
Random Forest Clustering Identifies Three Subgroups of β-Thalassemia with Distinct Clinical Severity


 ,
,  ,
, 
 ,
,  , , ,
, , ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Statistical Analysis
2.1.1. NbClust Procedure
2.1.2. Random Forest Clustering
2.1.3. Identification of the Most Important IPhS
2.1.4. Random Forest Using the Most Important IPhS
2.1.5. Other Statistical Methods
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galanello, R.; Origa, R. Beta-thalassemia. Orphanet J. Rare Dis. 2010, 5, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taher, A.T.; Musallam, K.M.; Cappellini, M.D. β-Thalassemias. N. Engl. J. Med. 2021, 384, 727–743. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Weatherall, D.J.; Cappellini, M.D. Thalassaemia. Lancet 2018, 391, 155–167. [Google Scholar] [CrossRef]
- Steinberg, M.H.; Forget, B.G.; Higgs, D.R.; Weatherall, D.J. Disorders of Hemoglobin: Genetics Pathophysiology, and Clinical Management, 2nd ed.; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Musallam, K.M.; Rivella, S.; Vichinsky, E.; Rachmilewitz, E.A. Non-transfusion-dependent thalassemias. Haematologica 2013, 98, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Musallam, K.M.; Cappellini, M.D.; Viprakasit, V.; Kattamis, A.; Rivella, S.; Taher, A.T. Revisiting the non-transfusion-dependent (NTDT) vs. transfusion-dependent (TDT) thalassemia classification 10 years later. Am. J. Hematol. 2021, 96, E54–E56. [Google Scholar] [CrossRef]
- Vichinsky, E. Non-transfusion-dependent thalassemia and thalassemia intermedia: Epidemiology, complications, and management. Curr. Med. Res. Opin. 2016, 32, 191–204. [Google Scholar] [CrossRef]
- Viprakasit, V.; Tyan, P.; Rodmai, S.; Taher, A.T. Identification and key management of non-transfusion-dependent thalassaemia patients: Not a rare but potentially under-recognised condition. Orphanet J. Rare Dis. 2014, 9, 131. [Google Scholar] [CrossRef] [Green Version]
- Kattamis, A.; Forni, G.L.; Aydinok, Y.; Viprakasit, V. Changing patterns in the epidemiology of beta-thalassemia. Eur. J. Haematol. 2020, 105, 692–703. [Google Scholar] [CrossRef]
- Vitrano, A.; Calvaruso, G.; Lai, E.; Colletta, G.; Quota, A.; Gerardi, C.; Concetta Rigoli, L.; Pitrolo, L.; Cuccia, L.; Gagliardotto, F.; et al. The era of comparable life expectancy between thalassaemia major and intermedia: Is it time to revisit the major-intermedia dichotomy? Br. J. Haematol. 2017, 176, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Taher, A.; Musallam, K.; Cappellini, M.D. Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT); Thalassaemia International Federation: Nicosia, Cyprus, 2017; Volume 2. [Google Scholar]
- Vitrano, A.; Meloni, A.; Addario Pollina, W.; Karimi, M.; El-Beshlawy, A.; Hajipour, M.; Di Marco, V.; Hussain Ansari, S.; Filosa, A.; Ricchi, P.; et al. A complication risk score to evaluate clinical severity of thalassaemia syndromes. Br. J. Haematol. 2021, 192, 626–633. [Google Scholar] [CrossRef]
- Shi, T.; Horvath, S. Unsupervised learning with random forest predictors. J. Comput. Graph. Stat. 2006, 15, 118–138. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Lear. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.; Horvath, S. Using random forest similarities in unsupervised learning: Applications to microarray data. In Atlantic Symposium on Computational Biology and Genome Informatics (CBGI’03); The Association of Intelligent Machinery: Durham, NC, USA, 2003. [Google Scholar]
- Lesmeister, C. Mastering machine learning with R. In Advanced Prediction, Algorithms, and Learning Methods with R, 2nd ed.; Packt Publishing: Birmingham, UK, 2017. [Google Scholar]
- Shi, T.; Seligson, D.; Belldegrun, A.S.; Palotie, A.; Horvath, S. Tumor classification by tissue microarray profiling: Random forest clustering applied to renal cell carcinoma. Mod. Pathol. 2005, 18, 547–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, L.; Rousseeuw, P.J. Finding Groups in Data: An. Introduction to Cluster Analysis; Wiley: New York, NY, USA, 1990. [Google Scholar]
- Chen, X.; Ishwaran, H. Random forests for genomic data analysis. Genomics 2012, 99, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Charrad, M.; Ghazzali, N.; Boiteau, V.; Niknafs, A. NbClust: An R Package for Determining the Relevant Number of Clusters in a Data Set. J. Stat. Softw. 2014, 61, 1–36. [Google Scholar] [CrossRef] [Green Version]
- Ward, J.H. Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 1963, 58, 236–244. [Google Scholar] [CrossRef]
- Florek, K.; Lukaszewicz, J.; Perkal, J.; Zubrzycki, S. Sur la Liaison et la Division des Points d’un Ensemble Fini. Colloq. Math. 1951, 2, 282–285. [Google Scholar] [CrossRef]
- Sokal, R.; Michener, C. A statistical method for evaluating systematic relationships. Univ. Kans. Sci. Bull. 1958, 38, 1409–1438. [Google Scholar]
- Sorensen, T.A. A method of establishing groups of equal amplitude in plant sociology based on similarity of species and its application to analyses of the vegetation on danish commons. Biol. Skr. 1948, 5, 1–34. [Google Scholar]
- McQuitty, L.L. Similarity analysis by reciprocal pairs for discrete and continuous data. Educ. Psychol. Meas. 1966, 26, 825–831. [Google Scholar] [CrossRef]
- Gower, J.C. A comparison of some methods of cluster analysis. Biometrics 1967, 23, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Pepe, A.; Pistoia, L.; Gamberini, M.R.; Cuccia, L.; Lisi, R.; Cecinati, V.; Maggio, A.; Sorrentino, F.; Filosa, A.; Rosso, R.; et al. National networking in rare diseases and reduction of cardiac burden in thalassemia major. Eur. Heart J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Vitrano, A.; Meloni, A.; Pollina, S.A.; Karimi, M.; El-Beshlawy, A.; Hajipour, M.; Di Marco, V.; Ansari, S.H.; Filosa, A.; et al. Survival and causes of death in 2,033 patients with non-transfusion-dependent beta-thalassemia. Haematologica 2021, 106, 2489–2492. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Cappellini, M.D.; Daar, S.; Taher, A.T. Morbidity-free survival and hemoglobin level in non-transfusion-dependent beta-thalassemia: A 10-year cohort study. Ann. Hematol. 2021. [Google Scholar] [CrossRef]
- Musallam, K.M.; Vitrano, A.; Meloni, A.; Pollina, S.A.; Karimi, M.; El-Beshlawy, A.; Hajipour, M.; Di Marco, V.; Ansari, S.H.; Filosa, A.; et al. Risk of mortality from anemia and iron overload in nontransfusion-dependent beta-thalassemia. Am. J. Hematol. 2021. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Variable | Type |
|---|---|
| Age, years | Continuous |
| Age at diagnosis, months | Continuous |
| Age at first transfusion, months | Continuous |
| Age at first iron chelation, months | Continuous |
| Sex (Femal/Male) | Dichotomous |
| Transfusion (Yes/No) | Dichotomous |
| Mean SF, ng/mL | Continuous |
| No. of complications | Counting |
| Cancer (Yes/No) | Dichotomous |
| Cardiac complications (Yes/No) | Dichotomous |
| Diabetes (Yes/No) | Dichotomous |
| Hypogonadism (Yes/No) | Dichotomous |
| Hypoparathyroidism (Yes/No) | Dichotomous |
| Hypothyroidism (Yes/No) | Dichotomous |
| Infections (Yes/No) | Dichotomous |
| Liver complications (Yes/No) | Dichotomous |
| Osteoporosis (Yes/No) | Dichotomous |
| Splenectomy (Yes/No) | Dichotomous |
| Status of death (Yes/No) | Dichotomous |
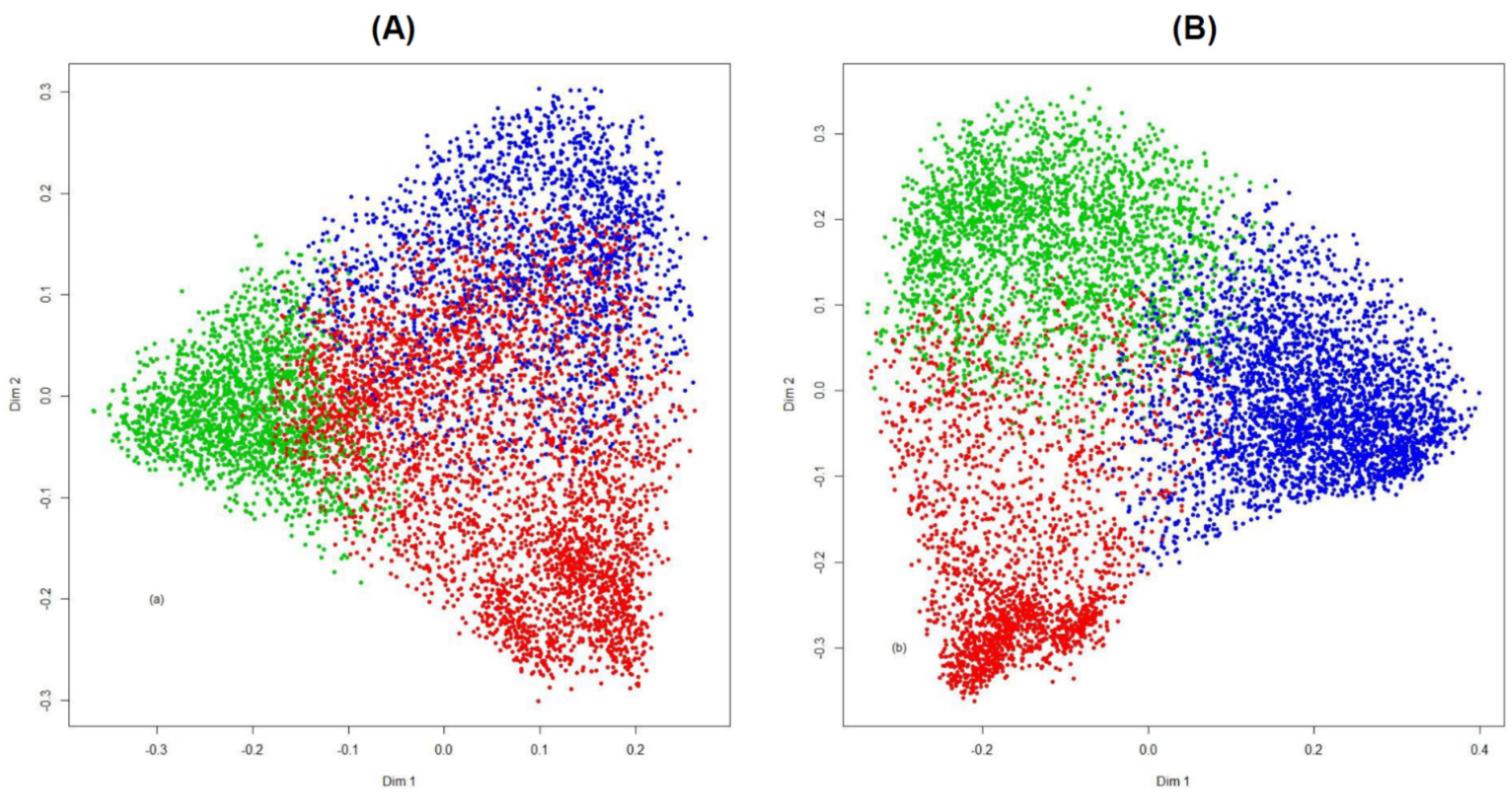
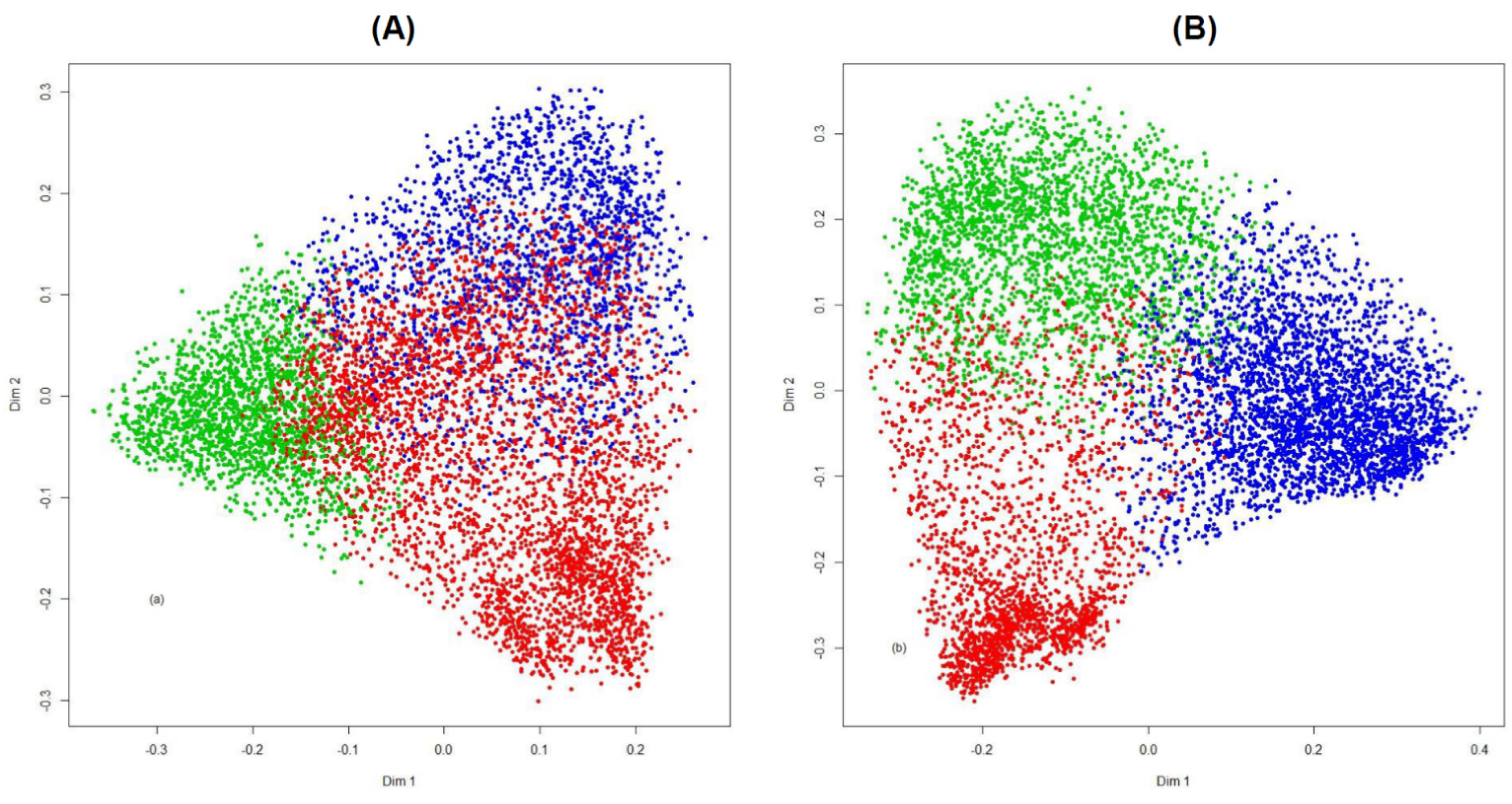
| PAM-RF Distance * | Supervised RF | ||
|---|---|---|---|
| Class 1 | Class 2 | Class 3 | |
| Cluster 1 | 1403 | 79 | 37 |
| Cluster 2 | 33 | 1638 | 46 |
| Cluster 3 | 13 | 29 | 2259 |
| No. misclassified = 237, error rate 4.3% | |||
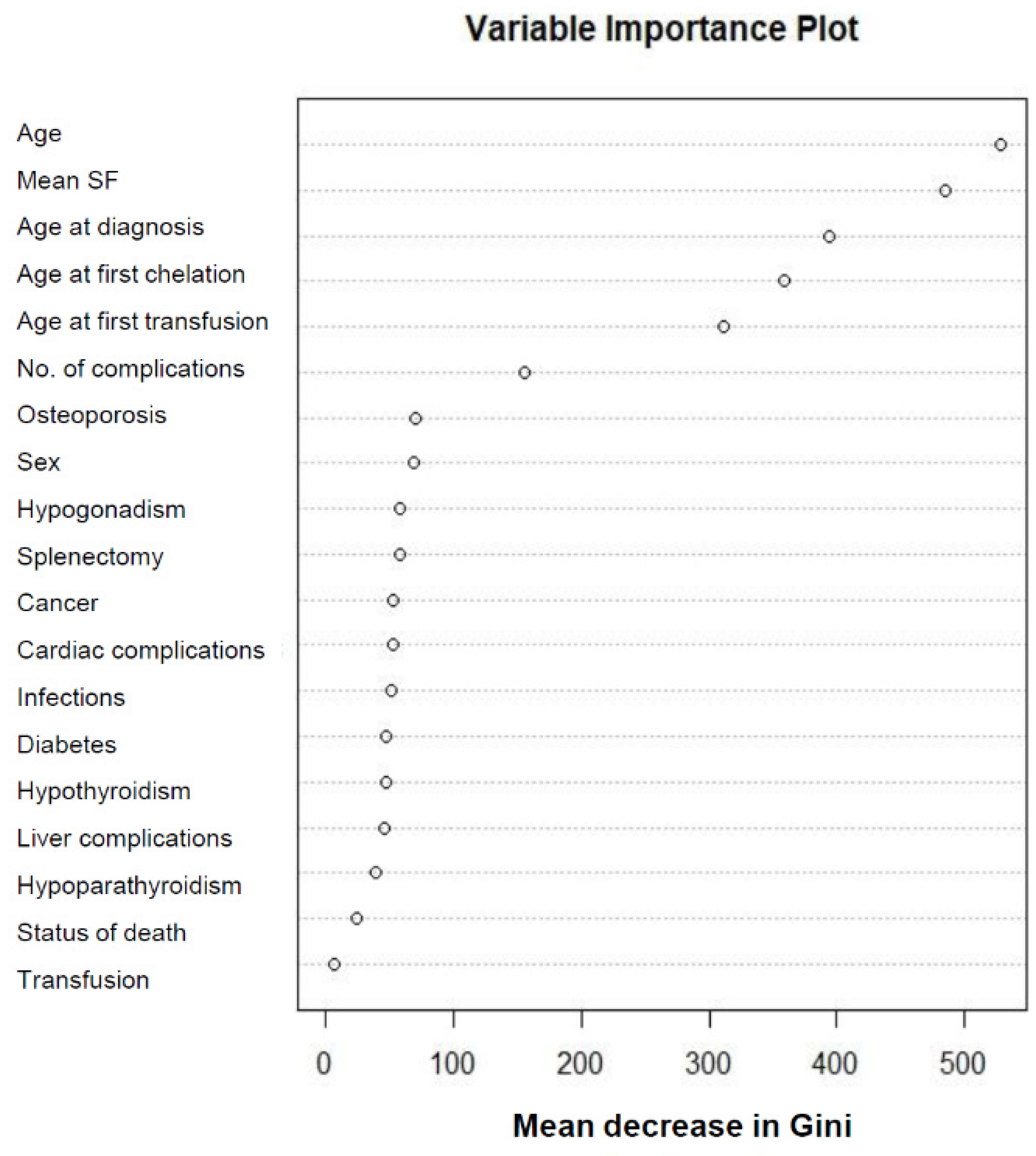
| IPhS | Cluster 1 (n = 2156) | Cluster 2 (n = 2454) | Cluster 3 (n = 3300) | p-Value |
|---|---|---|---|---|
| Age, years | * 39.5 (15.6) * | * 38.9 (7.0) * | 20.3 (7.9) | <0.001 |
| Age at diagnosis, months | 106.1 (125.0) | 12.6 (5.5) | 13.2 (9.4) | <0.001 |
| Age at first transfusion, months | 151.3 (143.6) | 19.2 (11.4) | 17.6 (18.3) | <0.001 |
| Age at first chelation, months | 244.1 (139.2) | 59.5 (30.2) | 51.5 (32.2) | <0.001 |
| Mean SF, ng/mL | 1184.0 (1533.0) * | 1183.0 (715.0) * | 3124.0 (2240.0) | <0.001 |
| No. of complications | 1.8 (1.7) | 2.3 (1.6) | 1.0 (1.2) | <0.001 |
| IPhS | Cluster 1 (n = 99) | Cluster 2 (n = 58) | Cluster 3 (n = 289) | p-Value |
|---|---|---|---|---|
| Heart failure, n (%) | 23 (23.2) | 19 (32.8) | 94 (32.5) | 0.017 |
| Liver damage, n (%) | 7 (7.1) | 7 (12.1) | 2 (0.7) | <0.0001 |
| Hepatocellular carcinoma, n (%) | 14 (14.1) | 7 (12.1) | 5 (1.7) | <0.0001 |
| Other cancers, n (%) | 19 (19.2) | 6 (10.3) | 2 (0.7) | <0.0001 |
| Infections, n (%) | 10 (10.1) | 8 (13.8) | 16 (5.5) | 0.282 |
| Other complications, n (%) | 26 (26.3) | 11 (19.0) | 170 (58.8) | <0.0001 |
| Age at death (years), mean (SD) | 47.3 (17.4) | 39.2 (6.4) | 20.4 (6.8) | <0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitrano, A.; Musallam, K.M.; Meloni, A.; Addario Pollina, S.; Karimi, M.; El-Beshlawy, A.; Hajipour, M.; Di Marco, V.; Ansari, S.H.; Filosa, A.; et al. Random Forest Clustering Identifies Three Subgroups of β-Thalassemia with Distinct Clinical Severity. Thalass. Rep. 2022, 12, 14-23. https://doi.org/10.3390/thalassrep12010004
Vitrano A, Musallam KM, Meloni A, Addario Pollina S, Karimi M, El-Beshlawy A, Hajipour M, Di Marco V, Ansari SH, Filosa A, et al. Random Forest Clustering Identifies Three Subgroups of β-Thalassemia with Distinct Clinical Severity. Thalassemia Reports. 2022; 12(1):14-23. https://doi.org/10.3390/thalassrep12010004
Chicago/Turabian StyleVitrano, Angela, Khaled M. Musallam, Antonella Meloni, Sebastiano Addario Pollina, Mehran Karimi, Amal El-Beshlawy, Mahmoud Hajipour, Vito Di Marco, Saqib Hussain Ansari, Aldo Filosa, and et al. 2022. "Random Forest Clustering Identifies Three Subgroups of β-Thalassemia with Distinct Clinical Severity" Thalassemia Reports 12, no. 1: 14-23. https://doi.org/10.3390/thalassrep12010004
APA StyleVitrano, A., Musallam, K. M., Meloni, A., Addario Pollina, S., Karimi, M., El-Beshlawy, A., Hajipour, M., Di Marco, V., Ansari, S. H., Filosa, A., Ricchi, P., Ceci, A., Daar, S., Vlachaki, E., Singer, S. T., Naserullah, Z. A., Pepe, A., Scondotto, S., Dardanoni, G., ... International Working Group on Thalassemia (IWG-THAL). (2022). Random Forest Clustering Identifies Three Subgroups of β-Thalassemia with Distinct Clinical Severity. Thalassemia Reports, 12(1), 14-23. https://doi.org/10.3390/thalassrep12010004