1. Introduction
The use of comprehensive genomic profiling (CGP) to inform diagnosis, prognosis, and therapeutic selection has become increasingly common in the management of myeloid and advanced solid malignancies [
1]. Biomarker-driven precision medicine strategies have transformed the therapeutic landscape of these cancers and prompted the adaptation of novel clinical trial platforms [
2,
3]. However, the clinical utility of CGP in non-myeloid hematologic malignancies remains controversial, in part due to the limited number of biomarker-driven drugs available in this setting.
Although studies suggest that CGP frequently identifies potentially actionable variants in patients with non-myeloid hematologic malignancies, the definition of “potentially actionable” varies among studies, and the identified variants do not necessarily lead to changes in clinical practice [
4,
5,
6,
7]. For instance, one study found that 82% of patients with lymphoid malignancies who underwent CGP had a variant that could potentially guide therapy, but only 5% had a variant for which there was a United States Food and Drug Administration (FDA)-approved therapy for their diagnosis [
5]. Another study found that CGP identified potentially actionable variants in 52% of patients with lymphoma who underwent CGP to assess for targetable alterations, but none of these patients received a targeted therapy based on the CGP results [
6].
FoundationOne Heme is a commercially available DNA- and RNA-based CGP platform utilized for the sequencing of hematologic malignancies using formalin-fixed paraffin-embedded (FFPE) tissue samples, whole blood samples, or bone marrow aspirate samples [
8]. When RNA sequencing is required, this CGP panel may also be used for sarcomas and other solid malignancies. The sequencing panel includes 406 DNA-sequenced genes and 265 RNA-sequenced genes, including known pathogenic gene rearrangements and fusions. In addition to clinical care, CGP platforms such as these have been adopted for use in large clinical trial efforts for acute myeloid leukemia (AML), including the Beat AML Master Trial which studied the use of CGP-directed care in untreated AML and demonstrated the feasibility of rapid personalized medicine in these populations [
9].
In this study, we determined the rate of variants with potential diagnostic, prognostic, or therapeutic significance that were identified by CGP using FoundationOne Heme in patients with non-myeloid hematologic malignancies at our institution. To assess the clinical impact of these potentially actionable variants, we categorized the identified variants by evidence level and determined the rate at which variants found by CGP led to changes in clinical practice.
2. Materials and Methods
We identified patients who received care for non-myeloid hematologic malignancies at Duke University Medical Center (Durham, NC, USA) and who had CGP sent between October 2014 and November 2021 as part of routine clinical care. CGP was performed as part of routine clinical care by Foundation Medicine (Cambridge, MA, USA) using the FoundationOne Heme panel, which uses hybrid capture-based next-generation sequencing for the DNA sequencing of 406 genes and RNA sequencing of 265 genes [
8]. Diagnoses were made by hematopathologists and clinical oncologists based on the 2016 World Health Organization classifications [
10]. Patient characteristics, CGP results, and details on medical management as a result of CGP were abstracted from a retrospective review of medical records with the data cutoff date 4 February 2022. In addition, provider documentation of changes in medical care as a result of CGP data, including diagnostic and therapeutic management, or the consideration of a biomarker-driven clinical trial, were captured. Approval of this retrospective study was granted by the Duke University Institutional Review Board (protocol numbers Pro00102515 and Pro00101796) and included a waiver of informed consent.
CGP testing results at Duke University are stored and categorized in a secure and searchable clinical database called the Duke Molecular Registry of Tumors (MRT) as part of standard clinical operating protocol [
11]. Patients who have undergone CGP testing are then reviewed at a weekly hematology-specialized multidisciplinary molecular tumor board (MTB). MTB meetings routinely analyze the significant findings within next-generation sequencing reports, review potential clinical applications among a group of disease experts, and subsequently share these discussion findings with the primary oncology team to assist in patient care.
In this study, variants of clinical significance were identified with the aid of the OncoKB precision oncology knowledge base (
https://www.oncokb.org (accessed on 1 February 2022)) and classified by the level of evidence per the 2017 Joint Consensus Recommendations of the Association of Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists [
12,
13,
14]. According to this classification, variants were categorized as having evidence level A (FDA-approved or included in professional guidelines), B (supported by expert consensus), C (investigational or FDA-approved for different tumor type), or D (supported only by preclinical data or case reports) based on information at the time of data abstraction. Variants with therapy sensitizing, diagnostic, or prognostic significance were classified as having evidence level A, B, or C. Therapy sensitizing variants with evidence level C were further subdivided into levels C1 (investigational) or C2 (FDA-approved for different tumor type). Variants associated with therapy resistance were classified as having evidence level A or D. Variants of clinical significance were defined as mutations with therapy sensitizing, diagnostic, or prognostic significance with evidence level A–C, as well as variants related to treatment resistance with evidence level A or D. The original literature supporting the evidence level of each variant is cited on the OncoKB website.
3. Results
3.1. Patients Undergoing Comprehensive Genomic Profiling
A total of 105 samples from 101 patients with non-myeloid hematologic malignancies were sent for comprehensive genomic profiling between 2014 and 2021 (
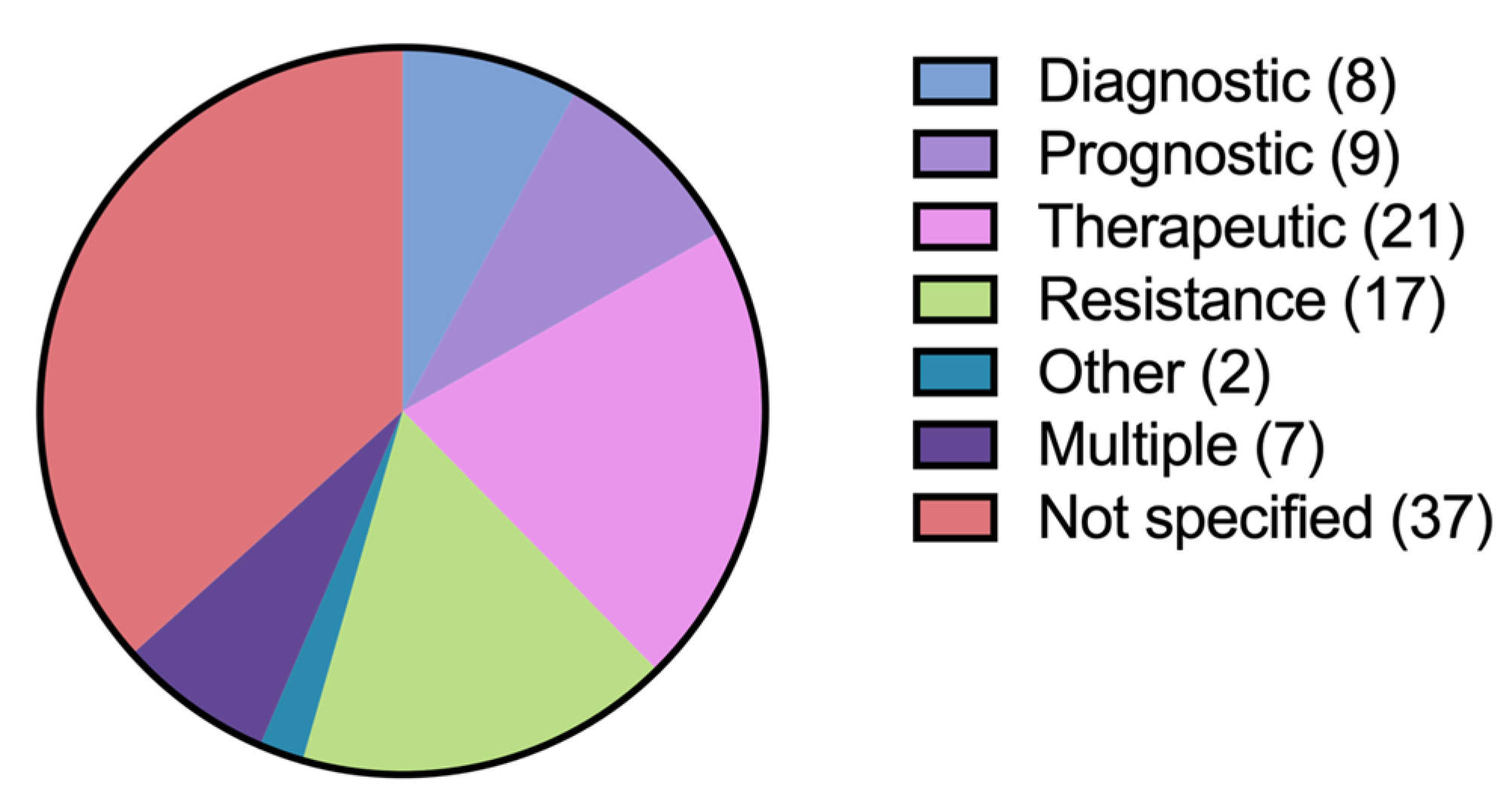
Table 1). In four patients, CGP was sent twice. The median age of tested patients was 60 years. The most common diagnoses among tested patients were chronic lymphocytic leukemia (CLL), B-cell non-Hodgkin lymphoma (NHL), T-cell NHL, histiocytic neoplasms, and plasma cell neoplasms. Justification for sending CGP was often not specified in clinical documentation (
Figure 1). When specified, the most common clinician-stated rationale for sending CGP were to direct therapy selection and to uncover resistance mutations.
The majority of specimens sent for CGP were bone marrow or peripheral blood (
Table 2). Among the 105 samples used to perform CGP, 92 (88%) identified one or more pathogenic or likely pathogenic mutation. The most commonly altered genes were
TP53 (30 patients),
PCLO (20 patients), and
NOTCH1 (20 patients). Notably, CGP data were only reported after 12 patients had already died, and within 30 days of death in an additional 11 patients (
Table 1). Among 101 patients, 73 (72%) had at least one variant of clinical significance associated with diagnosis, prognosis, therapy sensitization, or therapy resistance.
3.2. Variants Associated with Therapy Sensitization or Therapy Resistance
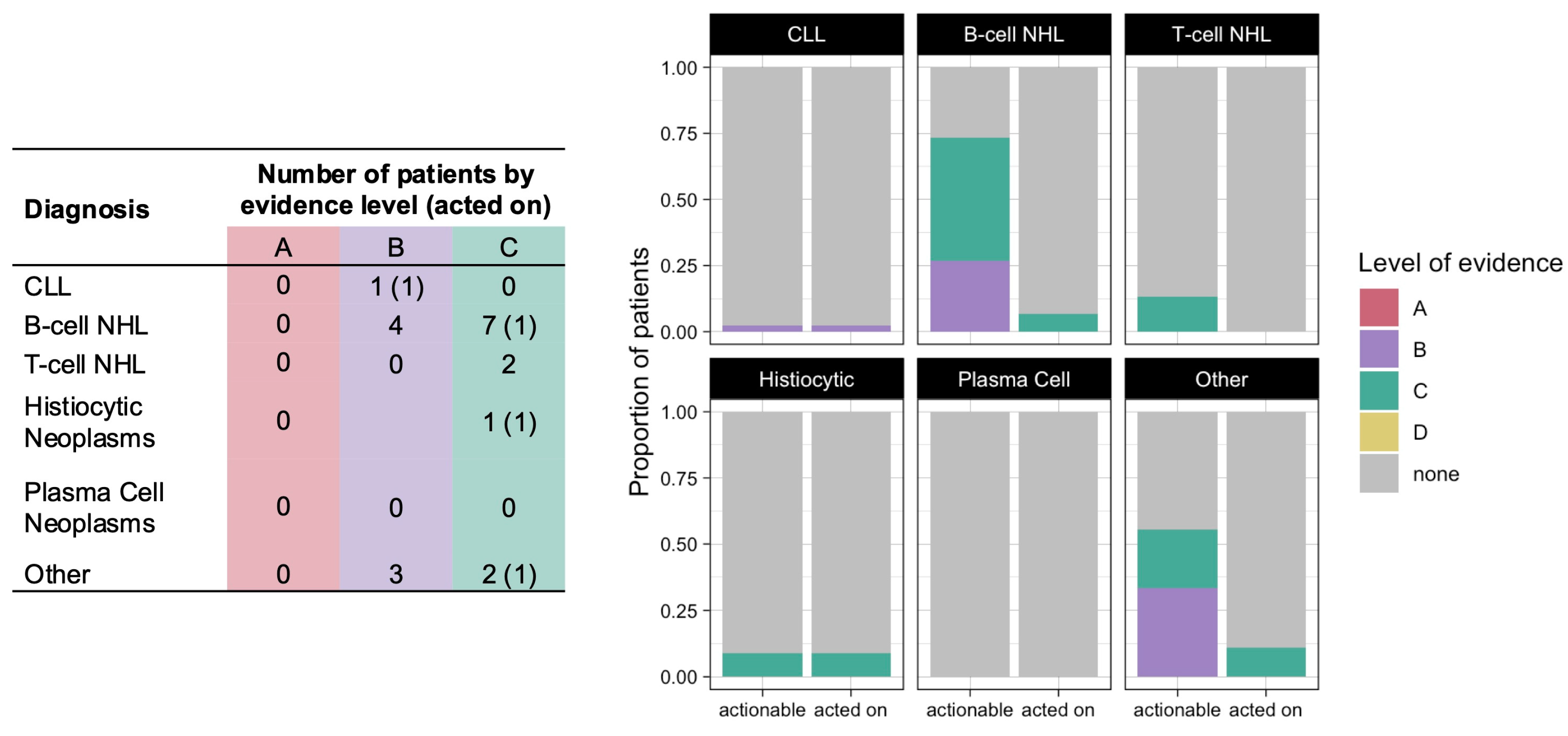
Among the 101 patients who underwent CGP, 61 patients (60%) had a therapy sensitizing variant, all with level of evidence B or C (
Figure 2). The four variants with level B evidence were
KRAS or
MAP2K1 mutations found in patients with histiocytic neoplasms, for which MEK inhibitors such as cobimetinib or trametinib could be utilized. The other variants had either level C1 evidence (investigational) in 42 patients or level C2 evidence (FDA-approved for a different tumor type) in 16 patients. Among the 61 patients with therapy sensitizing variants, 6 patients (10%) were offered a CGP-guided treatment regimen (
Figure 2 and
Table 3), of whom 3 patients actually received the therapy.
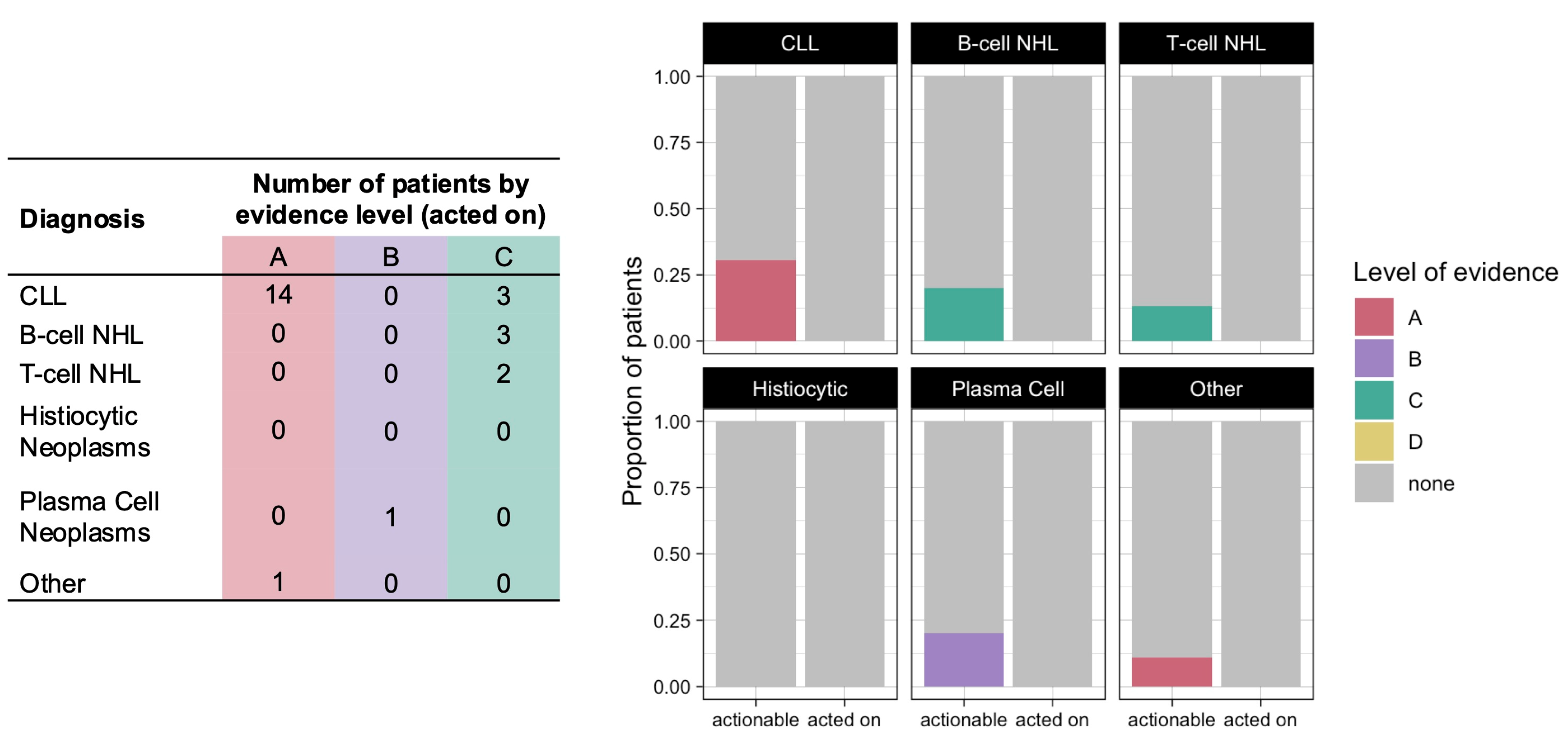
Variants conferring therapy resistance were identified in 13 patients (13%), all of whom had a diagnosis of CLL (
Figure 3). The only identified resistance mutation with level A evidence was the
BTK C481S mutation, which confers resistance to covalent Bruton tyrosine kinase (BTK) inhibitors. Variants with level D evidence included the
BTK C481R mutation and
PCL2G mutations, which confer resistance to BTK inhibitors, as well as the
BCL2 G101V mutation, which confers resistance to the B-cell lymphoma 2 (BCL2) inhibitor venetoclax [
15,
16]. Whereas therapy sensitizing variants rarely led to changes in clinical practice, the discovery of a resistance variant resulted in the suspension of the active treatment or guided subsequent treatment choice in 9 of the 13 patients (69%): 6 patients discontinued ibrutinib, 1 patient discontinued venetoclax, and 2 patients were treated with the non-covalent BTK inhibitor nemtabrutinib based on these CGP results. The absence of a resistance variant identified by CGP affected the treatment selection for another four patients.
3.3. Variants with Diagnostic, Prognostic, or Genetic Significance
Among the 101 patients who underwent CGP, 20 patients (20%) had a variant of diagnostic significance, all with level of evidence B or C (
Figure 4). However, CGP results were used to clarify the diagnosis in only 4 (20%) of these 20 patients: 1 patient with
SOCS1 truncation who was diagnosed with diffuse large B-cell lymphoma, 1 patient with
STAT3 Y640R mutation who was diagnosed with T-cell large granular lymphocytic leukemia, 1 patient with
ETV3–NCOA2 fusion who was diagnosed with indeterminate cell histiocytosis, and 1 patient with
MPL and
CALR mutations who carried a diagnosis of CLL but was additionally diagnosed with myelofibrosis based in part on these results.
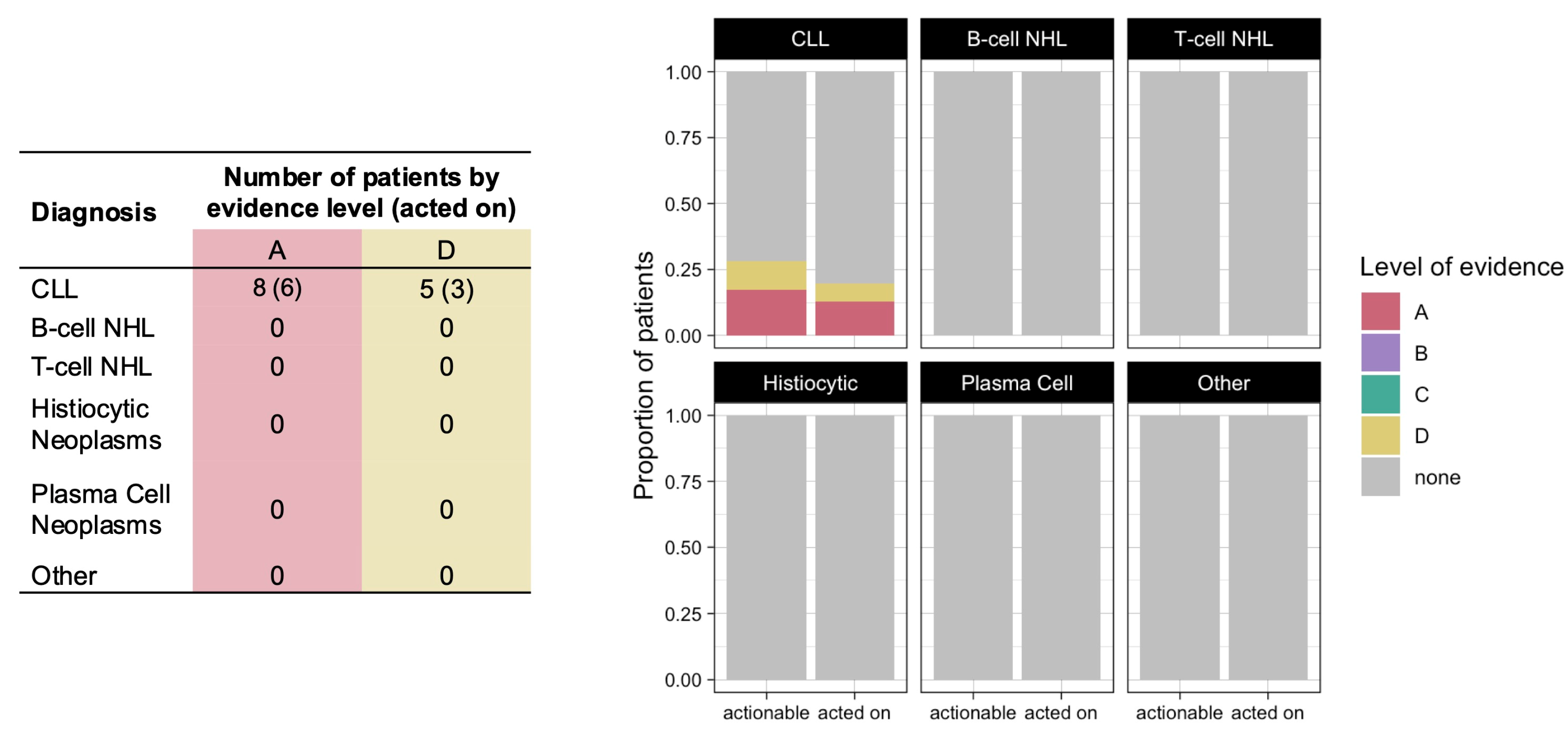
Variants of prognostic significance were identified in 21 patients (21%), but none of the identified variants led to changes in clinical practice (
Figure 5). CGP additionally resulted in genetic counseling consultation for three patients: one patient with CLL who was found to have a
BRCA2 mutation, one patient with CLL who was found to have an
MSH6 mutation, and one patient with marginal zone lymphoma who was found to have a
VHL mutation.
4. Discussion
With advances in our understanding of the diagnostic, prognostic, and therapeutic significance of genomic alterations, as well as the increasing availability of biomarker-directed therapies, comprehensive genomic profiling plays an increasingly important role in clinical oncology, particularly for myeloid and solid malignancies. In non-myeloid hematologic malignancies, the clinical utility of CGP remains in question given that there are a limited number of variants supported by a high level of evidence and few FDA-approved biomarker-directed therapies.
In this retrospective analysis of patients with non-myeloid hematologic malignancies who underwent CGP in a large academic institution, we found that CGP frequently identified variants of potential clinical significance (72%), but only a fraction of identified variants led to changes in clinical practice (22%). Variants conferring therapy resistance were more likely to influence treatment decisions (69%) compared to therapy sensitizing variants (10%), while variants of diagnostic significance helped clarify a diagnosis in 20% of cases, and variants of prognostic significance did not change clinical practice in any cases. The frequency of practice-changing findings on CGP also varied depending on the type of malignancy. While patients with plasma cell neoplasms were identified as having prognostic and therapy sensitizing variants, none of these CGP findings led to changes in clinical practice. Notably, CGP was often sent late in the disease course, with CGP resulting within 30 days of death or after death in 23% of patients. This observation raises the possibility that CGP could change clinical practice in a larger proportion of patients if it is sent earlier in the clinical course.
Notably, variants conferring therapy resistance were only identified in patients with chronic lymphocytic leukemia, but not in any other diagnoses. Moreover, resistance mutations were only identified in a few genes (BTK, PLC2G, and BCL2), suggesting that a targeted mutation panel might be a more cost-effective approach that would achieve the same results as CGP when the goal is to identify variants that confer therapy resistance in CLL. A formal cost-effectiveness analysis in this setting may help guide future use of this technology. Furthermore, with the advancement of medical technology, both the cost and efficiency of genomic assays may improve dramatically leading to greater utilization of this product. The importance of establishing a multi-disciplinary molecular tumor board within a cancer center to facilitate interpretation and clinical management based on CGP results will likely increase as this technology continues to develop.
5. Conclusions
CGP impacts clinical decision making for a subset of patients with non-myeloid hematologic malignancies. However, the current status of the evidence for variants with significance for diagnosis, prognosis, or therapy may not support the universal use of CGP for non-myeloid hematologic malignancies, but suppinstead suggests that CGP should be individualized to the appropriate clinical setting, whereas other approaches such as targeted mutation panels may be more appropriate in certain circumstances. The utility of CGP will need to be re-evaluated periodically as the landscape of genomic biomarkers and biomarker-directed therapies for non-myeloid hematologic malignancies evolves.
Author Contributions
Conceptualization, C.L. and M.S.M.; Investigation, C.L. and K.I.Z.; Methodology, all authors; Data curation, C.L. and K.I.Z.; Visualization, C.L, K.I.Z., B.A.C., J.H.S. and M.B.D.; Project Administration, M.F.G.; Resources, M.F.G., B.A.C., J.H.S., M.B.D. and M.S.M.; Software, M.B.D.; Writing—original draft preparation, C.L. and K.I.Z.; Writing—review and editing, all authors; Supervision, M.S.M. All authors have read and agreed to the published version of the manuscript.
Funding
C.L. and K.I.Z. were supported by the Duke Hematology & Transfusion Medicine Training Program (T32 HL007057).
Institutional Review Board Statement
This study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of Duke Cancer Institute (protocol number Pro00102515 and Pro00101796 determined exempt).
Informed Consent Statement
Patient consent was waived since this study only involved retrospective review of existing archival clinical data.
Data Availability Statement
Please reach out to the corresponding author for data requests.
Conflicts of Interest
The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results. C.L. has performed consulting for Rigel and ad board for Autolus and ADC Therapeutics. B.A.C. has received honoraria from Foundation Medicine and served on an ad board for Guardant. M.F.G is employed by Labcorp. J.S. performs consulting for Abbvie, Astellas, AstraZeneca, Bayer, Beigene, Daiichi-Sankyo, Eli Lilly, GE Healthcare, GSK, Ipsen, Johnson and Johnson, Jazz Pharmaceuticals, Merck, Natera, Pfizer, Roche/Genentech, Regeneron, Sanofi, Taiho, Takeda, and Xilio Therapeutics, owns stock options in Triumvira Immunologics, and receives research funding from Abbvie, Amgen, Apollo Therapeutics, AStar D3, Bayer, Beigene, Curegenix, Daiichi-Sankyo, Eli Lilly, Erasca, GSK, Leap Therapeutics, Novartis, Pfizer, Quanta Therapeutics, Revolution Medicines, and Roche/Genentech. The remaining authors have no relevant disclosures.
References
- Duncavage, E.J.; Bagg, A.; Hasserjian, R.P.; DiNardo, C.D.; Godley, L.A.; Iacobucci, I.; Jaiswal, S.; Malcovati, L.; Vannucchi, A.M.; Patel, K.P.; et al. Genomic Profiling for Clinical Decision Making in Myeloid Neoplasms and Acute Leukemia. Blood 2022, 140, 2228–2247. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Tan, D.S.W. Targeted Therapies for Lung Cancer Patients with Oncogenic Driver Molecular Alterations. J. Clin. Oncol. 2022, 40, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, J.D.; Meric-Bernstam, F.; Swanton, C.; Hurwitz, H.; Spigel, D.R.; Sweeney, C.; Burris, H.A.; Bose, R.; Yoo, B.; Stein, A.; et al. Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results from MyPathway, an Open-Label, Phase IIa Multiple Basket Study. J. Clin. Oncol. 2018, 36, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Joffe, E.; Batlevi, C.L.; Hilden, P.; He, J.; Seshan, V.E.; Zelenetz, A.D.; Palomba, M.L.; Moskowitz, C.H.; Portlock, C.; et al. Integrated DNA/RNA Targeted Genomic Profiling of Diffuse Large B-Cell Lymphoma Using a Clinical Assay. Blood Cancer J. 2018, 8, 60. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Choi, M.; Wieduwilt, M.; Mulroney, C.; Costello, C.; Frampton, G.; Miller, V.; Kurzrock, R. Next Generation Sequencing Reveals Potentially Actionable Alterations in the Majority of Patients with Lymphoid Malignancies. JCO Precis. Oncol. 2017, 1, P16.00004. [Google Scholar] [CrossRef] [PubMed]
- Pillonel, V.; Juskevicius, D.; Bihl, M.; Stenner, F.; Halter, J.P.; Dirnhofer, S.; Tzankov, A. Routine next Generation Sequencing of Lymphoid Malignancies: Clinical Utility and Challenges from a 3-Year Practical Experience. Leuk. Lymphoma 2020, 61, 2568–2583. [Google Scholar] [CrossRef] [PubMed]
- Ikbal Atli, E.; Gurkan, H.; Atli, E.; Yalcintepe, S.; Demir, S.; Eker, D.; Kalkan, R.; Muzaffer Demir, A. Targeted Massively Parallel Sequencing in the Management of Cytogenetically Normal Lymphoid Malignancies. J. BUON 2021, 26, 1540–1548. [Google Scholar] [PubMed]
- FoundationOne. FoundationOne Heme Technical Specifications 2024. Available online: https://www.foundationmedicine.com/sites/default/files/media/documents/2024-04/FoundationOne_Heme_Technical_Specifications.pdf (accessed on 13 September 2024).
- Burd, A.; Levine, R.L.; Ruppert, A.S.; Mims, A.S.; Borate, U.; Stein, E.M.; Patel, P.A.; Baer, M.R.; Stock, W.; Deininger, M.W.; et al. Precision Medicine Treatment in Older AML: Results of Beat AML Master Trial. Blood 2019, 134, 175. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- Green, M.F.; Bell, J.L.; Hubbard, C.B.; McCall, S.J.; McKinney, M.S.; Riedel, J.E.; Menendez, C.S.; Abbruzzese, J.L.; Strickler, J.H.; Datto, M.B. Implementation of a Molecular Tumor Registry to Support the Adoption of Precision Oncology within an Academic Medical Center: The Duke University Experience. JCO Precis. Oncol. 2021, 5, PO.21.00030. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017, PO.17.00011. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [PubMed]
- Suehnholz, S.P.; Nissan, M.H.; Zhang, H.; Kundra, R.; Nandakumar, S.; Lu, C.; Carrero, S.; Dhaneshwar, A.; Fernandez, N.; Xu, B.W.; et al. Quantifying the Expanding Landscape of Clinical Actionability for Patients with Cancer. Cancer Discov. 2024, 14, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Ruppert, A.S.; Guinn, D.; Lehman, A.; Blachly, J.S.; Lozanski, A.; Heerema, N.A.; Zhao, W.; Coleman, J.; Jones, D.; et al. BTKC481S-Mediated Resistance to Ibrutinib in Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2017, 35, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Blombery, P.; Anderson, M.A.; Gong, J.; Thijssen, R.; Birkinshaw, R.W.; Thompson, E.R.; Teh, C.E.; Nguyen, T.; Xu, Z.; Flensburg, C.; et al. Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia. Cancer Discov. 2019, 9, 342–353. [Google Scholar] [CrossRef] [PubMed]
| Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}