
Genotype–Phenotype Correlation in a Couple in Which the Wife Is a Carrier of the Beta-Thalassemia Trait and the Husband Is a Carrier of a Mutation in the ALAS2 Gene: Both Gene Defects Are Associated with Non-Iron-Deficiency Microcytic Anemia

 , , , and
, , , and
Abstract
1. Introduction
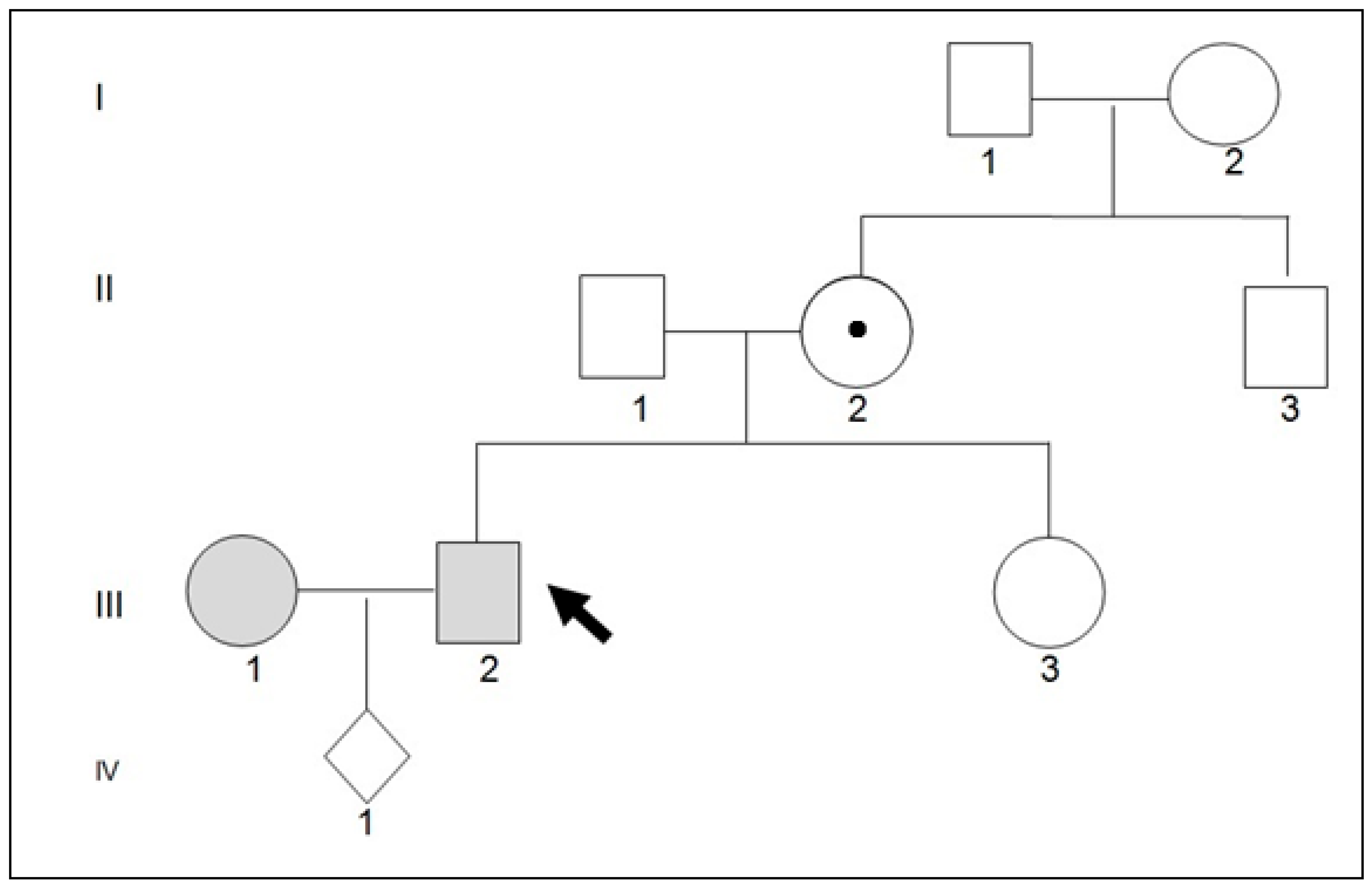
2. Detailed Case Description
3. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ekpe, A.C.; Adefemi, S.A.; Pemi, M.D. Predictors of Anaemia among Pregnant Women Booking for Antenatal Care at Federal Medical Centre, Bida, Niger State, Nigeria. West Afr. J. Med. 2023, 40, 831–837. [Google Scholar] [PubMed]
- Ducamp, S.; Fleming, M.D. The molecular genetics of sideroblastic anemia. Blood 2019, 133, 59–69. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, F.; Sheane, R.; Reynaud, N.; McAuliffe, F.M.; Walsh, J.M. Screening and treatment of iron deficiency anemia in pregnancy: A review and appraisal of current international guidelines. Int. J. Gynaecol. Obstet. 2023, 9, 214–227. [Google Scholar] [CrossRef] [PubMed]
- Dell’Edera, D.; Pacella, E.; Epifania, A.A.; Benedetto, M.; Tinelli, A.; Mazzone, E.; Laterza, F.; Malvasi, A. Importance of molecular biology in the characterization of beta-thalassemia carriers. Eur. Rev. Med. Pharmacol. Sci. 2011, 15, 79–86. [Google Scholar] [PubMed]
- Dell’Edera, D.; Vitucci, A.; Andrisani, G.; Epifania, A.A. Limitations of the biochemical research in the identification of the thalassemic trait carrier. Minerva Ginecol. 2016, 68, 478–479. [Google Scholar] [PubMed]
- Ono, K.; Fujiwara, T.; Saito, K.; Nishizawa, H.; Takahashi, N.; Suzuki, C.; Ochi, T.; Kato, H.; Ishii, Y.; Onodera, K.; et al. Congenital sideroblastic anemia model due to ALAS2 mutation is susceptible to ferroptosis. Sci. Rep. 2022, 12, 9024. [Google Scholar] [CrossRef] [PubMed]
- Rollón, N.; Fernández-Jiménez, M.C.; Moreno-Carralero, M.I.; Murga-Fernández, M.J.; Morán-Jiménez, M.J. Microcytic anemia in a pregnant woman: Beyond iron deficiency. Int. J. Hematol. 2015, 101, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Matthes, T.; Rustin, P.; Trachsel, H.; Darbellay, R.; Costaridou, S.; Xaidara, A.; Rideau, A.; Beris, P. Different pathophysiological mechanisms of intramitochondrial iron accumulation in acquired and congenital sideroblastic anemia caused by mitochondrial DNA deletion. Eur. J. Haematol. 2006, 77, 169–174. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Blood Levels | ||
|---|---|---|
| Normal Values | ||
| Erythrocytes 106/µL | 5.87↑ | 4.0–5.60 |
| Hb g/dL | 12.4 | 12.8–16.5 |
| MCV fl | 69.9↓ | 80–98 |
| MCH pg | 21.1↓ | 27–32 |
| MCHC g/dL | 30.2↓ | 31.0–37.0 |
| HCT % | 41.1 | 41–53 |
| PLT 103/µL | 116.0↓ | 150–450 |
| RDW-CV % | 33.8↑ | 11.0–15.0 |
| HbA2 % | 2.2 | <3.2 |
| HbF % | 0.7 | <1.0 |
| Serum ferritin ng/mL | 170.0 | 22–322 |
| serum iron µg/dL | 261.0↑ | Male: 80–180 Female: 60–160 |
| transferrin mg/dL | 170 | 200–360 |
| % transferrin saturation | 57↑ | 15–55 |
| Blood Levels | ||||
|---|---|---|---|---|
| Proband: Pe. Gi.(III2) | Father (II1) | Mother (II3) | Normal Values | |
| Erythrocytes 106/µL | 5.87↑ | 5.03 | 5.15 | 4.0–5.60 |
| Hb g/dL | 12.4 | 15.0 | 13.40 | 12.8–16.5 |
| MCV fl | 69.9↓ | 93.0 | 83.4 | 80–98 |
| MCH pg | 21.1↓ | 29.7 | 26.0↓ | 27–32 |
| MCHC g/dL | 30.2↓ | 32.0 | 31.2 | 31.0–37.0 |
| HCT % | 41.1 | 46.8 | 42.9 | 41–53 |
| PLT 103/µL | 116.0↓ | 145.0↓ | 203.0 | 150–450 |
| HbA2 % | 2.2 | 2.6 | 2.5 | <3.2 |
| HbF % | 0.6 | 0.3 | 0.4 | <1.0 |
| Serum ferritin ng/mL | 170.0 | 133.0 | 83.0 | 22–322 |
| Serum iron µg/dL | 261.0↑ | 83.0 | 140.0 | Male: 80–180 Female: 60–160 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dell’Edera, D.; Centoducati, C.; Allegretti, A.; La Rocca, F.; Persia, B. Genotype–Phenotype Correlation in a Couple in Which the Wife Is a Carrier of the Beta-Thalassemia Trait and the Husband Is a Carrier of a Mutation in the ALAS2 Gene: Both Gene Defects Are Associated with Non-Iron-Deficiency Microcytic Anemia. Thalass. Rep. 2024, 14, 118-121. https://doi.org/10.3390/thalassrep14040012
Dell’Edera D, Centoducati C, Allegretti A, La Rocca F, Persia B. Genotype–Phenotype Correlation in a Couple in Which the Wife Is a Carrier of the Beta-Thalassemia Trait and the Husband Is a Carrier of a Mutation in the ALAS2 Gene: Both Gene Defects Are Associated with Non-Iron-Deficiency Microcytic Anemia. Thalassemia Reports. 2024; 14(4):118-121. https://doi.org/10.3390/thalassrep14040012
Chicago/Turabian StyleDell’Edera, Domenico, Carmela Centoducati, Arianna Allegretti, Francesco La Rocca, and Brunilde Persia. 2024. "Genotype–Phenotype Correlation in a Couple in Which the Wife Is a Carrier of the Beta-Thalassemia Trait and the Husband Is a Carrier of a Mutation in the ALAS2 Gene: Both Gene Defects Are Associated with Non-Iron-Deficiency Microcytic Anemia" Thalassemia Reports 14, no. 4: 118-121. https://doi.org/10.3390/thalassrep14040012
APA StyleDell’Edera, D., Centoducati, C., Allegretti, A., La Rocca, F., & Persia, B. (2024). Genotype–Phenotype Correlation in a Couple in Which the Wife Is a Carrier of the Beta-Thalassemia Trait and the Husband Is a Carrier of a Mutation in the ALAS2 Gene: Both Gene Defects Are Associated with Non-Iron-Deficiency Microcytic Anemia. Thalassemia Reports, 14(4), 118-121. https://doi.org/10.3390/thalassrep14040012