

Lymphocytic Lymphoma Transforming into Hodgkin Lymphoma in Sub-Saharan Africa: Case Report and Literature Review

 ,
, {kind=link}
Abstract
1. Introduction
2. Material and Methods
- -
- Slides were prepared by cutting the blocks with a microtome into 4-micron strips, then spread out on super-adhesive slides incubated at 37° for 1 h.
- -
- Dewaxing of preparations was performed by successive immersion in solvents (xylene 2, absolute ethanol, ethanol 80%, ethanol 70%, tap water and distilled water).
- -
- Heat unmasking was performed by immersing the preparations in a citrate buffer of pH 6.1 (K802321-2 DAKO, Agilent, Santa Clara, CA, USA). After cooling to room temperature, the slides were immersed in distilled water.
- -
- The slides were rinsed and marked by placing them in the wet chamber and rinsing them once with PBS buffer. The slides were drained and dried before being marked with a hydrophobic marker (PAPEN).
- -
- Epitopes were blocked by immersing the slides in a solution of hydrogen peroxide (H2O2). Rinsing was performed using PBS 3 times.
- -
- Primary antibodies were applied to the slides and incubated overnight in a humidity chamber. There was no dilution because the antibodies were ready to use. Two control slides were used for each antibody. After incubation, the deposited antibodies were rinsed with PBS buffer 3 times for 5 min.
- -
- The deposition of peroxidase-labelled secondary antibody (K802321-2 DAKO Envision Flex high PH kit) was performed. Incubation lasted 20 min in a humid chamber, followed by rinsing with PBS buffer 3 times for 5 min each.
- -
- Revealing antigen–antibody bonds: chromogen substrate (DAB: 3,3′-diaminobenzidine) was applied to the tissue in the darkroom for 10 min. The slides were rinsed with distilled water for 15 s.
- -
- Counter-staining was performed by staining preparations with a drop of MAYER hematoxylin solution for 30 s, then rinsing them with distilled water before being rehydrated and mounted between slides and coverslips.
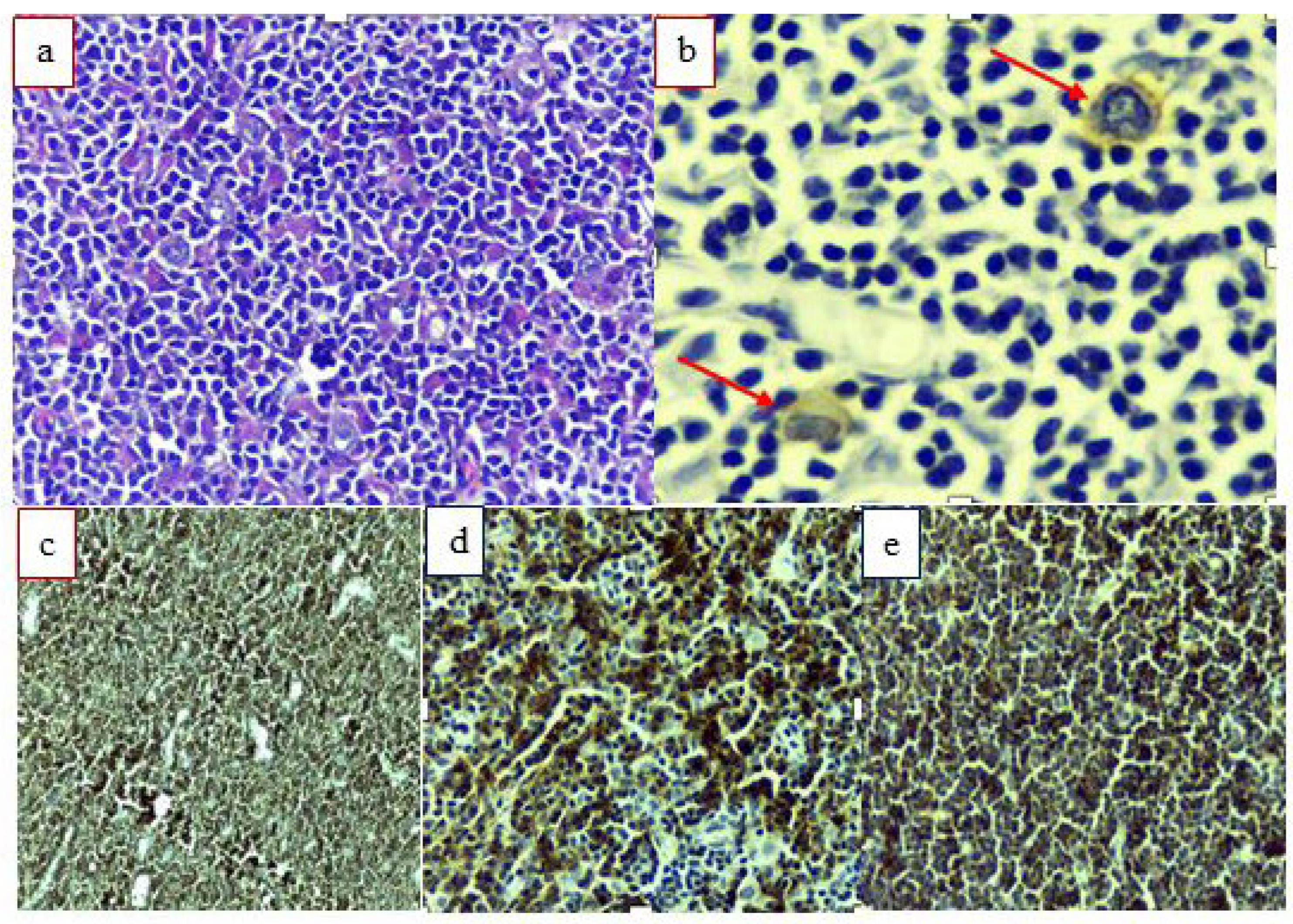
3. Case Description
4. Discussion
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Buckley, T.; Inamdar, A.; Mikhail, N.H.; Loo, A.; Cohen, S. Rare Richter Transformation of Chronic Lymphocytic Lymphoma to Hodgkin Lymphoma. Am. J. Case Rep. 2021, 22, e932904. [Google Scholar] [CrossRef] [PubMed]
- Jamroziak, K.; Grzybowska-Izydorczyk, O.; Jesionek-Kupnicka, D.; Gora-Tybor, J.; Robak, T. Poor prognosis of Hodgkin variant of Richter transformation in chronic lymphocytic leukemia treated with cladribine. Br. J. Haematol. 2012, 158, 286–288. [Google Scholar] [CrossRef]
- Tadmor, T.; Levy, I. Richter Transformation in Chronic Lymphocytic Leukemia: Update in the Era of Novel Agents. Cancers 2021, 13, 5141. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.A.; Siddiqi, T. Treatment of Richter’s syndrome. Hematol. Am. Soc. Hematol. Educ. Program 2022, 2022, 329–336. [Google Scholar] [CrossRef]
- Jain, N.; Keating, M.J. Richter transformation of CLL. Expert Rev. Hematol. 2016, 9, 793–801. [Google Scholar] [CrossRef]
- Tzankov, A.; Fong, D. Hodgkin’s disease variant of Richter’s syndrome clonally related to chronic lymphocytic leukemia arises in ZAP-70 negative mutated CLL. Med. Hypotheses 2006, 66, 577–579. [Google Scholar] [CrossRef]
- Parikh, S.A.; Habermann, T.M.; Chaffee, K.G.; Call, T.G.; Ding, W.; Leis, J.F.; Macon, W.R.; Schwager, S.M.; Ristow, K.M.; Porrata, L.F.; et al. Hodgkin transformation of chronic lymphocytic leukemia: Incidence, outcomes, and comparison to de novo Hodgkin lymphoma: Hodgkin transformation of CLL. Am. J. Hematol. 2015, 90, 334–338. [Google Scholar] [CrossRef]
- Padaro, E.; Layibo, Y.; Kueviakoe, I.D.M.; Agbétiafa, K.; Magnang, H.; Koudokpo, N.D.A.; Mawussi, K.; Vovor, A. Caractéristiques de la leucémie lymphoïde chronique au Togo. Pan. Afr. Med. J. 2019, 34, 84. [Google Scholar] [CrossRef]
- Innocenti, I.; Benintende, G.; Tomasso, A.; Fresa, A.; Autore, F.; Larocca, L.M.; Laurenti, L. Richter transformation in Chronic Lymphocytic Leukemia. Hematol. Oncol. 2023, 41, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Travis, L.B.; Curtis, R.E.; Hankey, B.F.; Fraumeni, J.F. Second Cancers in patients With Chronic Lymphocytic Leukemia. JNCI J. Natl. Cancer Inst. 1992, 84, 1422–1427. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; O’Brien, S.; Kantarjian, H.M.; Koller, C.; Hagemeister, F.B.; Fayad, L.; Lerner, S.; Bueso-Ramos, C.E.; Keating, M.J. Hodgkin transformation of chronic lymphocytic leukemia: The M. D. Anderson Cancer Center experience. Cancer 2006, 107, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- Bockorny, B.; Codreanu, I.; Dasanu, C.A. Hodgkin lymphoma as Richter transformation in chronic lymphocytic leukaemia: A retrospective analysis of world literature: Hodgkin Lymphoma as Richter transformation. Br. J. Haematol. 2012, 156, 50–66. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.A.; Slager, S.L.; Rabe, K.G.; Kay, N.E.; Cerhan, J.R.; Shanafelt, T.D. Heritable Predisposition To Richter Syndrome in Patients With Chronic Lymphocytic Leukemia. Blood 2013, 122, 2867. [Google Scholar] [CrossRef]
- Kanzler, H.; Küppers, R.; Helmes, S.; Wacker, H.H.; Chott, A.; Hansmann, M.L.; Rajewsky, K. Hodgkin and Reed-Sternberg–like cells in B-cell chronic lymphocytic leukemia represent the outgrowth of single germinal-center B-cell–derived clones: Potential precursors of Hodgkin and Reed-Sternberg cells in Hodgkin’s disease. Blood 2000, 95, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Momose, H.; Jaffe, E.S.; Shin, S.S.; Chen, Y.Y.; Weiss, L.M. Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma with Reed-Sternberg–Like Cells and Possible Transformation to Hodgkin’s Disease: Mediation by Epstein-Barr Virus. Am. J. Surg. Pathol. 1992, 16, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Tadmor, T.; Shvidel, L.; Goldschmidt, N.; Ruchlemer, R.; Fineman, R.; Bairey, O.; Rahimi-Levene, N.; Herishanu, Y.; Yuklea, M.; Arad, A.; et al. Hodgkin’s variant of Richter transformation in chronic lymphocytic leukemia; a retrospective study from the Israeli CLL study group. Anticancer. Res. 2014, 34, 785–790. [Google Scholar]
- Adiga, G.U.; Abebe, L.; Wiernik, P.H. Partially successful treatment of a patient with chronic lymphocytic leukemia and Hodgkin’s disease: Case report and literature review. Am. J. Hematol. 2003, 72, 267–273. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A.; Gross, A. Persistence of the Epstein–Barr Virus and the Origins of Associated Lymphomas. N. Engl. J. Med. 2004, 350, 1328–1337. [Google Scholar] [CrossRef]
- Dolcetti, R.; Masucci, M.G. Epstein-Barr virus: Induction and control of cell transformation. J. Cell. Physiol. 2003, 196, 207–218. [Google Scholar] [CrossRef]
- Ghosh, S.K.; Perrine, S.P.; Faller, D.V. Advances in Virus-Directed Therapeutics against Epstein-Barr Virus-Associated Malignancies. Adv. Virol. 2012, 2012, 509296. [Google Scholar] [CrossRef]
- Sigmund, A.M.; Kittai, A.S. Richter’s Transformation. Curr. Oncol. Rep. 2022, 24, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Touré, S.A.; Niang, D.; Gueye, S.M.; Keita, M.; Diallo, A.B.; Bousso, E.S.; Dieng, F.; Faye, B.F.; Seck, M.; Diop, S. Lymphocytic Lymphoma Transforming into Hodgkin Lymphoma in Sub-Saharan Africa: Case Report and Literature Review. Hematol. Rep. 2024, 16, 523-528. https://doi.org/10.3390/hematolrep16030050
Touré SA, Niang D, Gueye SM, Keita M, Diallo AB, Bousso ES, Dieng F, Faye BF, Seck M, Diop S. Lymphocytic Lymphoma Transforming into Hodgkin Lymphoma in Sub-Saharan Africa: Case Report and Literature Review. Hematology Reports. 2024; 16(3):523-528. https://doi.org/10.3390/hematolrep16030050
Chicago/Turabian StyleTouré, Sokhna Aïssatou, Dibor Niang, Serigne Mourtalla Gueye, Mohamed Keita, Alioune Badara Diallo, Elimane Seydi Bousso, Fatma Dieng, Blaise Felix Faye, Moussa Seck, and Saliou Diop. 2024. "Lymphocytic Lymphoma Transforming into Hodgkin Lymphoma in Sub-Saharan Africa: Case Report and Literature Review" Hematology Reports 16, no. 3: 523-528. https://doi.org/10.3390/hematolrep16030050
APA StyleTouré, S. A., Niang, D., Gueye, S. M., Keita, M., Diallo, A. B., Bousso, E. S., Dieng, F., Faye, B. F., Seck, M., & Diop, S. (2024). Lymphocytic Lymphoma Transforming into Hodgkin Lymphoma in Sub-Saharan Africa: Case Report and Literature Review. Hematology Reports, 16(3), 523-528. https://doi.org/10.3390/hematolrep16030050