
HbAdrian (α1:c.251del, p.Leu84Argfs*19)—A Novel Pathogenic Variant in the α1-Globin Gene Associated with Microcytosis from the North of Iran


Abstract
1. Introduction
2. Materials and Methods
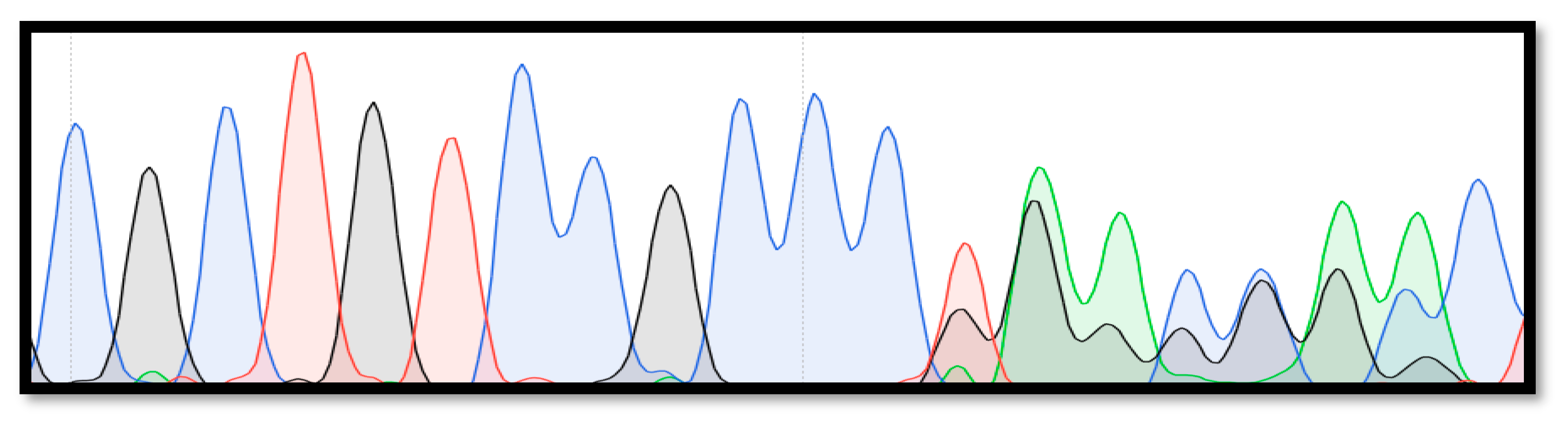
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Higgs, D.R.; Engel, J.D.; Stamatoyannopoulos, G. Thalassaemia. Lancet 2012, 379, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Weatherall, D.J.; Cappellini, M.D. Thalassaemia. Lancet 2018, 391, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Mettananda, S.; Higgs, D.R. Molecular basis and genetic modifiers of thalassemia. Hematol./Oncol. Clin. 2018, 32, 177–191. [Google Scholar] [CrossRef]
- Mettananda, S.; Gibbons, R.J.; Higgs, D.R. α-Globin as a molecular target in the treatment of β-thalassemia. Blood J. Am. Soc. Hematol. 2015, 125, 3694–3701. [Google Scholar] [CrossRef]
- Galanello, R.; Cao, A. Alpha-thalassemia. Genet. Med. 2011, 13, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Jalali, H.; Mahdavi, M.R.; Roshan, P.; Kosaryan, M.; Karami, H.; Mahdavi, M. Alpha thalassemia gene mutations in neonates from Mazandaran, Iran, 2012. Hematology 2014, 19, 192–195. [Google Scholar] [CrossRef]
- Farashi, S.; Harteveld, C.L. Molecular basis of α-thalassemia. Blood Cells Mol. Dis. 2018, 70, 43–53. [Google Scholar] [CrossRef]
- Giardine, B.; van Baal, S.; Kaimakis, P.; Riemer, C.; Miller, W.; Samara, M.; Kollia, P.; Anagnou, N.P.; Chui, D.H.; Wajcman, H.; et al. HbVar database of human hemoglobin variants and thalassemia mutations: 2007 update. Hum. Mutat. 2007, 28, 206. [Google Scholar] [CrossRef]
- Grosso, M.; Sessa, R.; Puzone, S.; Storino, M.R.; Izzo, P. Molecular basis of Thalassemia. Anemia 2012, 2012, 341–358. [Google Scholar]
- Weatherall, D.J.; Clegg, J.B. The Thalassaemia Syndromes; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Hashemieh, M.; Sessa, R.; Puzone, S.; Storino, M.R.; Izzo, P. The Iran thalassemia prevention program: Success or failure? Iran. J. Pediatr. Hematol. Oncol. 2015, 5, 161. [Google Scholar]
- Chong, S.S.; Boehm, C.D.; Higgs, D.R.; Cutting, G.R. Single-tube multiplex-PCR screen for common deletional determinants of α-thalassemia. Blood J. Am. Soc. Hematol. 2000, 95, 360–362. [Google Scholar]
- Tamaddoni, A.; Hadavi, V.; Nejad, N.H.; Khosh-Ain, A.; Siami, R.; Aghai-Meibodi, J.; Almadani, N.; Oberkanins, C.; Law, H.Y.; Najmabadi, H. α-Thalassemia mutation analyses in Mazandaran province, North Iran. Hemoglobin 2009, 33, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, M.R.; Jalali, H.; Kosaryan, M.; Roshan, P.; Mahdavi, M. β-Globin gene cluster haplotypes of Hb D-Los Angeles in Mazandaran province, Iran. Genes Genet. Syst. 2015, 90, 55–57. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mahdavi, M.R.; Bayat, N.; Hadavi, V.; Karami, H.; Roshan, P.; Najmabadi, H.; Rohanizadeh, H. Report of haemoglobin J-Toronto and alpha thalassemia in a family from North of Iran. JPMA-J. Pak. Med. Assoc. 2012, 62, 396. [Google Scholar] [PubMed]
- Mahdavi, M.R.; Karimi, M.; Yavarian, M.; Roshan, P.; Kosaryan, M.; Siami, R. Detection of Hb Setif in north Iran and the question of its origin: Iranian or multiethnic? Hemoglobin 2011, 35, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Jalali, H.; Rasouli, S.; Najafi, M.; Karami, H.; Mahdavi, M. A report of Hb Fontainebleau [α21 (B2) Ala> Pro] as a result of founder effect phenomenon. Gene Rep. 2020, 19, 100587. [Google Scholar] [CrossRef]
- Jalali, H.; Mahdavi, M.R.; Karami, H. Hemoglobin Daneshgah-Tehran (HBA1: c. 218A> G p. His72Arg): A Rare α1-Globin Variant from Iran. Iran. J. Pediatr. Hematol. Oncol. 2020, 10, 200–202. [Google Scholar]
- Aghajani, F.; Mahdavi, M.R.; Kosaryan, M.; Mahdavi, M.; Hamidi, M.; Jalali, H. Identification of β-globin haplotypes linked to sickle hemoglobin (Hb S) alleles in Mazandaran province, Iran. Genes Genet. Syst. 2016, 91, 311–313. [Google Scholar] [CrossRef]
- Jalali, H.; Mahdavi, M.R.; Mahdavi, M.; Abbasi, A. Hb Mazandaran (α1) α51 Gly> Cys (CE9), c. 154 GGC> TGC: A Novel Haemoglobin Variant of α1-Globin Gene. Thalass. Rep. 2022, 12, 51–54. [Google Scholar]
- Jalali, H.; Karami, H.; Mahdavi, M.R.; Mahdavi, M. Co-Inheritance of Heterozygous β0-Thalassemia with Single Functional α-Globin Gene: Challenges of Carrier Detection in Pre-Marital Screening Program for Thalassemia. Thalass. Rep. 2022, 12, 101–104. [Google Scholar] [CrossRef]
- Fucharoen, S.; Viprakasit, V. Hb H disease: Clinical course and disease modifiers. ASH Educ. Program Book 2009, 2009, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Papassotiriou, I.; Traeger-Synodinos, J.; Kanavakis, E.; Karagiorga, M.; Stamoulakatou, A.; Kattamis, C. Erythroid marrow activity and hemoglobin H levels in hemoglobin H disease. J. Pediatr. Hematol./Oncol. 1998, 20, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Kalle Kwaifa, I.; Lai, M.I.; Md Noor, S. Non-deletional alpha thalassaemia: A review. Orphanet J. Rare Dis. 2020, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Age (y) | RBC (×106/µL) | Hb (g/dL) | MCV (fl) | MCH (pg) | Hb-A (%) | Hb-A2 (%) | Hb-F (%) | |
|---|---|---|---|---|---|---|---|---|
| Subject | 29 | 5.11 | 11.9 | 74 | 23.3 | 97.4 | 2.6 | <0.5 |
| Mother | 51 | 3.83 | 10 | 74 | 23 | 97.2 | 2.8 | <0.5 |
| Father | 54 | 4.93 | 13 | 90.1 | 26.5 | 97.3 | 2.7 | <0.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jalali, H.; Karami, H.; Mahdavi, M.; Mahdavi, M.R. HbAdrian (α1:c.251del, p.Leu84Argfs*19)—A Novel Pathogenic Variant in the α1-Globin Gene Associated with Microcytosis from the North of Iran. Thalass. Rep. 2023, 13, 152-156. https://doi.org/10.3390/thalassrep13020014
Jalali H, Karami H, Mahdavi M, Mahdavi MR. HbAdrian (α1:c.251del, p.Leu84Argfs*19)—A Novel Pathogenic Variant in the α1-Globin Gene Associated with Microcytosis from the North of Iran. Thalassemia Reports. 2023; 13(2):152-156. https://doi.org/10.3390/thalassrep13020014
Chicago/Turabian StyleJalali, Hossein, Hossein Karami, Mahan Mahdavi, and Mohammad Reza Mahdavi. 2023. "HbAdrian (α1:c.251del, p.Leu84Argfs*19)—A Novel Pathogenic Variant in the α1-Globin Gene Associated with Microcytosis from the North of Iran" Thalassemia Reports 13, no. 2: 152-156. https://doi.org/10.3390/thalassrep13020014
APA StyleJalali, H., Karami, H., Mahdavi, M., & Mahdavi, M. R. (2023). HbAdrian (α1:c.251del, p.Leu84Argfs*19)—A Novel Pathogenic Variant in the α1-Globin Gene Associated with Microcytosis from the North of Iran. Thalassemia Reports, 13(2), 152-156. https://doi.org/10.3390/thalassrep13020014