 0.9684 | 1.0000 | 1.21 | - |
| Oropharynx | −2.7034 | 1.4854 | 0.0699 | 0.4192 | 54.33 | - |
| Gallbladder and Biliary | −1.1202 | 0.3420 | 0.0011 | 0.0098 | 487.89 | - |
| Larynx | −1.3647 | 0.3578 | 1.42 × 10−4 | 0.0018 | 27.57 | - |
| Stomach | −0.8809 | 0.2164 | 4.92 × 10−5 | 6.89 × 10−4 | 53.25 | - |
| Cancer | β-Estimate | Std. Error | p-Value | P. Adj. Holm | E-Value Estimate | E-Value Lower Bound |
|---|---|---|---|---|---|---|
| All Cancers nNMSC | 1.1789 | 0.0622 | 2.82 × 10−70 | 8.46 × 10−69 | 2.22 × 105 | 6.70 × 104 |
| Melanoma | 2.8370 | 0.2030 | 1.36 × 10−41 | 3.93 × 10−40 | 795.41 | 343.77 |
| All Cancers | 1.0317 | 0.0758 | 7.34 × 10−37 | 2.05 × 10−35 | 2065.96 | 761.63 |
| Pancreas | 1.5037 | 0.1432 | 6.91 × 10−25 | 1.80 × 10−23 | 185.22 | 79.36 |
| Breast | 0.7634 | 0.0813 | 2.34 × 10−20 | 5.86 × 10−19 | 142.23 | 58.19 |
| Anus | 1.1325 | 0.1299 | 1.12 × 10−17 | 2.69 × 10−16 | 159.29 | 59.29 |
| Non-Hodgkin’s Lymphoma | 1.4279 | 0.1679 | 4.80 × 10−17 | 1.10 × 10−15 | 101.29 | 40.73 |
| Lung | 0.7312 | 0.0921 | 4.22 × 10−15 | 9.29 × 10−14 | 71.04 | 29.12 |
| Kidney | 0.9545 | 0.1363 | 3.85 × 10−12 | 8.08 × 10−11 | 41.16 | 17.33 |
| Oesophagus | 1.2442 | 0.2143 | 7.86 × 10−9 | 1.42 × 10−7 | 23.50 | 9.87 |
| Testis | 0.8143 | 0.1502 | 7.38 × 10−8 | 1.26 × 10−6 | 33.71 | 11.76 |
| Oropharynx | 2.3818 | 0.5030 | 3.70 × 10−6 | 5.92 × 10−5 | 69.39 | 15.59 |
| Oropharynx_Broad | 1.1861 | 0.2844 | 3.67 × 10−5 | 4.78 × 10−4 | 24.73 | 7.15 |
| Bladder | 0.5357 | 0.1597 | 8.18-04 | 0.0098 | 7.87 | 3.04 |
| Hodgkin’s | 0.3397 | 0.1236 | 0.0061 | 0.0608 | 6.91 | 2.28 |
| Brain | 0.5346 | 0.1948 | 0.0061 | 0.0608 | 5.20 | 2.05 |
| Thyroid | 0.3513 | 0.1603 | 0.0287 | 0.2006 | 5.58 | 1.51 |
| Prostate | 0.3118 | 0.1532 | 0.0420 | 0.2521 | 4.40 | 1.23 |
| Kaposi | 0.1992 | 0.5144 | 0.6998 | 1.0000 | 9.45 | 1.00 |
| Myeloma | 0.0387 | 0.1039 | 0.7096 | 1.0000 | 1.70 | 1.00 |
| Colorectum | 0.0315 | 0.0960 | 0.7429 | 1.0000 | 1.58 | 1.00 |
| Corpus Uteri | −0.3324 | 0.2662 | 0.2119 | 0.8477 | 2.82 | - |
| Leukaemia—Myeloid | −0.4144 | 0.2523 | 0.1020 | 0.5100 | 5.73 | - |
| Liver | −0.3425 | 0.1377 | 0.0130 | 0.1044 | 7.02 | - |
| Leukaemia—Lymphoid | −0.4516 | 0.1521 | 0.0033 | 0.0366 | 15.46 | - |
| Cervix | −0.8035 | 0.1902 | 2.54 × 10−5 | 3.56 × 10−4 | 12.79 | - |
| Ovary | −0.8408 | 0.1827 | 4.57 × 10−6 | 6.86 × 10−5 | 14.58 | - |
| Gallbladder and Biliary | −0.8315 | 0.1407 | 4.78 × 10−9 | 9.08 × 10−8 | 116.77 | - |
| Larynx | −1.2162 | 0.2026 | 2.47 × 10−9 | 4.94 × 10−8 | 21.12 | - |
| Stomach | −1.3226 | 0.1114 | 4.89 × 10−31 | 1.32 × 10−29 | 352.51 | - |
| Herb. THC | Resin. THC | Daily Interpolated | Last Month’s Cannabis |
|---|---|---|---|
| All Cancers | All Cancers | All Cancers | |
| All Cancers nNMSC | All Cancers nNMSC | All Cancers nNMSC | All Cancers nNMSC |
| Anus | Anus | Anus | Anus |
| Bladder | Bladder | Bladder | |
| Brain | Brain | ||
| Breast | Breast | Breast | Breast |
| Cervix | |||
| Colorectum | Colorectum | Colorectum | |
| Corpus Uteri | |||
| Gallbladder and Biliary | |||
| Hodgkin’s | Hodgkin’s | Hodgkin’s | |
| Kaposi | Kaposi | Kaposi | |
| Kidney | Kidney | Kidney | Kidney |
| Larynx | Larynx | ||
| Leukaemia | |||
| Leukaemia—Lymphoid | Leukaemia—Lymphoid | ||
| Leukaemia—Myeloid | Leukaemia—Myeloid | Leukaemia—Myeloid | |
| Liver | Liver | Liver | |
| Lung | Lung | Lung | Lung |
| Melanoma | Melanoma | Melanoma | |
| Mesothelioma | |||
| Myeloma | Myeloma | Myeloma | |
| Non-Hodgkin’s Lymphoma | Non-Hodgkin’s Lymphoma | Non-Hodgkin’s Lymphoma | Non-Hodgkin’s Lymphoma |
| Oesophagus | Oesophagus | Oesophagus | Oesophagus |
| Oropharynx | Oropharynx | ||
| Oropharynx_Broad | Oropharynx_Broad | Oropharynx_Broad | Oropharynx_Broad |
| Pancreas | Pancreas | Pancreas | |
| Prostate | Prostate | ||
| Stomach | |||
| Testis | Testis | ||
| Thyroid | Thyroid | Thyroid | Thyroid |
| Vulva and Vagina | Vulva and Vagina | Vulva and Vagina |
| Cancer | p-Value | RR (C.I.) | AFE (C.I.) | PAF (C.I.) |
|---|---|---|---|---|
| Oropharynx | 0.0000 | 3.627 (3.6028, 3.6514) | 0.7243 (0.7224, 0.7261) | 0.5043 (0.5019, 0.5066) |
| Cervix | 0.0000 | 1.9962 (1.9932, 1.9992) | 0.499 (0.4983, 0.4998) | 0.2763 (0.2757, 0.2769) |
| Stomach | 0.0000 | 1.8241 (1.8216, 1.8266) | 0.4518 (0.451, 0.4525) | 0.2468 (0.2463, 0.2474) |
| Kidney | 0.0000 | 1.7574 (1.7549, 1.7599) | 0.431 (0.4302, 0.4318) | 0.2315 (0.2309, 0.2321) |
| Prostate | 0.0000 | 1.6336 (1.6328, 1.6344) | 0.3879 (0.3876, 0.3882) | 0.1964 (0.1962, 0.1966) |
| Pancreas | 0.0000 | 1.601 (1.5985, 1.6035) | 0.3754 (0.3744, 0.3764) | 0.1929 (0.1923, 0.1935) |
| Corpus Uteri | 0.0000 | 1.5789 (1.577, 1.5808) | 0.3666 (0.3659, 0.3674) | 0.1758 (0.1753, 0.1763) |
| Leukaemia | 0.0000 | 1.5676 (1.565, 1.5701) | 0.3621 (0.361, 0.3631) | 0.1615 (0.1609, 0.1621) |
| All Cancers | 0.0000 | 1.5195 (1.5186, 1.5203) | 0.3419 (0.3415, 0.3423) | 0.2234 (0.2231, 0.2237) |
| Ovary | 0.0000 | 1.4957 (1.4936, 1.4978) | 0.3314 (0.3305, 0.3323) | 0.1596 (0.1591, 0.1602) |
| Larynx | 0.0000 | 1.453 (1.4495, 1.4565) | 0.3118 (0.3101, 0.3134) | 0.1549 (0.1539, 0.1559) |
| Lung | 0.0000 | 1.442 (1.4409, 1.4431) | 0.3065 (0.306, 0.3071) | 0.1479 (0.1476, 0.1482) |
| Oesophagus | 0.0000 | 1.3267 (1.3236, 1.3298) | 0.2462 (0.2445, 0.248) | 0.115 (0.114, 0.1159) |
| All Cancers nNMSC | 0.0000 | 1.3089 (1.3086, 1.3093) | 0.236 (0.2358, 0.2362) | 0.0995 (0.0994, 0.0996) |
| Oropharynx_Broad | 0.0000 | 1.2962 (1.2905, 1.302) | 0.2285 (0.2251, 0.232) | 0.1432 (0.1408, 0.1456) |
| Brain | 0.0000 | 1.0472 (1.0452, 1.0492) | 0.0451 (0.0432, 0.0469) | 0.0178 (0.017, 0.0185) |
| Colorectum | 0.0000 | 1.0408 (1.0402, 1.0415) | 0.0392 (0.0386, 0.0398) | 0.0155 (0.0152, 0.0157) |
| Breast | 0.0000 | 1.0281 (1.0275, 1.0286) | 0.0273 (0.0268, 0.0278) | 0.011 (0.0107, 0.0112) |
| Liver | 2.25 × 10−34 | 1.0122 (1.0102, 1.0141) | 0.012 (0.0101, 0.0139) | 0.0049 (0.0041, 0.0056) |
| Bladder | 5.05 × 10−45 | 1.0089 (1.0076, 1.0101) | 0.0088 (0.0076, 0.01) | 0.0035 (0.003, 0.004) |
| Hepatocellular | 1.75 × 10−2 | 1.0937 (1.0063, 1.1888) | 0.0857 (0.0062, 0.1588) | 0.0081 (0.0003, 0.0159) |
| Penis | 7.09 × 10−13 | 0.9807 (0.9754, 0.986) | −0.0197 (−0.0252, −0.0142) | −0.0068 (−0.0087, −0.0049) |
| Melanoma | 0.0000 | 0.9646 (0.9632, 0.966) | −0.0367 (−0.0382, −0.0352) | −0.0139 (−0.0144, −0.0133) |
| Gallbladder and Biliary | 1.33 × 10−284 | 0.939 (0.9358, 0.9422) | −0.065 (−0.0686, −0.0613) | −0.0216 (−0.0227, −0.0204) |
| Leukaemia—Lymphoid | 4.55 × 10−161 | 0.9207 (0.9152, 0.9262) | −0.0861 (−0.0927, −0.0796) | −0.045 (−0.0482, −0.0417) |
| Hodgkin’s | 0.0000 | 0.9043 (0.9017, 0.9069) | −0.1059 (−0.109, −0.1027) | −0.0383 (−0.0393, −0.0372) |
| Anus | 0.0000 | 0.8729 (0.8682, 0.8776) | −0.1456 (−0.1518, −0.1395) | −0.0522 (−0.0542, −0.0501) |
| Thyroid | 0.0000 | 0.8523 (0.8508, 0.8538) | −0.1733 (−0.1754, −0.1712) | −0.0617 (−0.0624, −0.061) |
| Myeloma | 0.0000 | 0.8404 (0.8381, 0.8427) | −0.1899 (−0.1931, −0.1867) | −0.0669 (−0.0679, −0.0659) |
| Testis | 0.0000 | 0.8054 (0.8038, 0.807) | −0.2416 (−0.2441, −0.2392) | −0.0796 (−0.0803, −0.0789) |
| Non-Hodgkin’s Lymphoma | 0.0000 | 0.8028 (0.8015, 0.8041) | −0.2456 (−0.2477, −0.2436) | −0.0724 (−0.0729, −0.0719) |
| Vulva and Vagina | 0.0000 | 0.7813 (0.7773, 0.7853) | −0.28 (−0.2866, −0.2734) | −0.084 (−0.0857, −0.0823) |
| Vagina | 1.84 × 10−85 | 0.7031 (0.6786, 0.7285) | −0.4223 (−0.4737, −0.3727) | −0.065 (−0.0708, −0.0592) |
| Leukaemia—Myeloid | 0.0000 | 0.6386 (0.6329, 0.6443) | −0.566 (−0.5801, −0.5521) | −0.2466 (−0.2514, −0.2417) |
| Vulva | 0.0000 | 0.591 (0.5808, 0.6015) | −0.6919 (−0.7217, −0.6626) | −0.0926 (−0.0951, −0.09) |
| Cancer | p-Value | RR (C.I.) | AFE (C.I.) | PAF (C.I.) |
|---|---|---|---|---|
| Kaposi | 1.86 × 10−170 | 2.081 (1.9739, 2.1939) | 0.5195 (0.4934, 0.5442) | 0.2573 (0.2376, 0.2765) |
| Liver | 0.0000 | 1.7627 (1.7556, 1.7698) | 0.4327 (0.4304, 0.435) | 0.4077 (0.4055, 0.4099) |
| Thyroid | 0.0000 | 1.6921 (1.6861, 1.6981) | 0.409 (0.4069, 0.4111) | 0.385 (0.383, 0.3871) |
| Stomach | 0.0000 | 1.6847 (1.68, 1.6893) | 0.4064 (0.4048, 0.408) | 0.3827 (0.3811, 0.3843) |
| Oropharynx_Broad | 0.0000 | 1.6204 (1.6104, 1.6304) | 0.3829 (0.3791, 0.3866) | 0.3306 (0.3271, 0.3342) |
| Larynx | 0.0000 | 1.5906 (1.5822, 1.5991) | 0.3713 (0.368, 0.3746) | 0.3524 (0.3492, 0.3557) |
| Breast | 0.0000 | 1.4899 (1.4882, 1.4915) | 0.3288 (0.3281, 0.3296) | 0.3095 (0.3088, 0.3102) |
| All Cancers | 0.0000 | 1.4057 (1.4047, 1.4067) | 0.2886 (0.2881, 0.2891) | 0.2411 (0.2406, 0.2415) |
| Hodgkin’s | 0.0000 | 1.2985 (1.2918, 1.3054) | 0.2299 (0.2259, 0.2339) | 0.213 (0.2092, 0.2168) |
| Bladder | 0.0000 | 1.2896 (1.2866, 1.2925) | 0.2246 (0.2228, 0.2263) | 0.2077 (0.206, 0.2094) |
| Kidney | 0.0000 | 1.287 (1.2836, 1.2903) | 0.223 (0.221, 0.225) | 0.2062 (0.2043, 0.2081) |
| Pancreas | 0.0000 | 1.2859 (1.2822, 1.2895) | 0.2223 (0.2201, 0.2245) | 0.2056 (0.2035, 0.2077) |
| Prostate | 0.0000 | 1.274 (1.2728, 1.2751) | 0.2151 (0.2144, 0.2158) | 0.1984 (0.1978, 0.1991) |
| Lung | 0.0000 | 1.2704 (1.2685, 1.2724) | 0.2129 (0.2116, 0.2141) | 0.1992 (0.198, 0.2004) |
| Leukaemia | 0.0000 | 1.2471 (1.2436, 1.2507) | 0.1981 (0.1959, 0.2004) | 0.1819 (0.1798, 0.1841) |
| Colorectum | 0.0000 | 1.2186 (1.2173, 1.22) | 0.1794 (0.1785, 0.1803) | 0.1646 (0.1638, 0.1655) |
| All Cancers nNMSC | 0.0000 | 1.2121 (1.2115, 1.2127) | 0.175 (0.1746, 0.1754) | 0.1607 (0.1603, 0.1611) |
| Gallbladder and Biliary | 1.91 × 10−252 | 1.109 (1.1023, 1.1156) | 0.0982 (0.0928, 0.1036) | 0.0905 (0.0855, 0.0955) |
| Myeloma | 0.0000 | 1.1072 (1.1022, 1.1123) | 0.0968 (0.0927, 0.101) | 0.0884 (0.0846, 0.0922) |
| Leukaemia—Myeloid | 6.17 × 10−64 | 1.0916 (1.0806, 1.1028) | 0.084 (0.0746, 0.0933) | 0.0655 (0.0581, 0.073) |
| Leukaemia—Lymphoid | 9.73 × 10−101 | 1.0805 (1.0728, 1.0883) | 0.0745 (0.0679, 0.0811) | 0.0582 (0.0529, 0.0634) |
| Corpus Uteri | 0.0000 | 1.0657 (1.0638, 1.0676) | 0.0616 (0.06, 0.0633) | 0.0543 (0.0528, 0.0557) |
| Cervix | 0.0000 | 1.056 (1.0534, 1.0586) | 0.053 (0.0507, 0.0554) | 0.0481 (0.046, 0.0503) |
| Testis | 1.76 × 10−119 | 1.0386 (1.0353, 1.042) | 0.0372 (0.0341, 0.0403) | 0.0337 (0.0309, 0.0365) |
| Ovary | 2.24 × 10−92 | 1.0245 (1.0222, 1.0269) | 0.024 (0.0217, 0.0262) | 0.0217 (0.0196, 0.0238) |
| Anus | 1.47 × 10−8 | 1.0254 (1.0164, 1.0346) | 0.0248 (0.0161, 0.0334) | 0.0225 (0.0146, 0.0303) |
| Non-Hodgkin’s Lymphoma | 1.19 × 10−70 | 0.9781 (0.9757, 0.9805) | −0.0224 (−0.0249, −0.0199) | −0.02 (−0.0223, −0.0178) |
| Melanoma | 6.32 × 10−297 | 0.9571 (0.9549, 0.9594) | −0.0448 (−0.0472, −0.0424) | −0.0403 (−0.0425, −0.0381) |
| Hepatocellular | 0.0175 | 0.9143 (0.8412, 0.9938) | −0.0937 (−0.1888, −0.0063) | −0.0848 (−0.1699, −0.006) |
| Oesophagus | 0.0000 | 0.8962 (0.8929, 0.8996) | −0.1158 (−0.12, −0.1116) | −0.1037 (−0.1074, −0.0999) |
| Brain | 0.0000 | 0.7878 (0.7855, 0.79) | −0.2694 (−0.273, −0.2658) | −0.2371 (−0.2402, −0.234) |
| Penis | 0.0000 | 0.7663 (0.7604, 0.7722) | −0.305 (−0.3152, −0.2949) | −0.2659 (−0.2745, −0.2574) |
| Vulva | 0.0000 | 0.7187 (0.7103, 0.7271) | −0.3915 (−0.4078, −0.3753) | −0.2291 (−0.2375, −0.2207) |
| Vagina | 2.72 × 10−162 | 0.7082 (0.6907, 0.7262) | −0.412 (−0.4478, −0.377) | −0.2403 (−0.2585, −0.2223) |
| Vulva and Vagina | 0.0000 | 0.6967 (0.6918, 0.7017) | −0.4353 (−0.4456, −0.4251) | −0.3788 (−0.3874, −0.3703) |
| Oropharynx | 0.0000 | 0.6292 (0.6245, 0.6339) | −0.5894 (−0.6012, −0.5776) | −0.3675 (−0.3739, −0.3612) |
| Cancer | Term | β-Estimate | Std. Error | p-Value | Adj. P. FDR | Adj. P. Holm | E-Value Estimate | E-Value 95% Lower Bound |
|---|---|---|---|---|---|---|---|---|
| All Cancers nNMSC | LM.Cannabis | 28.793 | 2.305 | 7.96 × 10−26 | 5.58 × 10−25 | 4.86 × 10−24 | 3.50 × 1060 | 1.29 × 1051 |
| Myeloma | LM.Cannabis | 18.834 | 2.110 | 1.64 × 10−16 | 7.15 × 10−16 | 8.99 × 10−15 | 1.79 × 1043 | 6.91 × 1033 |
| Lung | LM.Cannabis | 27.039 | 2.293 | 1.32 × 10−25 | 8.38 × 10−25 | 7.90 × 10−24 | 7.26 × 1039 | 2.00 × 1033 |
| Kidney | LM.Cannabis | 34.251 | 2.951 | 4.67 × 10−25 | 2.73 × 10−24 | 2.76 × 10−23 | 1.70 × 1039 | 4.68 × 1032 |
| Pancreas | LM.Cannabis | 29.502 | 2.956 | 7.13 × 10−20 | 3.57 × 10−19 | 4.07 × 10−18 | 5.97 × 1033 | 1.65 × 1027 |
| Leukaemia—Lymphoid | LM.Cannabis | 9.998 | 2.584 | 3.66 × 10−4 | 5.56 × 10−4 | 9.14 × 10−3 | 1.09 × 1045 | 2.64 × 1022 |
| All Cancers nNMSC | THC.Herb | 10.642 | 0.521 | 1.15 × 10−47 | 1.34 × 10−46 | 7.48 × 10−46 | 3.69 × 1022 | 2.73 × 1020 |
| Non-Hodgkin’s Lymphoma | LM.Cannabis | 26.819 | 3.429 | 1.75 × 10−13 | 6.45 × 10−13 | 9.10 × 10−12 | 3.42 × 1026 | 9.39 × 1019 |
| Colorectum | LM.Cannabis | 18.142 | 2.303 | 1.13 × 10−13 | 4.38 × 10−13 | 5.97 × 10−12 | 5.83 × 1025 | 2.76 × 1019 |
| Prostate | LM.Cannabis | 22.525 | 3.112 | 6.20 × 10−12 | 1.97 × 10−11 | 3.04 × 10−10 | 3.77 × 1024 | 1.04 × 1018 |
| All Cancers | LM.Cannabis | 17.153 | 4.312 | 1.51 × 10−4 | 2.40 × 10−4 | 4.07 × 10−3 | 4.43 × 1034 | 5.71 × 1017 |
| Pancreas | THC.Herb | 15.302 | 0.584 | 1.27 × 10−72 | 8.88 × 10−71 | 8.88 × 10−71 | 4.61 × 1017 | 2.33 × 1016 |
| Hodgkin’s | LM.Cannabis | 13.231 | 2.507 | 2.91 × 10−7 | 6.56 × 10−7 | 1.16 × 10−5 | 3.05 × 1025 | 1.41 × 1016 |
| Stomach | LM.Cannabis | 20.239 | 3.032 | 1.67 × 10−10 | 4.88 × 10−10 | 7.87 × 10−9 | 4.86 × 1022 | 1.34 × 1016 |
| Stomach | THC.Herb | 15.302 | 0.599 | 1.20 × 10−70 | 4.21 × 10−69 | 8.29 × 10−69 | 1.68 × 1017 | 8.51 × 1015 |
| Prostate | THC.Herb | 15.119 | 0.616 | 2.58 × 10−67 | 6.01 × 10−66 | 1.75 × 10−65 | 3.94 × 1016 | 1.98 × 1015 |
| Breast | LM.Cannabis | 13.958 | 2.225 | 1.66 × 10−9 | 4.29 × 10−9 | 7.28 × 10−8 | 2.18 × 1021 | 5.99 × 1014 |
| Kidney | THC.Herb | 13.038 | 0.583 | 8.04 × 10−61 | 1.41 × 10−59 | 5.38 × 10−59 | 1.32 × 1015 | 6.66 × 1013 |
| Lung | THC.Herb | 9.751 | 0.453 | 8.43 × 10−58 | 1.18 × 10−56 | 5.56 × 10−56 | 3.69 × 1014 | 1.86 × 1013 |
| All Cancers | THC.Herb | 9.102 | 1.851 | 4.52 × 10−6 | 8.78 × 10−6 | 1.58 × 10−4 | 3.37 × 1018 | 1.89 × 1011 |
| Breast | THC.Herb | 7.398 | 0.440 | 2.23 × 10−42 | 2.23 × 10−41 | 1.43 × 10−40 | 2.83 × 1011 | 1.43 × 1010 |
| Melanoma | LM.Cannabis | 19.128 | 4.101 | 5.13 × 10−6 | 9.71 × 10−6 | 1.74 × 10−4 | 8.80 × 1015 | 2.42 × 109 |
| Non-Hodgkin’s Lymphoma | THC.Herb | 10.009 | 0.679 | 2.68 × 10−35 | 2.35 × 10−34 | 1.69 × 10−33 | 1.24 × 1010 | 6.20 × 108 |
| Oropharynx | THC.Herb | 17.962 | 5.844 | 3.42 × 10−3 | 4.35 × 10−3 | 5.47 × 10−2 | 4.75 × 1019 | 2.22 × 107 |
| Corpus Uteri | THC.Herb | 10.573 | 0.871 | 7.86 × 10−27 | 6.12 × 10−26 | 4.88 × 10−25 | 2.06 × 108 | 1.06 × 107 |
| Cervix | THC.Herb | 7.048 | 0.692 | 1.79 × 10−20 | 9.64 × 10−20 | 1.04 × 10−18 | 1.15 × 107 | 5.77 × 105 |
| Oropharynx | THC.Resin | 7.056 | 1.081 | 3.30 × 10−8 | 7.96 × 10−8 | 1.38 × 10−6 | 8.17 × 107 | 4.28 × 105 |
| Colorectum | THC.Herb | 4.636 | 0.467 | 1.03 × 10−19 | 4.79 × 10−19 | 5.75 × 10−18 | 6.43 × 106 | 3.36 × 105 |
| Myeloma | THC.Herb | 4.293 | 1.067 | 7.80 × 10−5 | 1.33 × 10−4 | 2.34 × 10−3 | 1.24 × 1010 | 2.16 × 105 |
| Bladder | LM.Cannabis | 12.723 | 3.734 | 7.68 × 10−4 | 1.12 × 10−3 | 1.77 × 10−2 | 5.35 × 1011 | 1.47 × 105 |
| Larynx | LM.Cannabis | 21.062 | 6.232 | 8.46 × 10−4 | 1.21 × 10−3 | 1.86 × 10−2 | 4.31 × 1011 | 1.19 × 105 |
| Oesophagus | LM.Cannabis | 21.862 | 6.703 | 1.27 × 10−3 | 1.74 × 10−3 | 2.53 × 10−2 | 1.73 × 1011 | 4.78 × 104 |
| Ovary | THC.Herb | 6.596 | 0.867 | 6.26 × 10−13 | 2.09 × 10−12 | 3.13 × 10−11 | 2.24 × 105 | 1.13 × 104 |
| Larynx | THC.Herb | 8.671 | 1.231 | 1.95 × 10−11 | 5.93 × 10−11 | 9.35 × 10−10 | 9.27 × 104 | 4.69 × 103 |
| Liver | THC.Herb | 6.224 | 1.927 | 1.44 × 10−3 | 1.94 × 10−3 | 2.74 × 10−2 | 1.64 × 108 | 2.64 × 103 |
| Melanoma | THC.Herb | 5.221 | 0.810 | 6.37 × 10−10 | 1.72 × 10−9 | 2.87 × 10−8 | 3.72 × 104 | 1.88 × 103 |
| Oropharynx | Income | 2.004 | 0.345 | 4.46 × 10−7 | 9.75 × 10−7 | 1.74 × 10−5 | 2.90 × 102 | 53.68 |
| Bladder | THC.Herb | 2.976 | 0.738 | 7.35 × 10−5 | 1.29 × 10−4 | 2.28 × 10−3 | 9.41 × 102 | 47.12 |
| Liver | LM.Cannabis | 8.543 | 3.824 | 2.66 × 10−2 | 2.86 × 10−2 | 0.1792 | 1.45 × 1011 | 44.49 |
| Oesophagus | THC.Herb | 5.217 | 1.324 | 1.07 × 10−4 | 1.74 × 10−4 | 0.0031 | 814.30 | 40.70 |
| Testis | Income | 0.568 | 0.090 | 1.72 × 10−9 | 4.30 × 10−9 | 7.40 × 10−8 | 18.99 | 9.07 |
| Brain | THC.Herb | 3.806 | 1.278 | 3.20 × 10−3 | 4.14 × 10−3 | 0.0543 | 188.66 | 9.00 |
| Gallbladder and Biliary | Income | 0.600 | 0.123 | 2.23 × 10−6 | 4.59 × 10−6 | 8.24 × 10−5 | 23.49 | 8.33 |
| Thyroid | THC.Resin | 1.567 | 0.533 | 3.62 × 10−3 | 4.52 × 10−3 | 0.0547 | 61.00 | 5.75 |
| Anus | Income | 0.347 | 0.069 | 1.09 × 10−6 | 2.30 × 10−6 | 4.12 × 10−5 | 11.84 | 5.52 |
| Myeloma | THC.Resin | 0.632 | 0.222 | 4.79 × 10−3 | 5.68 × 10−3 | 0.0575 | 54.80 | 5.11 |
| Gallbladder and Biliary | Alcohol | 0.296 | 0.037 | 1.09 × 10−13 | 4.38 × 10−13 | 5.87 × 10−12 | 6.27 | 4.48 |
| Ovary | LM.Cannabis | 9.010 | 4.381 | 4.08 × 10−2 | 4.14 × 10−2 | 0.1792 | 1.58 × 107 | 3.76 |
| Myeloma | Income | 0.272 | 0.069 | 1.06 × 10−4 | 1.74 × 10−4 | 0.0031 | 7.82 | 3.53 |
| Oropharynx | Tobacco | 0.298 | 0.037 | 1.99 × 10−10 | 5.58 × 10−10 | 9.17 × 10−9 | 3.61 | 2.89 |
| All Cancers nNMSC | THC.Resin | 0.643 | 0.290 | 2.78 × 10−2 | 2.95 × 10−2 | 0.1792 | 43.75 | 2.24 |
| Leukaemia—Myeloid | Alcohol | 0.164 | 0.062 | 1.15 × 10−2 | 1.32 × 10−2 | 0.1155 | 5.24 | 1.96 |
| Testis | Alcohol | 0.126 | 0.035 | 3.89 × 10−4 | 5.80 × 10−4 | 0.0093 | 2.70 | 1.83 |
| Hodgkin’s | THC.Resin | 0.564 | 0.263 | 3.29 × 10−2 | 3.38 × 10−2 | 0.1792 | 23.15 | 1.80 |
| Prostate | Tobacco | 0.089 | 0.011 | 2.47 × 10−13 | 8.66 × 10−13 | 1.26 × 10−11 | 1.80 | 1.64 |
| Corpus Uteri | Tobacco | 0.085 | 0.016 | 1.05 × 10−7 | 2.45 × 10−7 | 4.31 × 10−6 | 1.59 | 1.43 |
| Breast | Income | 0.145 | 0.060 | 0.0172 | 0.0192 | 0.1379 | 2.69 | 1.42 |
| Myeloma | Tobacco | 0.029 | 0.006 | 2.86 × 10−6 | 5.72 × 10−6 | 1.03 × 10−4 | 1.60 | 1.42 |
| Anus | Alcohol | 0.066 | 0.027 | 0.0140 | 0.0158 | 0.1263 | 2.18 | 1.36 |
| Breast | Tobacco | 0.037 | 0.008 | 1.21 × 10−5 | 2.24 × 10−5 | 4.01 × 10−4 | 1.53 | 1.36 |
| Hodgkin’s | Income | 0.176 | 0.082 | 0.0325 | 0.0338 | 0.1792 | 3.75 | 1.35 |
| Kidney | Tobacco | 0.045 | 0.011 | 4.32 × 10−5 | 7.75 × 10−5 | 0.0014 | 1.50 | 1.32 |
| Lung | Tobacco | 0.032 | 0.008 | 0.0002 | 0.0003 | 0.0046 | 1.47 | 1.29 |
| Hodgkin’s | Tobacco | 0.023 | 0.007 | 0.0011 | 0.0016 | 0.0237 | 1.45 | 1.25 |
| Non-Hodgkin’s Lymphoma | Tobacco | 0.041 | 0.013 | 0.0016 | 0.0022 | 0.0294 | 1.42 | 1.23 |
| Pancreas | Tobacco | 0.031 | 0.011 | 0.0039 | 0.0048 | 0.0551 | 1.39 | 1.19 |
| Colorectum | Tobacco | 0.025 | 0.009 | 0.0041 | 0.0050 | 0.0551 | 1.39 | 1.19 |
| All Cancers | Tobacco | 0.039 | 0.017 | 0.0256 | 0.0280 | 0.1792 | 1.68 | 1.19 |
| Stomach | Tobacco | 0.030 | 0.011 | 0.0077 | 0.0089 | 0.0842 | 1.37 | 1.17 |
| Ovary | Tobacco | 0.033 | 0.016 | 0.0418 | 0.0418 | 0.1792 | 1.31 | 1.05 |
| Term | Count | Negative Total of p-Value Exponents | Mean of the Negative p-Value Exponents | Median of the Negative p-Value Exponents | Total of the Lower E-Value Exponents | Mean of the Lower E-Value Exponents | Median of the Lower E-Value Exponents |
|---|---|---|---|---|---|---|---|
| Last Month’s Cannabis | 19 | 189 | 9.95 | 8 | 341 | 17.95 | 17 |
| Herb. THC | 21 | 551 | 26.24 | 18 | 165 | 7.86 | 7 |
| Resin. THC | 5 | 13 | 2.6 | 2 | 5 | 1.00 | 0 |
| Income | 7 | 29 | 4.14 | 5 | 1 | 0.14 | 0 |
| Alcohol | 4 | 17 | 4.25 | 2 | 0 | 0 | 0 |
| Tobacco | 14 | 55 | 3.93 | 2.5 | 0 | 0 | 0 |
| Cancer | Term | β-Estimate | Std. Error | p-Value | Adj. P. FDR | Adj. P. Holm | E-Value Estimate | E-Value 95% Lower Bound |
|---|---|---|---|---|---|---|---|---|
| Colorectum | Herb. THC | 55.387 | 5.081 | 1.17 × 10−26 | 1.13 × 10−25 | 3.14 × 10−24 | 8.51 × 1030 | 2.72 × 1025 |
| Breast | Herb. THC | 37.005 | 3.771 | 4.69 × 10−22 | 3.59 × 10−21 | 1.22 × 10−19 | 7.65 × 1027 | 2.43 × 1022 |
| Gallbladder and Biliary | Herb. THC | 20.744 | 2.892 | 1.41 × 10−12 | 6.28 × 10−12 | 3.28 × 10−10 | 1.84 × 1026 | 1.53 × 1019 |
| Oropharynx_Broad | Herb. THC | 23.856 | 4.274 | 4.15 × 108 | 1.24 × 10−7 | 8.26 × 10−6 | 1.31 × 1026 | 1.17 × 1017 |
| All Cancers | Herb. THC | 12.034 | 2.019 | 4.45 × 109 | 1.46 × 10−8 | 9.26 × 10−7 | 1.41 × 1023 | 4.48 × 1015 |
| Thyroid | Herb. THC | 20.386 | 3.101 | 7.61 × 10−11 | 2.80 × 10−10 | 1.67 × 10−8 | 2.72 × 1021 | 1.40 × 1015 |
| Anus | Herb. THC | 13.789 | 2.229 | 8.74 × 10−10 | 3.03 × 10−9 | 1.86 × 10−7 | 1.74 × 1020 | 8.63 × 1013 |
| Testis | Herb. THC | 33.843 | 6.241 | 7.22 × 108 | 2.03 × 10−7 | 1.39 × 10−5 | 5.43 × 1017 | 2.80 × 1011 |
| Stomach | Herb. THC | 24.735 | 4.252 | 7.31 × 109 | 2.28 × 10−8 | 1.49 × 10−6 | 4.63 × 1016 | 1.46 × 1011 |
| Oropharynx | Resin. THC | 7.349 | 0.625 | 4.34 × 10−22 | 3.40 × 10−21 | 1.13 × 10−19 | 1.92 × 1011 | 2.86 × 109 |
| Corpus Uteri | Herb. THC | 25.436 | 5.293 | 1.70 × 106 | 4.16 × 10−6 | 3.01 × 10−4 | 6.37 × 1013 | 2.03 × 108 |
| Prostate | Herb. THC | 24.791 | 5.498 | 7.03 × 106 | 1.65 × 10−5 | 1.21 × 10−3 | 9.35 × 1012 | 2.98 × 107 |
| Oesophagus | Herb. THC | 13.982 | 3.228 | 1.58 × 105 | 3.58 × 10−5 | 2.65 × 10−3 | 3.05 × 1012 | 9.59 × 106 |
| Leukaemia—Lymphoid | LM. Cannabis: Herb. THC | 7.397 | 3.030 | 1.57 × 102 | 2.59 × 10−2 | 1.0000 | 2.60 × 1023 | 7.91 × 104 |
| Melanoma | Herb. THC | 10.965 | 3.151 | 5.17 × 104 | 1.03 × 10−3 | 7.76 × 10−2 | 1.26 × 1010 | 3.90 × 104 |
| Cervix | Herb. THC | 13.081 | 5.359 | 1.48 × 102 | 2.46 × 10−2 | 1.00 | 1.47 × 107 | 45.71 |
| Oesophagus | Resin. THC | 1.763 | 0.155 | 1.11 × 10−28 | 1.27 × 10−27 | 3.04 × 10−26 | 68.18 | 36.84 |
| All Cancers nNMSC | Tobacco: Herb. THC | 0.527 | 0.059 | 9.85 × 10−19 | 5.87 × 10−18 | 2.45 × 10−16 | 48.59 | 23.92 |
| Oropharynx | Income | 1.024 | 0.191 | 3.81 × 10−7 | 1.01 × 10−6 | 7.12 × 105 | 67.24 | 18.20 |
| Stomach | LM. Cannabis | 1.283 | 0.096 | 3.16 × 10−38 | 6.28 × 10−37 | 8.98 × 10−36 | 13.60 | 10.06 |
| Kidney | Herb. THC | 6.010 | 2.758 | 2.95 × 10−2 | 4.60 × 10−2 | 1.00 | 2.69 × 106 | 7.94 |
| Colorectum | LM. Cannabis | 1.323 | 0.115 | 2.41 × 10−29 | 3.27 × 10−28 | 6.69 × 10−27 | 10.26 | 7.56 |
| Myeloma | Resin. THC | 0.526 | 0.090 | 6.60 × 10−9 | 2.12 × 10−8 | 1.36 × 10−6 | 14.34 | 7.06 |
| Ovary | Herb. THC | 9.381 | 4.345 | 3.10 × 10−2 | 4.76 × 10−2 | 1.00 | 2.37 × 106 | 6.91 |
| Larynx | Resin. THC | 0.796 | 0.145 | 4.56 × 10−8 | 1.33 × 10−7 | 8.99 × 10−6 | 10.56 | 5.48 |
| Larynx | LM. Cannabis | 0.612 | 0.068 | 8.34 × 10−19 | 5.08 × 10−18 | 2.09 × 10−16 | 6.93 | 5.05 |
| Leukaemia—Lymphoid | Alcohol | 0.164 | 0.021 | 4.81 × 10−13 | 2.24 × 10−12 | 1.13 × 10−10 | 5.96 | 4.29 |
| Breast | LM. Cannabis | 0.628 | 0.086 | 3.61 × 10−13 | 1.71 × 10−12 | 8.51 × 10−11 | 5.32 | 3.83 |
| Thyroid | LM. Cannabis | 0.433 | 0.065 | 4.51 × 10−11 | 1.75 × 10−10 | 1.00 × 10−8 | 5.07 | 3.57 |
| Pancreas | Herb. THC | 5.546 | 2.709 | 4.09 × 10−2 | 6.06 × 10−2 | 1.00 | 1.14 × 106 | 3.00 |
| Hodgkin’s | LM. Cannabis | 0.253 | 0.044 | 1.54 × 10−8 | 4.64 × 10−8 | 3.08 × 10−6 | 4.28 | 2.98 |
| Gallbladder and Biliary | Tobacco: LM. Cannabis: Herb. THC | 0.266 | 0.034 | 7.85 × 10−15 | 4.10 × 10−14 | 1.90 × 10−12 | 3.72 | 2.95 |
| Leukaemia—Myeloid | LM. Cannabis: Herb. THC | 10.783 | 5.449 | 4.96 × 10−2 | 7.24 × 10−2 | 1.00 | 1.08 × 1019 | 2.67 |
| Oropharynx | Tobacco: Herb. THC | 0.509 | 0.187 | 7.31 × 10−3 | 1.27 × 10−2 | 9.36 × 10−1 | 11.00 | 2.66 |
| Leukaemia—Myeloid | Alcohol | 0.195 | 0.037 | 6.00 × 10−7 | 1.53 × 10−6 | 1.09 × 10−4 | 3.78 | 2.63 |
| Corpus Uteri | LM. Cannabis | 0.611 | 0.120 | 3.94 × 10−7 | 1.04 × 10−6 | 7.34 × 10−5 | 3.64 | 2.55 |
| Gallbladder and Biliary | LM. Cannabis | 0.252 | 0.055 | 5.50 × 10−6 | 1.31 × 10−5 | 9.57 × 104 | 3.55 | 2.40 |
| Colorectum | Tobacco: LM. Cannabis: Herb. THC | 0.453 | 0.069 | 5.71 × 10−11 | 2.18 × 10−10 | 1.26 × 10−8 | 2.96 | 2.37 |
| Myeloma | Income | 0.137 | 0.023 | 6.68 × 10−9 | 2.12 × 10−8 | 1.37 × 10−6 | 2.77 | 2.19 |
| Testis | Income | 0.440 | 0.076 | 7.35 × 10−9 | 2.28 × 10−8 | 1.49 × 10−6 | 2.76 | 2.18 |
| Prostate | LM. Cannabis | 0.512 | 0.125 | 4.20 × 10−5 | 9.14 × 10−5 | 6.81 × 10−3 | 3.05 | 2.08 |
| Prostate | Income | 0.372 | 0.054 | 9.01 × 10−12 | 3.63 × 10−11 | 2.03 × 10−9 | 2.47 | 2.08 |
| Stomach | Tobacco: LM. Cannabis: Herb. THC | 0.317 | 0.058 | 4.27 × 10−8 | 1.26 × 10−7 | 8.46 × 10−6 | 2.63 | 2.07 |
| Thyroid | Tobacco: LM. Cannabis: Herb. THC | 0.202 | 0.038 | 9.80 × 10−8 | 2.73 × 10−7 | 1.88 × 10−5 | 2.62 | 2.06 |
| All Cancers | Alcohol | 0.092 | 0.013 | 5.34 × 10−13 | 2.45 × 10−12 | 1.25 × 10−10 | 2.36 | 2.03 |
| Oropharynx | Tobacco | 0.148 | 0.033 | 1.44 × 10−5 | 3.27 × 10−5 | 2.42 × 10−3 | 2.71 | 2.00 |
| Breast | Tobacco: LM. Cannabis: Herb. THC | 0.267 | 0.051 | 1.98 × 10−7 | 5.47 × 10−7 | 3.78 × 10−5 | 2.54 | 2.00 |
| Anus | Tobacco: LM. Cannabis: Herb. THC | 0.138 | 0.027 | 4.91 × 10−7 | 1.26 × 10−6 | 8.99 × 10−5 | 2.55 | 1.98 |
| Breast | Income | 0.216 | 0.037 | 8.22 × 10−9 | 2.53 × 10−8 | 1.66 × 10−6 | 2.25 | 1.87 |
| Non-Hodgkin’s Lymphoma | Tobacco: Herb. THC | 0.314 | 0.103 | 2.26 × 10−3 | 4.19 × 10−3 | 3.12 × 10−1 | 3.15 | 1.82 |
| Lung | Tobacco: Herb. THC | 0.186 | 0.061 | 2.51 × 10−3 | 4.62 × 10−3 | 3.44 × 10−1 | 3.11 | 1.80 |
| Brain | Income | 0.136 | 0.027 | 7.04 × 10−7 | 1.76 × 10−6 | 1.27 × 10−4 | 2.09 | 1.72 |
| Gallbladder and Biliary | Tobacco | 0.072 | 0.005 | 1.16 × 10−41 | 2.89 × 10−40 | 3.34 × 10−39 | 1.76 | 1.68 |
| Larynx | Alcohol | 0.097 | 0.010 | 8.57 × 10−22 | 6.38 × 10−21 | 2.22 × 10−19 | 1.76 | 1.64 |
| Colorectum | Resin. THC | 0.604 | 0.242 | 1.28 × 10−2 | 2.14 × 10−2 | 1.00 | 3.74 | 1.64 |
| Prostate | Tobacco: LM. Cannabis: Herb. THC | 0.253 | 0.074 | 6.94 × 10−4 | 1.37 × 10−3 | 1.03 × 10−1 | 2.03 | 1.53 |
| Kidney | Tobacco: LM. Cannabis: Herb. THC | 0.127 | 0.037 | 7.28 × 10−4 | 1.43 × 10−3 | 1.07 × 10−1 | 2.03 | 1.52 |
| Colorectum | Tobacco | 0.104 | 0.007 | 1.16 × 10−-46 | 4.95 × 10−45 | 3.39 × 10−44 | 1.54 | 1.49 |
| Breast | Tobacco | 0.074 | 0.005 | 5.07 × 10−43 | 1.51 × 10−41 | 1.47 × 10−40 | 1.53 | 1.47 |
| Oesophagus | Tobacco: LM. Cannabis: Herb. THC | 0.136 | 0.044 | 1.86 × 10−3 | 3.48 × 10−3 | 2.60 × 10−1 | 1.96 | 1.45 |
| Stomach | Tobacco | 0.076 | 0.006 | 3.92 × 10−37 | 6.88 × 10−36 | 1.11 × 10−34 | 1.50 | 1.44 |
| Colorectum | Alcohol | 0.103 | 0.017 | 7.58 × 10−10 | 2.66 × 10−9 | 1.62 × 10−7 | 1.54 | 1.41 |
| Pancreas | Tobacco: LM. Cannabis: Herb. THC | 0.108 | 0.037 | 3.44 × 10−3 | 6.22 × 10−3 | 4.62 × 10−1 | 1.91 | 1.40 |
| Ovary | LM. Cannabis | 0.249 | 0.099 | 1.16 × 10−2 | 1.97 × 10−2 | 1.00 | 2.26 | 1.40 |
| Corpus Uteri | Tobacco | 0.077 | 0.007 | 1.75 × 10−25 | 1.58 × 10−24 | 4.66 × 10−23 | 1.43 | 1.37 |
| Oropharynx | Tobacco: LM. Cannabis | 0.060 | 0.020 | 2.75 × 10−3 | 5.02 × 10−3 | 3.74 × 10−1 | 1.76 | 1.37 |
| All Cancers nNMSC | Tobacco: LM. Cannabis | 0.014 | 0.001 | 1.10 × 10−28 | 1.27 × 10−27 | 3.01 × 10−26 | 1.41 | 1.36 |
| Ovary | Tobacco | 0.056 | 0.006 | 3.11 × 10−20 | 2.21 × 10−19 | 8.00 × 10−18 | 1.39 | 1.34 |
| Prostate | Tobacco | 0.068 | 0.008 | 7.41 × 10−19 | 4.60 × 10−18 | 1.86 × 10−16 | 1.38 | 1.33 |
| Corpus Uteri | Tobacco: LM. Cannabis: Herb. THC | 0.188 | 0.072 | 0.0085 | 0.0147 | 1.0000 | 1.83 | 1.32 |
| Hodgkin’s | Tobacco: LM. Cannabis: Herb. THC | 0.067 | 0.026 | 0.0091 | 0.0157 | 1.0000 | 1.84 | 1.31 |
| Bladder | Resin. THC | 0.266 | 0.125 | 0.0330 | 0.0505 | 1.0000 | 3.30 | 1.30 |
| Testis | Tobacco | 0.063 | 0.009 | 2.70 × 10−11 | 1.06 × 10−10 | 6.02 × 10−9 | 1.37 | 1.29 |
| Prostate | Alcohol | 0.075 | 0.018 | 3.02 × 10−5 | 6.71 × 10−5 | 0.0050 | 1.41 | 1.27 |
| Oesophagus | Alcohol | 0.041 | 0.011 | 1.26 × 10−4 | 2.63 × 10−4 | 0.0198 | 1.39 | 1.25 |
| Stomach | Alcohol | 0.053 | 0.014 | 1.41 × 10−4 | 2.92 × 10−4 | 0.0219 | 1.39 | 1.25 |
| Oropharynx_Broad | Tobacco | 0.037 | 0.012 | 0.0017 | 0.0033 | 0.2432 | 1.42 | 1.23 |
| Larynx | Tobacco | 0.024 | 0.004 | 9.70 × 10−9 | 2.95 × 10−8 | 1.95 × 10−6 | 1.29 | 1.22 |
| Melanoma | Tobacco: LM. Cannabis | 0.020 | 0.002 | 4.91 × 10−18 | 2.87 × 10−17 | 1.22 × 10−15 | 1.25 | 1.22 |
| Liver | Tobacco: LM. Cannabis | 0.018 | 0.002 | 3.69 × 10−16 | 2.03 × 10−15 | 9.03 × 10−14 | 1.25 | 1.21 |
| Cervix | Alcohol | 0.059 | 0.018 | 0.0008 | 0.0016 | 0.1181 | 1.36 | 1.21 |
| Breast | Alcohol | 0.041 | 0.012 | 0.0011 | 0.0020 | 0.1510 | 1.35 | 1.20 |
| Lung | Tobacco: LM. Cannabis | 0.011 | 0.001 | 5.55 × 10−15 | 2.95 × 10−14 | 1.35 × 10−12 | 1.23 | 1.20 |
| Melanoma | Income | 0.073 | 0.031 | 0.0183 | 0.0297 | 1.0000 | 1.60 | 1.19 |
| Bladder | LM. Cannabis | 0.126 | 0.059 | 0.0332 | 0.0505 | 1.0000 | 2.08 | 1.19 |
| Pancreas | Tobacco: LM. Cannabis | 0.014 | 0.002 | 1.62 × 10−12 | 7.01 × 10−12 | 3.73 × 10−10 | 1.22 | 1.18 |
| Melanoma | Tobacco: LM. Cannabis: Herb. THC | 0.095 | 0.043 | 0.0266 | 0.0420 | 1.0000 | 1.73 | 1.18 |
| Oesophagus | Tobacco: LM. Cannabis | 0.015 | 0.002 | 7.62 × 10−11 | 2.80 × 10−10 | 1.67 × 10−8 | 1.21 | 1.17 |
| Kidney | Tobacco: LM. Cannabis | 0.013 | 0.002 | 2.33 × 10−10 | 8.48 × 10−10 | 5.07 × 10−8 | 1.21 | 1.17 |
| Brain | Tobacco: LM. Cannabis: Herb. THC | 0.081 | 0.038 | 0.0308 | 0.0476 | 1.0000 | 1.71 | 1.15 |
| Thyroid | Alcohol | 0.027 | 0.011 | 0.0156 | 0.0258 | 1.0000 | 1.33 | 1.12 |
| Anus | Tobacco: LM. Cannabis | 0.006 | 0.001 | 6.92 × 10−5 | 0.0001 | 0.0110 | 1.16 | 1.11 |
| Non-Hodgkin’s Lymphoma | Tobacco: LM. Cannabis | 0.009 | 0.002 | 6.70 × 10−5 | 0.0001 | 0.0107 | 1.16 | 1.11 |
| Testis | Tobacco: LM. Cannabis: Herb. THC | 0.154 | 0.076 | 0.0422 | 0.0619 | 1.0000 | 1.69 | 1.09 |
| All Cancers | Tobacco: LM. Cannabis | 0.013 | 0.006 | 0.0307 | 0.0476 | 1.0000 | 1.31 | 1.08 |
| Cervix | Tobacco | 0.017 | 0.007 | 0.0192 | 0.0310 | 1.0000 | 1.17 | 1.06 |
| Term | Count | Negative Total of p-Value Exponents | Mean of the Negative p-Value Exponents | Median of the Negative p-Value Exponents | Total of the Lower E-Value Exponents | Mean of the Lower E-Value Exponents | Median of the Lower E-Value Exponents |
|---|---|---|---|---|---|---|---|
| Herb. THC | 17 | 128 | 7.53 | 7 | 174 | 10.24 | 11 |
| Resin. THC | 6 | 60 | 10.00 | 7.5 | 10 | 1.67 | 0 |
| Herb. THC: Resin. THC | 2 | 2 | 1.00 | 1 | 4 | 2.00 | 2 |
| Income | 7 | 48 | 6.86 | 8 | 1 | 0.14 | 0 |
| Last Month’s Cannabis | 11 | 124 | 11.27 | 7 | 1 | 0.09 | 0 |
| Tobacco: Herb. THC | 4 | 24 | 6.00 | 2 | 1 | 0.25 | 0 |
| Alcohol | 11 | 76 | 6.91 | 4 | 0 | 0 | 0 |
| Tobacco | 12 | 254 | 21.17 | 18.5 | 0 | 0 | 0 |
| Tobacco: Last Month’s Cann. | 11 | 115 | 10.45 | 10 | 0 | 0 | 0 |
| Tobacco: LM. Cann: Herb. THC | 15 | 67 | 4.47 | 3 | 0 | 0 | 0 |
| No. | Mixed_Effects | Panel_Additive | Panel_Interactive | Panel_2_Lags | Panel_4_Lags | Panel_6_Lags | All Models | 5/6 Models |
|---|---|---|---|---|---|---|---|---|
| 1 | All Cancers | All Cancers | All Cancers | All Cancers | All Cancers | All Cancers | 1 | |
| 2 | All Cancers nNMSC | All Cancers nNMSC | All Cancers nNMSC | All Cancers nNMSC | All Cancers nNMSC | All Cancers nNMSC | 1 | |
| 3 | Anus | Anus | Anus | Anus | ||||
| 4 | Bladder | Bladder | Bladder | Bladder | Bladder | Bladder | 1 | |
| 5 | Brain | Brain | Brain | Brain | Brain | 1 | ||
| 6 | Breast | Breast | Breast | Breast | Breast | Breast | 1 | |
| 7 | Cervix | Cervix | Cervix | Cervix | Cervix | 1 | ||
| 8 | Colorectum | Colorectum | Colorectum | Colorectum | Colorectum | Colorectum | 1 | |
| 9 | Corpus Uteri | Corpus Uteri | Corpus Uteri | Corpus Uteri | ||||
| 10 | Gallbladder and Biliary | Gallbladder and Biliary | Gallbladder and Biliary | Gallbladder and Biliary | ||||
| 11 | Hodgkin’s | Hodgkin’s | Hodgkin’s | Hodgkin’s | Hodgkin’s | Hodgkin’s | 1 | |
| 12 | Kidney | Kidney | Kidney | Kidney | Kidney | Kidney | 1 | |
| 13 | Larynx | Larynx | Larynx | Larynx | Larynx | Larynx | 1 | |
| 14 | Leukaemia—Lymphoid | Leukaemia—Lymphoid | Leukaemia—Lymphoid | |||||
| 15 | Leukaemia—Myeloid | Leukaemia—Myeloid | ||||||
| 16 | Liver | Liver | Liver | Liver | Liver | 1 | ||
| 17 | Lung | Lung | Lung | Lung | Lung | 1 | ||
| 18 | Melanoma | Melanoma | Melanoma | Melanoma | Melanoma | Melanoma | 1 | |
| 19 | Myeloma | Myeloma | Myeloma | Myeloma | Myeloma | Myeloma | 1 | |
| 20 | Non-Hodgkin’s Lymphoma | Non-Hodgkin’s Lymphoma | Non-Hodgkin’s Lymphoma | Non-Hodgkin’s Lymphoma | Non-Hodgkin’s Lymphoma | Non-Hodgkin’s Lymphoma | 1 | |
| 21 | Oesophagus | Oesophagus | Oesophagus | Oesophagus | Oesophagus | Oesophagus | 1 | |
| 22 | Oropharynx | Oropharynx | Oropharynx | Oropharynx | 1 | |||
| 23 | Oropharynx_Broad | Oropharynx_Broad | Oropharynx_Broad | Oropharynx_Broad | ||||
| 24 | Ovary | Ovary | Ovary | Ovary | Ovary | Ovary | 1 | |
| 25 | Pancreas | Pancreas | Pancreas | Pancreas | Pancreas | Pancreas | 1 | |
| 26 | Prostate | Prostate | Prostate | Prostate | Prostate | Prostate | 1 | |
| 27 | Stomach | Stomach | Stomach | Stomach | Stomach | Stomach | 1 | |
| 28 | Testis | Testis | Testis | Testis | Testis | 1 | ||
| 29 | Thyroid | Thyroid | Thyroid | Thyroid | Thyroid | Thyroid | 1 | |
| Totals | 17 | 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reece, A.S.; Bennett, K.; Hulse, G.K. Cannabis- and Substance-Related Carcinogenesis in Europe: A Lagged Causal Inferential Panel Regression Study. J. Xenobiot. 2023, 13, 323-385. https://doi.org/10.3390/jox13030024
Reece AS, Bennett K, Hulse GK. Cannabis- and Substance-Related Carcinogenesis in Europe: A Lagged Causal Inferential Panel Regression Study. Journal of Xenobiotics. 2023; 13(3):323-385. https://doi.org/10.3390/jox13030024
Chicago/Turabian StyleReece, Albert Stuart, Kellie Bennett, and Gary Kenneth Hulse. 2023. "Cannabis- and Substance-Related Carcinogenesis in Europe: A Lagged Causal Inferential Panel Regression Study" Journal of Xenobiotics 13, no. 3: 323-385. https://doi.org/10.3390/jox13030024
APA StyleReece, A. S., Bennett, K., & Hulse, G. K. (2023). Cannabis- and Substance-Related Carcinogenesis in Europe: A Lagged Causal Inferential Panel Regression Study. Journal of Xenobiotics, 13(3), 323-385. https://doi.org/10.3390/jox13030024