
Haemophilia A: A Review of Clinical Manifestations, Treatment, Mutations, and the Development of Inhibitors

 ,
,
Abstract
1. Methods
2. Physiology of the Blood Coagulation System
2.1. Haemostasis
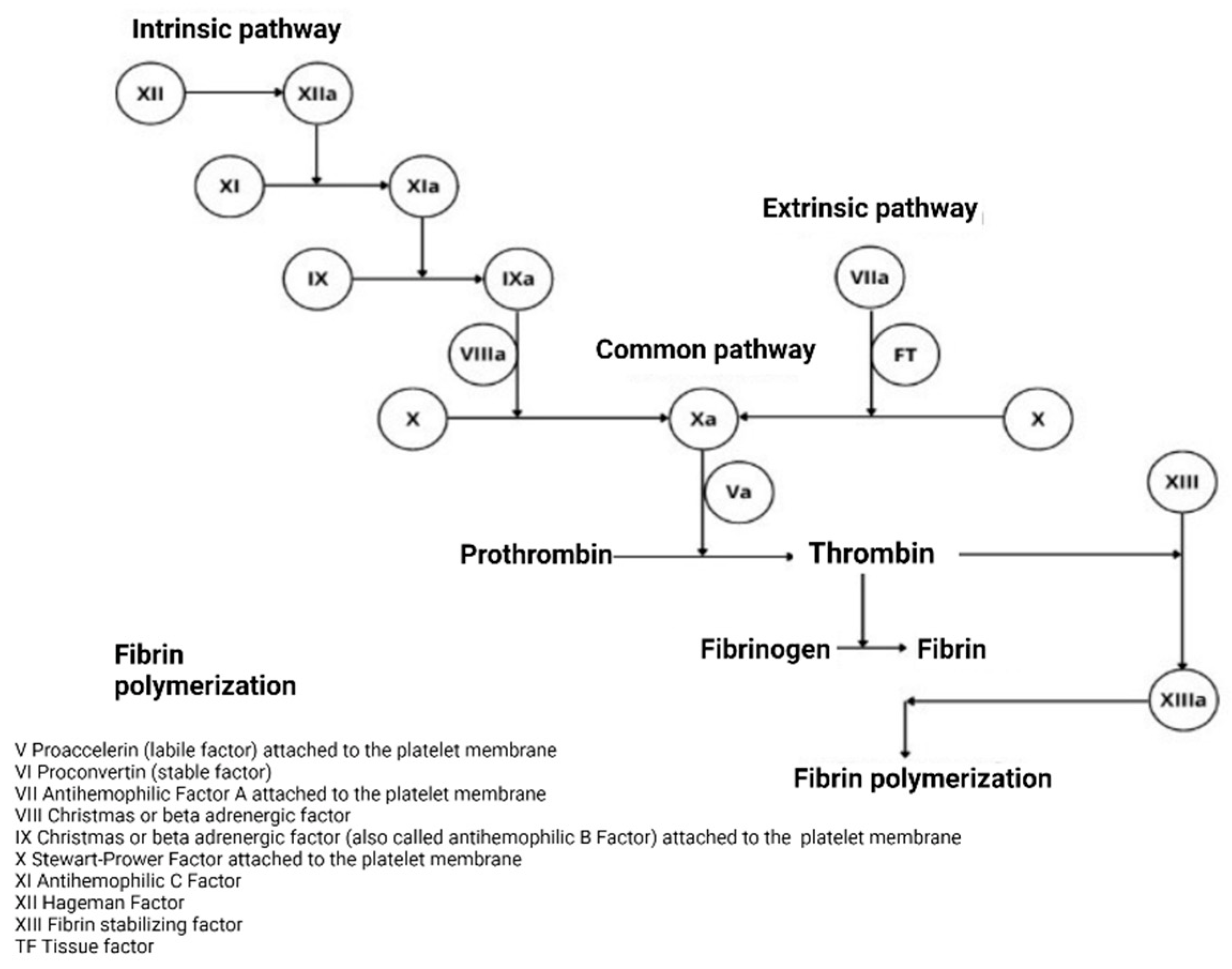
2.1.1. Classical Theory
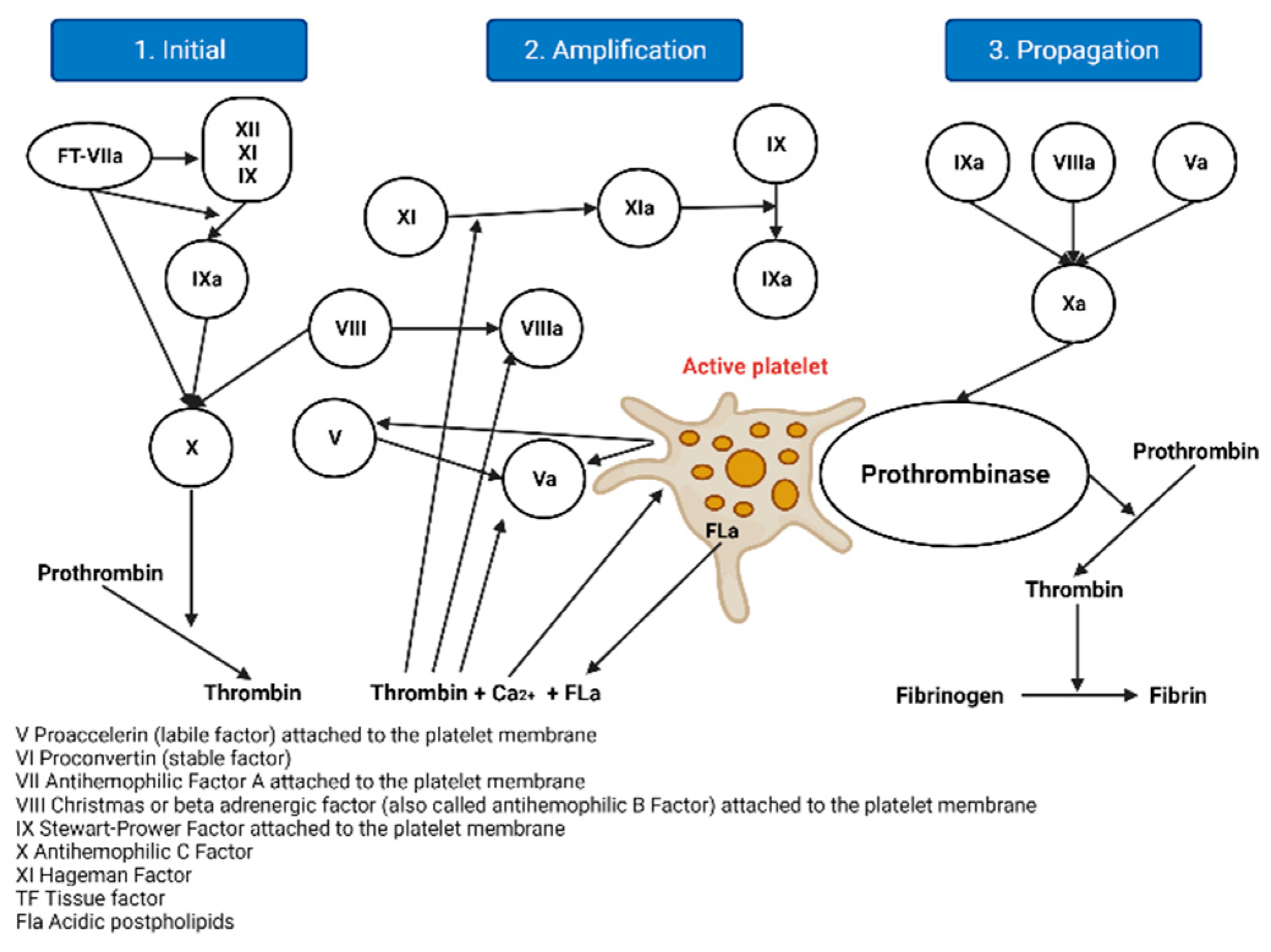
2.1.2. The Cellular Model of Thrombin Formation
- Initiation: Haemostasis is initiated by vascular rupture, giving rise to the exposure of the tissue factor (TF), which is not usually in contact with blood. When this union with FVIIa occurs, a tenase complex is formed that activates FX by joining FV, forming a new complex, enzyme prothrombinase, which generates thrombin. In addition to activating FV, it is also activated by FXI, which results in FXa and thrombin, but in minimal amounts, since it is inhibited by the thrombin-inhibiting factor [5]. The extrinsic pathway is now the initiating physiological pathway that produces small amounts of thrombin and activates platelets, as mentioned above [2].
- Amplification: This stage occurs through positive feedback on the intrinsic and common pathways, generating large amounts of thrombin. It is known as amplification [2], as described below. The reduced amounts of thrombin (FIIa) formed in the initiation phase activate the FV cofactors, FXI, FXIII, and platelets, through protease-activated receptors; FVIII is separated from VWF by thrombin and the other factors are activated for the next phase [6,7].
- Spread: At this stage, the phase of fibrinogenesis and platelet aggregation begins [2]. It occurs on the surfaces of previously activated platelets, leaving receptors exposed for the binding of activated coagulation factors. It binds FXI, activating more FIX molecules by binding to its cofactor FVIII and forming a more potent defence complex, forming more FXa. FXA binds the FV cofactor again, which leads to the generation of more thrombin, but it is no longer inhibited [6,7].
3. Haemophilia A
3.1. History
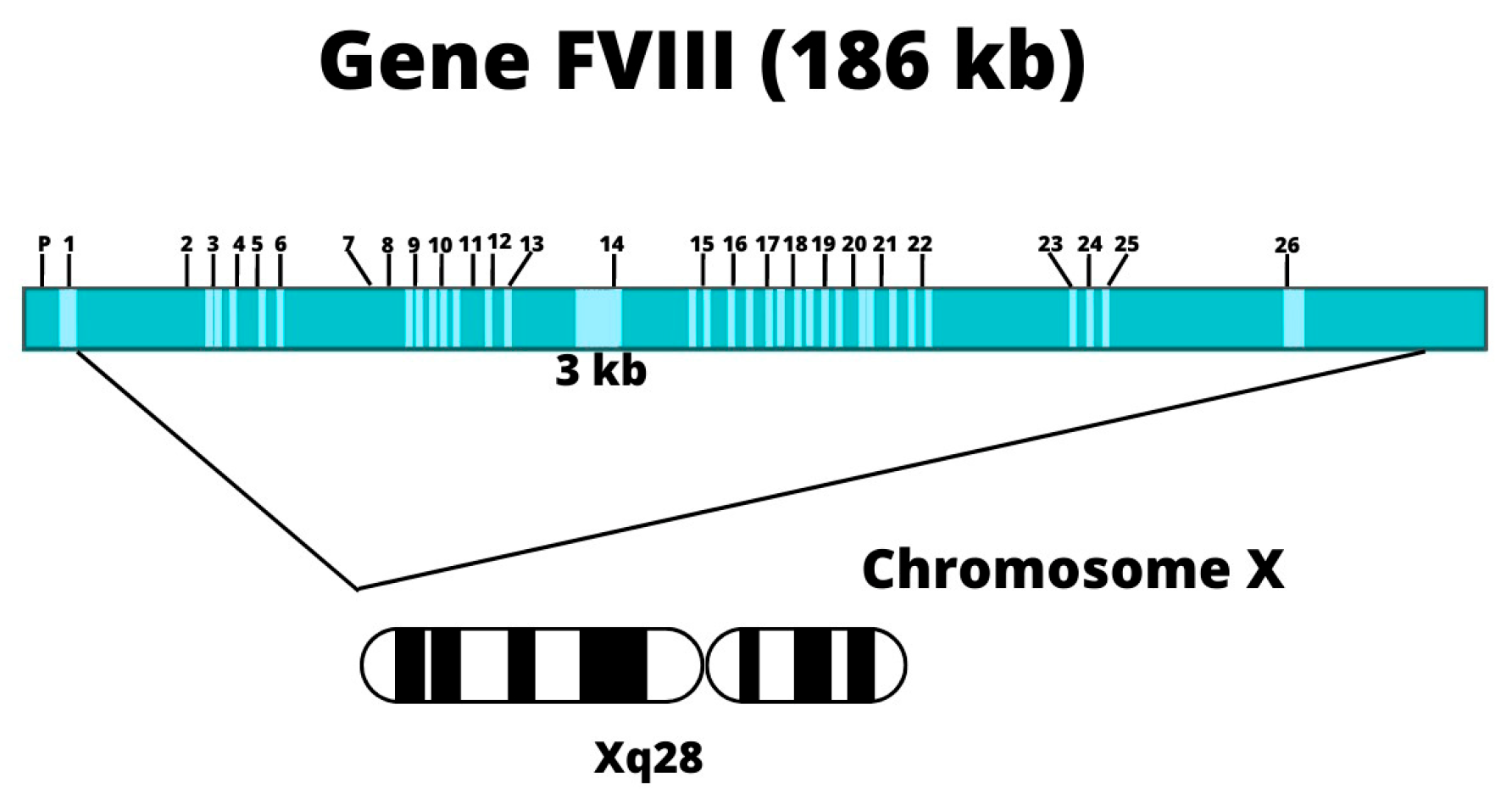
3.2. Genetic Etiology
3.3. Epidemiology
3.4. Clinical Features
- ▪
- Mild deficiency (5–40% FVIII activity): It usually only presents with bleeding after surgical procedures.
- ▪
- Moderate deficiency (1 to 5% FVIII activity).
- ▪
Bleeding in FVIII Deficiency
- Hemarthroses (70–80% incidence): the most common events occur in the knee, ankle, and elbow joints, and less frequently in the shoulders, wrists, and hips.
- Muscles: there is an incidence of 70–80%.
- Other important haemorrhages occur with an incidence of 5–10%.
- Central nervous system: there is an incidence of <5%. It is the main cause of mortality in severe haemophilia patients, but, fortunately, its incidence has been declining with the widespread introduction of prophylaxis. All head injuries that are accompanied by headache, drowsiness, or vomiting should be considered as possible intracranial bleeding and treated immediately. If the severe pain is at the level of the back, it may be a symptom of bleeding at the level of the spinal cord.
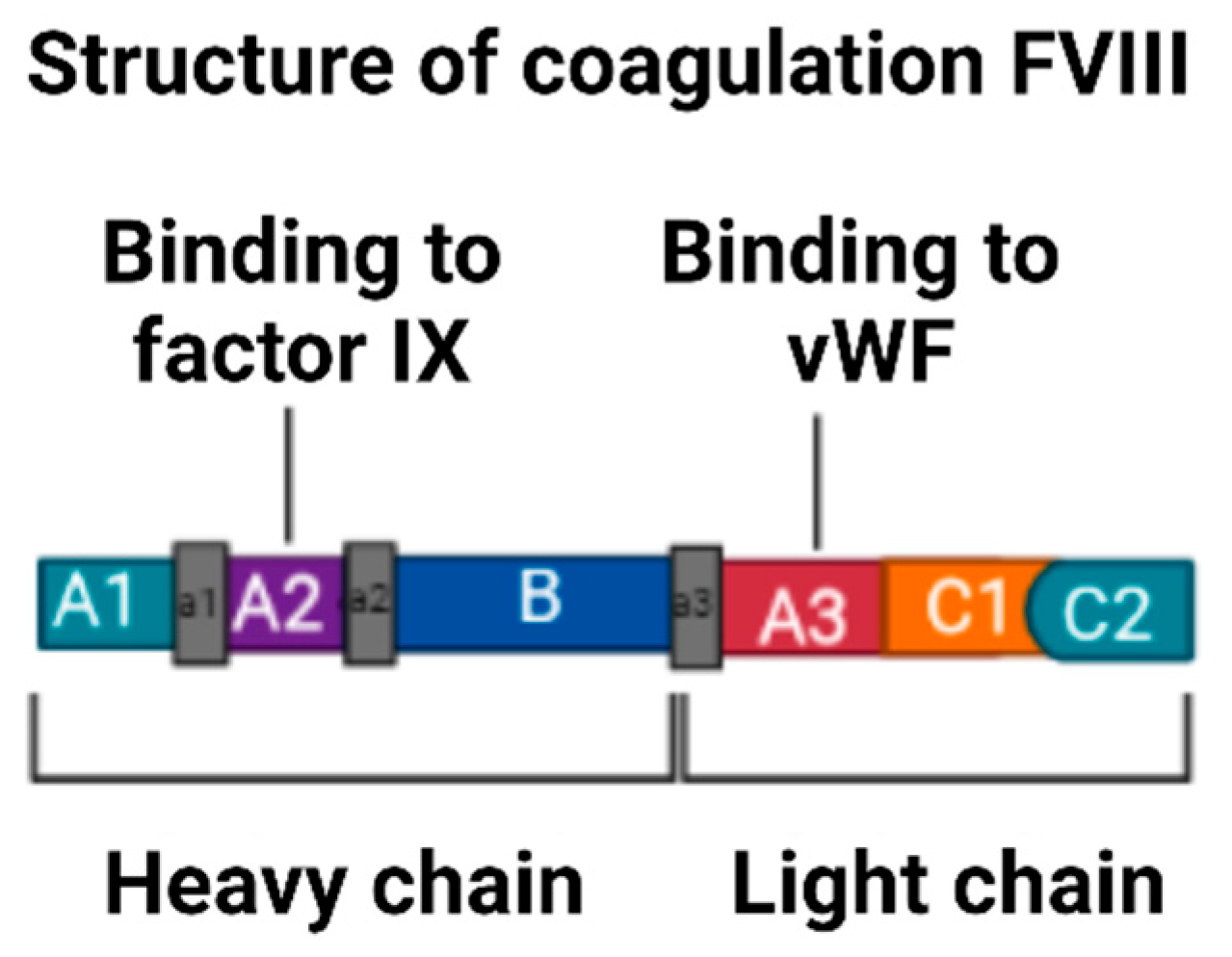
3.5. Description of Factor VIII
3.6. Inhibitors in Haemophilia
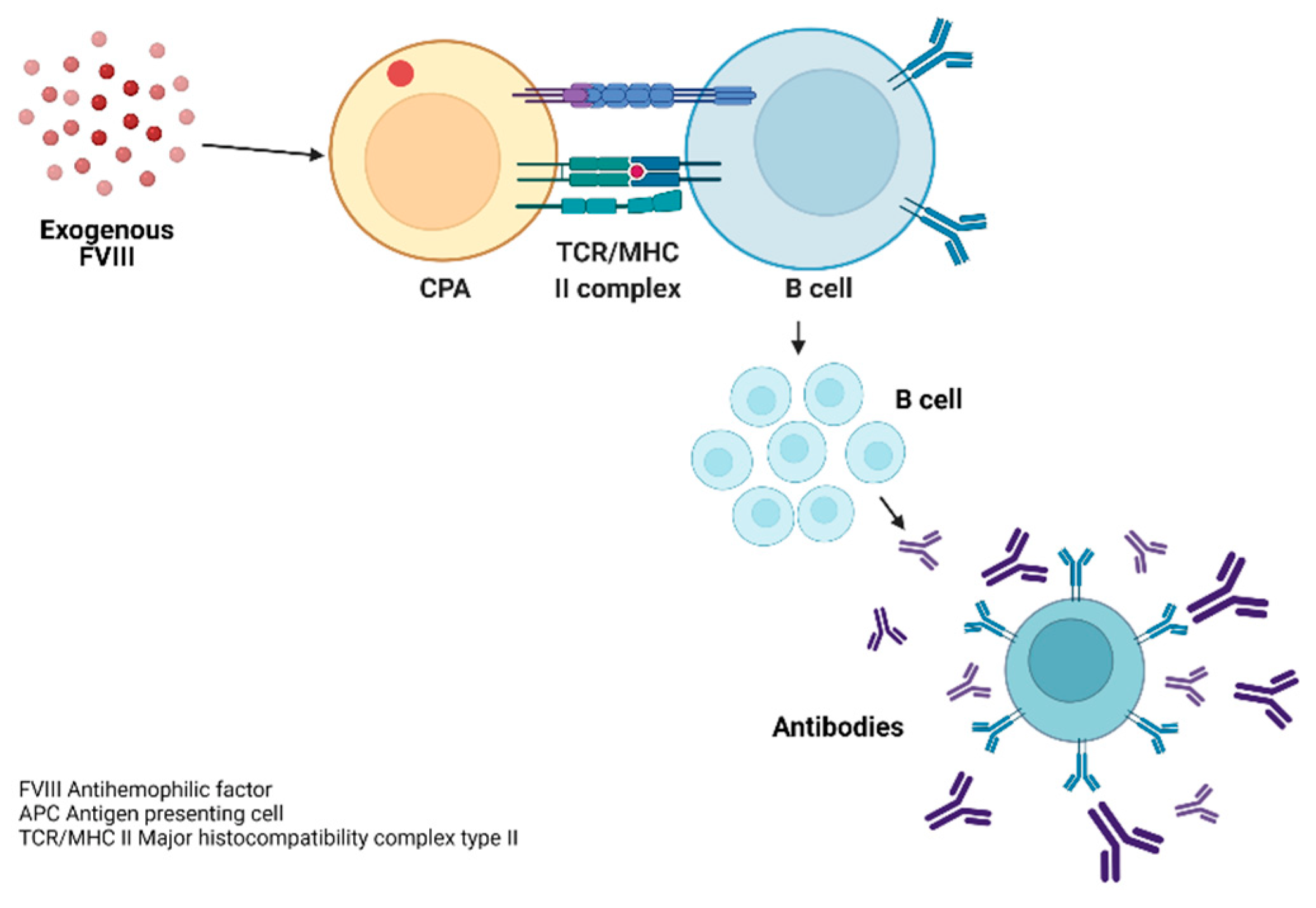
3.7. Pathophysiology of the Inmune Response to FVIII
3.8. Development of Inhibitors in Haemophilia A
- ▪
- Negative inhibitor titre: The titre is below the limit of detection of inhibitors in the local laboratory. If the reference value of the local laboratory is not available, any titre <0.6 Bethesda unit (BU) will be considered negative [27].
- ▪
- A low titre of inhibitor: Any titre that is between the limit of detection of inhibitors of the local laboratory and ≤5 BU will be considered a low-titre inhibitor. If the local laboratory reference value is not available, any titre ≥0.6 BU will be considered a low-titre inhibitor [27].
- ▪
- A high titre of inhibitor: Any titre ≥5 BU at any time after diagnosis will be considered a high-titre inhibitor [27].
3.8.1. Incidence of Inhibitors in a Previously Treated Patient (PTP) in Haemophilia
3.8.2. Incidence of Inhibitors in Treatment-Naive Patients (PUP)
3.9. Risk Factors Associated with the Development of Inhibitors
3.9.1. Immune Response in Haemophilia
3.9.2. Major Histocompatibility Complex in Haemophilia
3.9.3. TNF-α
3.9.4. Race and Ethnicity
3.10. Mutation Responsible for Haemophilia A
- Large deletions or insertions: These are those in which more than 50 bp [52] is added or lost, which alters the reading frame.
- Small deletions or insertions: These are defined by the loss of less than 50 base pairs (bp), generating changes in the reading (frameshift) and generating a premature stop codon [52]. This class of mutations is caused by polymerase slippage during the DNA replication phase [30]. Most patients with severe haemophilia A have large deletions and insertions [30].
- Mutations of amino acid change or missense:
- This produces a stop codon, where a termination codon is produced (TAA, TAG, TGA). It is responsible for a severe phenotype [22].
3.10.1. Background in the Literature on the Relationship between FVIII Mutations and Inhibitor Development
3.10.2. History of Variants or Mutations in Colombia
3.11. Laboratory Diagnostic Test
3.11.1. Methods That Measure the Functional Activity of FVIII
3.11.2. Chromogenic Method (Ccro) of FVIII
3.11.3. Coagulometric or One-Stage Assay
3.11.4. Quantitative Measurement of Factor VIII Inhibitors
3.12. Treatment
- ▪
- Primary: This begins before the second joint bleed, without the presence of joint damage, and prophylaxis treatment started before the age of three.
- ▪
- Secondary: This begins after two or more joint bleeds, but before joint damage is established.
- ▪
- Tertiary: This begins after joint damage has been confirmed by diagnostic means.
3.12.1. Pharmacokinetics (PK)
3.12.2. Population Pharmacokinetics
3.12.3. Types of FVIII Products Used in Haemophilia A
Plasma-Derived Factor
Recombinant Factor VIII Concentrates
3.12.4. Emicizumab
3.12.5. Treatment of Patients with Inhibitors
Immune Tolerance Regimens
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Initials | Definition |
| aa | Amino acid |
| ADN | Deoxyribonucleic acid |
| ARN | Ribonucleic acid |
| EDTA | Ethylenediaminetetraacetic acid |
| FVIII | Factor VIII |
| FIX | Factor IX |
| FvW | Von Willebrand factor |
| HAMSTeRS | The Haemophilia A Mutation, Structure, Test and Resource Site |
| INV1 | Intron-1 inversion |
| INV22 | Intron-22 inversion |
| ITI | Immunetolerance |
| Kb | Kilobase |
| Pb | Base pair |
| PCR | Polymerase chain reaction |
| BU | Bethesda units |
References
- Gray, C.R. On the coagulation of the blood: An elaboration of Lord Lister’s hypothesis and the four-factor model of Morawitz. Med. Hypotheses 1990, 33, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Alvarado Arteaga, I.M. Fisiología de la coagulación: Nuevos conceptos aplicados al cuidado perioperatorio. Univ. Médica 2013, 54, 338–352. [Google Scholar] [CrossRef]
- Available online: http://www.sah.org.ar/revista/numeros/vol21/extra/08-Vol%2021-extra.pdf (accessed on 10 November 2021).
- Mann, K.G.; Nesheim, M.E.; Church, W.R.; Haley, P.; Krishnaswamy, S. Surface-dependent reactions of the vitamin K-dependent enzyme complexes. Blood 1990, 76, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Iorio, A.; Halimeh, S.; Holzhauer, S.; Goldenberg, N.; Marchesini, E.; Marcucci, M.; Young, G.; Bidlingmaier, C.; Brandao, L.R.; Ettingshausen, C.E.; et al. Rate of inhibitor development in previously untreated hemophilia A patients treated with plasma-derived or recombinant factor VIII concentrates: A systematic review. J. Thromb. Haemost. 2010, 8, 1256–1265. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.; Monroe, D.M. A cell-based model of hemostasis. Thromb. Haemost. 2001, 85, 958–965. [Google Scholar] [PubMed]
- Hoffman, M.; Monroe, D.M. Coagulation 2006: A modern view of hemostasis. Hematol. Oncol. Clin. N. Am. 2007, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gouw, S.C.; van den Berg, H.M.; le Cessie, S.; van der Bom, J.G. Treatment characteristics and the risk of inhibitor development: A multicenter cohort study among previously untreated patients with severe hemophilia A. J. Thromb. Haemost. 2007, 5, 1383–1390. [Google Scholar] [CrossRef]
- Available online: https://www.minsalud.gov.co/sites/rid/Lists/BibliotecaDigital/RIDE/DE/CA/Protocolo-hemofilia-marzo-2015.pdf (accessed on 26 April 2021).
- Bolton-Maggs, P.H.B.; Pasi, K.J. Haemophilias A and B. Lancet 2003, 361, 1801–1819. [Google Scholar] [CrossRef]
- Srivastava, A.; Brewer, A.K.; Mauser-Bunschoten, E.P.; Key, N.S.; Kitchen, S.; Llinas, A.; Ludlam, C.A.; Mahlangu, J.N.; Mulder, K.; Poon, M.C.; et al. Guidelines for the management of hemophilia. Haemophilia 2013, 19, e1–e47. [Google Scholar] [CrossRef]
- Castro, H.E.; Briceño, M.F.; Casas, C.P.; Rueda, J.D. The history and evolution of the clinical effectiveness of haemophilia type a treatment: A systematic review. Indian J. Hematol. Blood Transfus. 2014, 30, 1–11. [Google Scholar] [CrossRef]
- Available online: http://www1.wfh.org/publications/files/pdf-1532.pdf (accessed on 5 October 2021).
- Tantawy, A.A.G. Molecular genetics of hemophilia A: Clinical perspectives. Egypt. J. Med. Hum. Genet. 2010, 11, 105–114. [Google Scholar] [CrossRef]
- Situación de la Hemofilia en Colombia, 2019. Cuenta de Alto Costo. 2020. Available online: https://cuentadealtocosto.org/site/hemofilia/situacion-de-la-hemofilia-en-colombia-2019/ (accessed on 22 April 2021).
- Spena, S.; Garagiola, I.; Cannavò, A.; Mortarino, M.; Mannucci, P.M.; Rosendaal, F.R.; Peyvandi, F.; Sippet Study Group. Prediction of factor VIII inhibitor development in the SIPPET cohort by mutational analysis and factor VIII antigen measurement. J. Thromb. Haemost. 2018, 16, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.W. Personalized prophylaxis. Haemophilia 2012, 18 (Suppl. 4), 131–135. [Google Scholar] [CrossRef] [PubMed]
- Importance of Pharmacokinetics in the Management of Hemophilia—Barnes—2013—Pediatric Blood & Cancer—Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/10.1002/pbc.24339 (accessed on 27 October 2021).
- Fuenmayor Castaño, A.; Jaramillo Restrepo, M.; Salinas Durán, F. Quality of life in a population with haemophilia: A cross-sectional study from a single haemophilia treatment center. Rev. Colomb. Reumatol. Engl. Ed. 2017, 24, 18–24. [Google Scholar] [CrossRef]
- Martínez-Sánchez, L.M.; Álvarez-Hernández, L.F.; Ruiz-Mejía, C.; Jaramillo-Jaramillo, L.I.; Builes-Restrepo, L.N.; Villegas-Álzate, J.D. Hemofilia: Abordaje diagnóstico y terapéutico. Rev. Fac. Nac. Salud Pública 2018, 36, 85–93. [Google Scholar] [CrossRef]
- Toole, J.J.; Knopf, J.L.; Wozney, J.M.; Sultzman, L.A.; Buecker, J.L.; Pittman, D.D.; Kaufman, R.J.; Brown, E.; Shoemaker, C.; Orr, E.C.; et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984, 312, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Bogdanova, N.; Markoff, A.; Eisert, R.; Wermes, C.; Pollmann, H.; Todorova, A.; Chlystun, M.; Nowak-Göttl, U.; Horst, J. Spectrum of molecular defects mutation detection rate in patients with severe hemophilia, A. Hum. Mutat. 2005, 26, 249–254. [Google Scholar] [CrossRef]
- Bases Moleculares de la Hemofilia A. Available online: http://www.hemobase.com/molecular_hemofilia/print/Hemofilia_A.htm (accessed on 16 September 2021).
- Castillo-González, D.D. Hemofilia II. Aspectos moleculares y de genética poblacional. Rev. Cuba. Hematol. Inmunol. Hemoter. 2012, 28, 111–119. [Google Scholar]
- Hernández-Carvajal, E.; Arce-Solano, S.; Mena-Aguilar, D.; Fuentes-Prior, P. Producción heteróloga y caracterización bioquímica del procoagulante humano Factor VIII para ensayos de cristalización de macromoléculas proteicas. Rev. Tecnol. Marcha 2016, 29, 78–91. [Google Scholar] [CrossRef]
- Available online: http://cidbimena.desastres.hn/RFCM/pdf/2010/pdf/RFCMVol7-2-2010-6.pdf (accessed on 22 October 2021).
- Carcao, M.; Goudemand, J. Los Inhibidores en la Hemofilia: Información Básica; Federación Mundial de Hemofilia: Montreal, QC, Canada, 2018; p. 24. [Google Scholar]
- Fondo Colombiano de Enfermedades de Alto Costo. Situacion de la Hemofilia en Colombia. 2019. Available online: https://cuentadealtocosto.org/site/wp-content/uploads/2020/02/cac-co_2020_1_30_libro_sit_hemofilia_2019.pdf (accessed on 9 August 2021).
- Hausl, C.; Ahmad, R.U.; Sasgary, M.; Doering, C.B.; Lollar, P.; Richter, G.; Schwarz, H.P.; Turecek, P.L.; Reipert, B.M. High-dose factor VIII inhibits factor VIII-specific memory B cells in hemophilia A with factor VIII inhibitors. Blood 2005, 106, 3415–3422. [Google Scholar] [CrossRef]
- Oldenburg, J.; El-Maarri, O.; Schwaab, R. Inhibitor development in correlation to factor VIII genotypes. Haemophilia 2002, 8 (Suppl. 2), 23–29. [Google Scholar] [CrossRef] [PubMed]
- Wight, J.; Paisley, S. The epidemiology of inhibitors in haemophilia A: A systematic review. Haemophilia 2003, 9, 418–435. [Google Scholar] [CrossRef] [PubMed]
- Astermark, J.; Oldenburg, J.; Carlson, J.; Pavlova, A.; Kavakli, K.; Berntorp, E.; Lefvert, A.K. Polymorphisms in the TNFA gene the risk of inhibitor development in patients with hemophilia, A. Blood 2006, 108, 3739–3745. [Google Scholar] [CrossRef] [PubMed]
- Frommel, D.; Allain, J.P. Genetic predisposition to develop factor VIII antibody in classic hemophilia. Clin. Immunol. Immunopathol. 1977, 8, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Lassila, R.; Peyvandi, F.; Calizzani, G.; Gatt, A.; Lambert, T.; Windyga, J.; Iorio, A.; Gilman, E.; Makris, M.; et al. Inhibitor development in haemophilia according to concentrate: Four-year results from the European Haemophilia Safety Surveillance (EUHASS) project. Thromb. Haemost. 2015, 113, 968–975. [Google Scholar]
- Astermark, J.; Berntorp, E.; White, G.C.; Kroner, B.L.; MIBS Study Group. The Malmö International Brother Study (MIBS): Further support for genetic predisposition to inhibitor development in hemophilia patients. Haemophilia 2001, 7, 267–272. [Google Scholar] [CrossRef]
- Santagostino, E.; Mancuso, M.E.; Rocino, A.; Mancuso, G.; Mazzucconi, M.G.; Tagliaferri, A.; Messina, M.; Mannucci, P.M. Environmental risk factors for inhibitor development in children with haemophilia A: A case–control study. Br. J. Haematol. 2005, 130, 422–427. [Google Scholar] [CrossRef]
- A Randomized Trial of Factor VIII and Neutralizing Antibodies in Hemophilia A|NEJM. Available online: https://www.nejm.org/doi/full/10.1056/nejmoa1516437 (accessed on 7 September 2021).
- Gouw, S.C.; van den Berg, H.M.; Oldenburg, J.; Astermark, J.; Groot, P.G.; Margaglione, M.; Thompson, A.R.; Van Heerde, W.; Boekhorst, J.; Miller, C.H.; et al. F8 gene mutation type and inhibitor development in patients with severe hemophilia A: Systematic review and meta-analysis. Blood 2012, 119, 2922–2934. [Google Scholar] [CrossRef]
- Oldenburg, J.; Pavlova, A. Genetic risk factors for inhibitors to factors VIII and IX. Haemophilia 2006, 12 (Suppl. 6), 15–22. [Google Scholar] [CrossRef]
- Oldenburg, J.; Picard, J.K.; Schwaab, R.; Brackmann, H.H.; Tuddenham, E.G.; Simpson, E. HLA genotype of patients with severe haemophilia A due to intron 22 inversion with without inhibitors of factor VIII. Thromb. Haemost. 1997, 77, 238–242. [Google Scholar] [CrossRef]
- Hay, C.R.; Ollier, W.; Pepper, L.; Cumming, A.; Keeney, S.; Goodeve, A.C.; Colvin, B.T.; Hill, F.G.; Preston, F.E.; Peake, I.R. HLA class II profile: A weak determinant of factor VIII inhibitor development in severe haemophilia A. UKHCDO Inhibitor Working Party. Thromb. Haemost. 1997, 77, 234–237. [Google Scholar] [PubMed]
- Kruse-Jarres, R.; Pajewski, N.M.; Leissinger, C.A. The role of race and ethnicity in the clinical outcomes of severe hemophilia a patients with inhibitors. Blood 2007, 110, 1163. [Google Scholar] [CrossRef]
- Rondón González, F.; Guillermo, B. Estructura genética, ancestralidad y su relación con los estudios en salud humana. Medicas UIS 2013, 26, 37–43. [Google Scholar]
- Carpenter, S.L.; Soucie, J.M.; Sterner, S.; Presley, R. Increased prevalence of inhibitors in Hispanic patients with severe haemophilia A enrolled in the Universal Data Collection database. Haemophilia 2012, 18, e260–e265. [Google Scholar] [CrossRef]
- Rossiter, J.P.; Young, M.; Kimberland, M.L.; Hutter, P.; Ketterling, R.P.; Gitschier, J.; Horst, J.; Morris, M.A.; Schaid, D.J.; De Moerloose, P.; et al. Factor VIII gene inversions causing severe hemophilia A originate almost exclusively in male germ cells. Hum. Mol. Genet. 1994, 3, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Yunis, L.K.; Linares, A.; Cabrera, E.; Yunis, J.J. Systematic molecular analysis of hemophilia A patients from Colombia. Genet. Mol. Biol. 2018, 41, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Carcao, M.D.; van den Berg, H.M.; Ljung, R.; Mancuso, M.E. Correlation between phenotype genotype in a large unselected cohort of children with severe hemophilia, A. Blood 2013, 121, 3946–3952. [Google Scholar] [CrossRef]
- Andrikovics, H.; Klein, I.; Bors, A.; Nemes, L.; Marosi, A.; Váradi, A.; Tordai, A. Analysis of large structural changes of the factor VIII gene involving intron, 1.; 22; in severe hemophilia, A. Haematologica 2003, 88, 778–784. [Google Scholar]
- Garcés, M.F.; Linares, A.; Sarmiento, I.C.; Caminos, J.E. Estudio molecular de la inversión de los intrones 1 y 22 del factor VIII de la coagulación en niños con hemofilia A severa utilizando técnica de PCR de larga distancia. Rev. Fac. Med. J. 2017, 65, 245–251. [Google Scholar] [CrossRef]
- Antonarakis, S.E.; Rossiter, J.P.; Young, M.; Horst, J.; de Moerloose, P.; Sommer, S.S.; Ketterling, R.P.; Kazazian, H.H., Jr.; Négrier, C.; Vinciguerra, C.; et al.; et al. Factor VIII gene inversions in severe hemophilia A: Results of an international consortium study. Blood 1995, 86, 2206–2212. [Google Scholar] [CrossRef] [PubMed]
- Bagnall, R.D.; Waseem, N.; Green, P.M.; Giannelli, F. Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A. Blood 2002, 99, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Nuttall, G.A. Hemostasis and Thrombosis: Basic Principles and Clinical Practice, 5th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; Volume 104, p. 1317. [Google Scholar]
- Nogami, K.; Zhou, Q.; Wakabayashi, H.; Fay, P.J. Thrombin-catalyzed activation of factor VIII with His substituted for Arg372 at the P1 site. Blood 2005, 105, 4362–4368. [Google Scholar] [CrossRef]
- Shima, M.; Ware, J.; Yoshioka, A.; Fukui, H.; Fulcher, C.A. An arginine to cysteine amino acid substitution at a critical thrombin cleavage site in a dysfunctional factor VIII molecule. Blood 1989, 74, 1612–1617. [Google Scholar] [CrossRef]
- Jacquemin, M.; Lavend’homme, R.; Benhida, A.; Vanzieleghem, B.; d’Oiron, R.; Lavergne, J.M.; Brackmann, H.H.; Schwaab, R.; VandenDriessche, T.; Chuah, M.K.; et al. A novel cause of mild/moderate hemophilia A: Mutations scattered in the factor VIII C1 domain reduce factor VIII binding to von Willebrand factor. Blood 2000, 96, 958–965. [Google Scholar] [CrossRef]
- Mantilla-Capacho, J.M.; Beltrán-Miranda, C.P.; Luna-Záizar, H.; Aguilar-López, L.; Esparza-Flores, M.A.; López-Guido, B.; Troyo-Sanromán, R.; Jaloma-Cruz, A.R. Frequency of intron, 1.; 22 inversions of Factor VIII gene in Mexican patients with severe hemophilia, A. Am. J. Hematol. 2007, 82, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Hu, S.H.; Cheng, S.N.; Chao, T.Y. Genetic analysis of haemophilia A in Taiwan. Haemophilia 2010, 16, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.K.; Laursen, A.L.; Poulsen, L.H.; Mogensen, T.H. Identification of a novel mutation in the factor VIII gene causing severe haemophilia A. BMC Hematol. 2018, 18, 17. [Google Scholar] [CrossRef]
- Albánez, S.; Ruiz-Sáez, A.; Boadas, A.; de Bosch, N.; Porco, A. Identification of factor VIII gene mutations in patients with severe haemophilia A in Venezuela: Identification of seven novel mutations. Haemophilia 2011, 17, e913–e918. [Google Scholar] [CrossRef]
- Miller, C.H.; Benson, J.; Ellingsen, D.; Driggers, J.; Payne, A.; Kelly, F.M.; Soucie, J.M.; Craig Hooper, W. F8 and F9 mutations in US haemophilia patients: Correlation with history of inhibitor and race/ethnicity. Haemophilia 2012, 18, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Murillo, C.I.I.I. Propuesta de recomendaciones para el diagnóstico y tratamiento en hemofilia. Gac. Médica México 2004, 140, 7. [Google Scholar]
- Sueldo, E.; Duboscq, C.; Arias, M. Validación del ensayo FVIII:C cromogénico en una plataforma automatizada. Acta Bioquímica Clínica Latinoam. 2020, 54, 135–143. [Google Scholar]
- Kitchen, S.; McCraw, A.; Echenagucia, M. Manual de Laboratorio. World Federation of Hemophilia. 2010. Available online: https://www1.wfh.org/publication/files/pdf-1283.pdf (accessed on 26 October 2021).
- Available online: http://www1.wfh.org/publication/files/pdf-1179.pdf (accessed on 26 October 2021).
- Available online: https://www.anmm.org.mx/GMM/2013/n3/GMM_149_2013_3_308-321.pdf (accessed on 12 October 2021).
- Van den Berg, H.M.; Fischer, K.; Mauser-Bunschoten, E.P.; Beek, F.J.; Roosendaal, G.; van der Bom, J.G.; Nieuwenhuis, H.K. Long-term outcome of individualized prophylactic treatment of children with severe haemophilia. Br. J. Haematol. 2001, 112, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Feldman, B.M.; Pai, M.; Rivard, G.E.; Israels, S.; Poon, M.C.; Demers, C.; Robinson, S.; Luke, K.H.; Wu, J.K.; Gill, K.; et al. Tailored prophylaxis in severe hemophilia A: Interim results from the first 5 years of the Canadian Hemophilia Primary Prophylaxis Study. J. Thromb. Haemost. 2006, 4, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Dargaud, Y.; Delavenne, X.; Hart, D.P.; Meunier, S.; Mismetti, P. Individualized PK-based prophylaxis in severe haemophilia. Haemophilia 2018, 24 (Suppl. 2), 3–17. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.H.; Whiting, B. Bayesian parameter estimation and population pharmacokinetics. Clin. Pharmacokinet. 1992, 22, 447–467. [Google Scholar] [CrossRef]
- Proyecto Wapps-Hemo: La Tecnología al Servicio del Paciente. Available online: http://www.eightfactor.com/es/proyecto-wapps-hemo-la-tecnologia-al-servicio-del-paciente/ (accessed on 20 October 2021).
- Björkman, S. Limited blood sampling for pharmacokinetic dose tailoring of FVIII in the prophylactic treatment of haemophilia A. Haemophilia 2010, 16, 597–605. [Google Scholar] [CrossRef]
- Carlsson, M.; Berntorp, E.; Björkman, S.; Lethagen, S.; Ljung, R. Improved cost-effectiveness by pharmacokinetic dosing of factor VIII in prophylactic treatment of haemophilia A. Haemophilia 1997, 3, 96–101. [Google Scholar] [CrossRef]
- Hemophilia, Historia de 40 Anys. Available online: https://www.hemofilia.cat/castellano/hemofilia/tractaments.html (accessed on 25 August 2022).
- History. National Hemophilia Foundation. Available online: https://www.hemophilia.org/bleeding-disorders-a-z/overview/history (accessed on 17 August 2022).
- CDC. Treatment of Hemophilia|CDC. Centers for Disease Control and Prevention. Available online: https://www.cdc.gov/ncbddd/hemophilia/treatment.html (accessed on 17 August 2022).
- Teitel, J. Los Agentes Transmisibles y la Seguridad de los Concentrados de Factores de la Coagulación. Serie Hechos y Cifras, número 4; Federacion Mundial de la Hemofilia. 1997. Available online: http://hemofiliagipuzkoa.org/upload/documentacion/hematologia/Agentes%20transmisibles%20y%20la%20seguridad%20de%20los%20concentrados.pdf (accessed on 25 August 2022).
- Available online: https://www.tdx.cat/bitstream/handle/10803/650828/rrr1de1.pdf?sequence=1 (accessed on 25 August 2022).
- Powell, J.S.; Josephson, N.C.; Quon, D.; Ragni, M.V.; Cheng, G.; Li, E.; Jiang, H.; Li, L.; Dumont, J.A.; Goyal, J.; et al. Safety and prolonged activity of recombinant factor VIII Fc fusion protein in hemophilia A patients. Blood 2012, 119, 3031–3037. [Google Scholar] [CrossRef]
- ASVAPAHE. Recomendaciones para el Tratamiento de la Hemofilia con INHIBIDOR—ASVAPAHE: Hemofilia Valladolid y Palencia. Available online: https://www.hemofiliavalladolidpalencia.org/2020/05/02/recomendaciones-para-el-tratamiento-de-la-hemofilia-con-inhibidor/ (accessed on 20 October 2021).
- Arbesú, G. Emicizumab: Nuevo tratamiento para personas con hemofilia A con y sin inhibidor. Rev. Hematol. 2019, 23, 81–90. [Google Scholar]
- Mancuso, M.E.; Callaghan, M.U.; Kruse-Jarres, R.; Kempton, C.L.; Xu, J.; Catalani, O.; Asikanius, E.; Levy, G.G.; Shima, M.; Young, G.; et al. Emicizumab prophylaxis in adolescent/adult patients with hemophilia A previously receiving episodic or prophylactic bypassing agent treatment: Updated analyses from the HAVEN 1 study. Blood 2017, 130 (Suppl. 1), 1071. [Google Scholar]
- Sampei, Z.; Igawa, T.; Soeda, T.; Okuyama-Nishida, Y.; Moriyama, C.; Wakabayashi, T.; Tanaka, E.; Muto, A.; Kojima, T.; Kitazawa, T.; et al. Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PLoS ONE 2013, 8, e57479. [Google Scholar] [CrossRef]
- Mahlangu, J.; Oldenburg, J.; Paz-Priel, I.; Negrier, C.; Niggli, M.; Mancuso, M.E.; Schmitt, C.; Jiménez-Yuste, V.; Kempton, C.; Dhalluin, C.; et al. Emicizumab prophylaxis in patients who have hemophilia A without inhibitors. N. Engl. J. Med. 2018, 379, 811–822. [Google Scholar] [CrossRef]
- Oldenburg, J.; Mahlangu, J.N.; Kim, B.; Schmitt, C.; Callaghan, M.U.; Young, G.; Santagostino, E.; Kruse-Jarres, R.; Negrier, C.; Kessler, C.; et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N. Engl. J. Med. 2017, 377, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Kruse-Jarres, R.; Callaghan, M.U.; Croteau, S.E.; Jimenez-Yuste, V.; Khoo, L.; Liesner, R.; Matshushida, T.; Recht, M.; Young, G.; Chag, T.; et al. Surgical experience in two multicenter, open-label phase 3 studies of emicizumab in persons with hemophilia A with inhibitors (HAVEN 1 and HAVEN 2). Blood 2017, 130, 89. [Google Scholar] [CrossRef]
- Young, G.; Liesner, R.; Sidonio, R.F., Jr.; Oldenburg, J.; Jimenez-Yuste, V.; Mahlangu, J.; Kruse-Jarres, R.; Wang, M.; Chang, T. Emicizumab prophylaxis provides flexible and effective bleed control in children with hemophilia A with inhibitors: Results from the HAVEN 2 study. Blood 2018, 132 (Suppl. 1), 632. [Google Scholar] [CrossRef]
- Tjønnfjord, G.E.; Andre Holme, P. Factor Eight Inhibitor Bypass Activity (FEIBA) in the management of bleeds in hemophilia patients with high-titer inhibitors. Vasc. Health Risk Manag. 2007, 3, 527–531. [Google Scholar]
- Dimichele, D. Inhibitors: Resolving diagnostic and therapeutic dilemmas. Haemophilia 2002, 8, 280–287. [Google Scholar] [CrossRef]
- Oldenburg, J.; Schwaab, R.; Brackmann, H.H. Induction of immune tolerance in haemophilia A inhibitor patients by the «Bonn Protocol»: Predictive parameter for therapy duration and outcome. Vox Sang. 1999, 77 (Suppl. 1), 49–54. [Google Scholar] [CrossRef]
- Mauser-Bunschoten, E.P.; Roosendaal, G.; van den Berg, H.M. Low-dose immune tolerance therapy: The van creveld model. Vox Sang. 1996, 70 (Suppl. 1), 66–67. [Google Scholar] [CrossRef]
- Freiburghaus, C.; Berntorp, E.; Ekman, M.; Gunnarsson, M.; Kjellberg, B.; Nilsson, I.M. Tolerance induction using the Malmö treatment model 1982–1995. Haemophilia 1999, 5, 32–39. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Risk Factors | Summary | Support Level | ||
|---|---|---|---|---|
| Non-Modifiable Genetic Risk Factors | ||||
| Type of FVIII mutation (null vs. non-null) and the position of the mutation | Type of mutation | Risk of inhibitor formation | Well established | |
| Null | Multidomain deletions | 75% | ||
| Light-chain missense mutations | 30–40% | |||
| Intron-22 inversion | 20–25% | |||
| Domain deletions unique | 15–25% | |||
| Small insertions/deletions in non-A zone | 15–20% | |||
| Heavy-chain missense mutations | 10–20% | |||
| Not null | FVIII missense mutations | <10% | ||
| Small insertions/deletions in zone A | <5% | |||
| Mutations at the splice site | <5% | |||
| Family history | Risk 3.2 times higher (95% CI 2.1–4.9) if there was a family member with inhibitors | Well established | ||
| Ethnicity | 1.9- to 4.7-fold increased risk in non-Caucasians (Black African > Latin American > Caucasian ancestry) | Established, but not well understood | ||
| TNF-α IL-10 CTLA-4 polymorphisms | TNF-α −308 A/A increases risk IL-10: allele 134 increases the risk CTLA-4: T-allele decreases risk | Some evidence, but not well understood | ||
| FVIII haplotypes | Haplotypes H3 or H4 have a higher risk of inhibitors since current FVIII products consist mainly of haplotypes H1 and H2 | Reports discordant | ||
| Class I/II MHC genes or HLA polymorphisms | 2-fold increased risk for HLADR15 and HLA-DQ6 and inhibitor formation | Reports discordant | ||
| Potentially Modifiable Environmental Risk Factors | ||||
| Trauma/surgeries | Major surgeries and trauma leading to treatment spikes increase the risk of inhibitor development | Established, but not well understood | ||
| Inflammation/infection | Could increase the formation of inhibitors | Established, but not well understood | ||
| Intense exposure, particularly at a young age | Increased risk of inhibitor formation | Established, but not well understood | ||
| Type of factor concentrate | Some studies suggest that conventional recombinant factor carries a higher risk of inhibitor formation than plasma-derived factor | Reports discordant | ||
| Early initiation of prophylaxis | Could confer true protection | No concrete evidence | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarmiento Doncel, S.; Díaz Mosquera, G.A.; Cortes, J.M.; Agudelo Rico, C.; Meza Cadavid, F.J.; Peláez, R.G. Haemophilia A: A Review of Clinical Manifestations, Treatment, Mutations, and the Development of Inhibitors. Hematol. Rep. 2023, 15, 130-150. https://doi.org/10.3390/hematolrep15010014
Sarmiento Doncel S, Díaz Mosquera GA, Cortes JM, Agudelo Rico C, Meza Cadavid FJ, Peláez RG. Haemophilia A: A Review of Clinical Manifestations, Treatment, Mutations, and the Development of Inhibitors. Hematology Reports. 2023; 15(1):130-150. https://doi.org/10.3390/hematolrep15010014
Chicago/Turabian StyleSarmiento Doncel, Samuel, Gina Alejandra Díaz Mosquera, Javier Mauricio Cortes, Carol Agudelo Rico, Francisco Javier Meza Cadavid, and Ronald Guillermo Peláez. 2023. "Haemophilia A: A Review of Clinical Manifestations, Treatment, Mutations, and the Development of Inhibitors" Hematology Reports 15, no. 1: 130-150. https://doi.org/10.3390/hematolrep15010014
APA StyleSarmiento Doncel, S., Díaz Mosquera, G. A., Cortes, J. M., Agudelo Rico, C., Meza Cadavid, F. J., & Peláez, R. G. (2023). Haemophilia A: A Review of Clinical Manifestations, Treatment, Mutations, and the Development of Inhibitors. Hematology Reports, 15(1), 130-150. https://doi.org/10.3390/hematolrep15010014