
The Global Antimicrobial Resistance Trends of Staphylococcus aureus and Influencing Factors

Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Phylogenetic and Comparative Genomic Analysis
2.3. Genome Annotation
2.4. Statistical Analysis and Visualization
3. Results and Discussion
3.1. Global Distribution of Collected S. aureus Strains
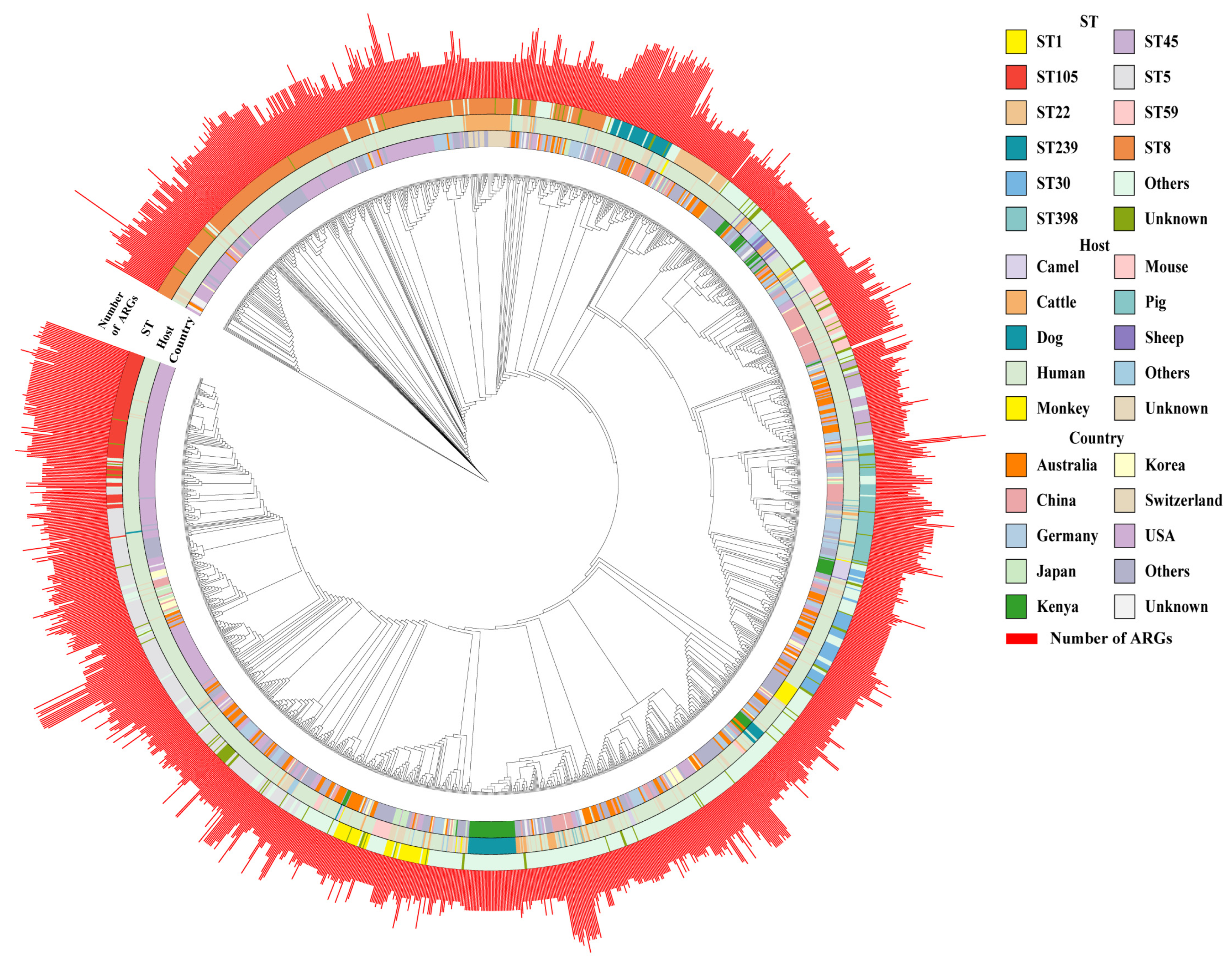
3.2. Phylogenetic Tree and Genomic Characterization of S. aureus
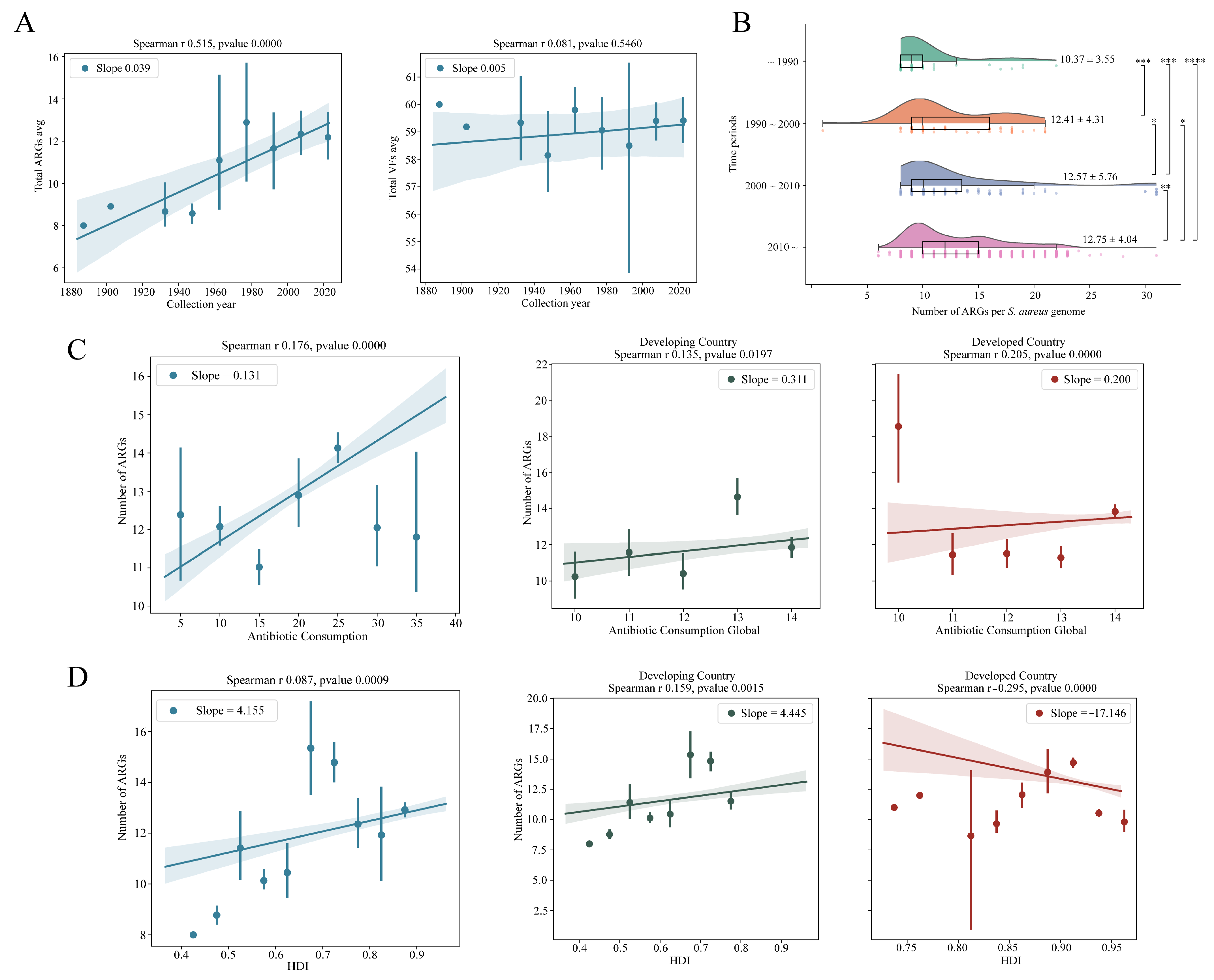
3.3. Monitoring the Antimicrobial Resistance Trends of S. aureus
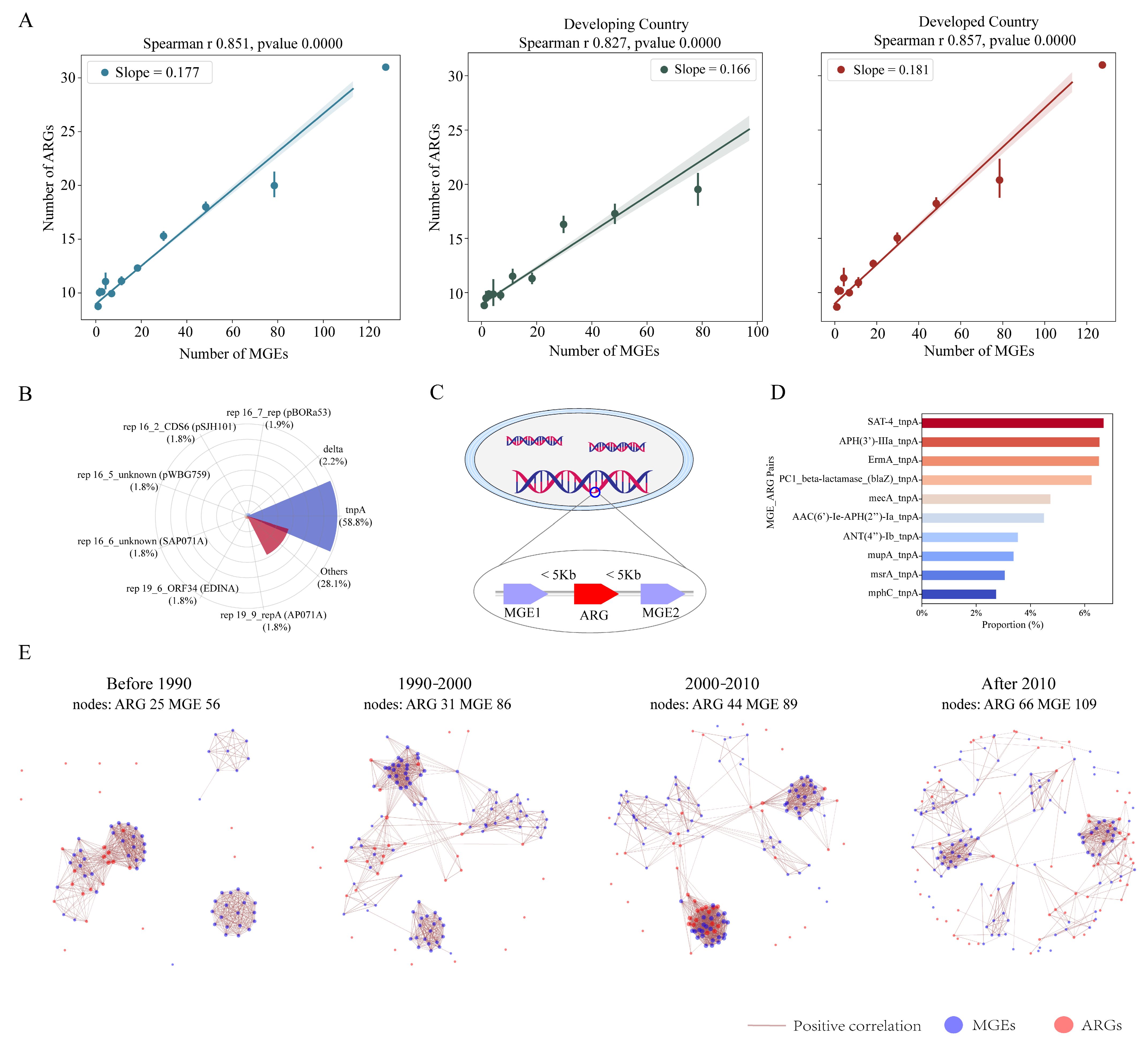
3.4. The Intrinsic Driving Effect of MGEs on ARGs
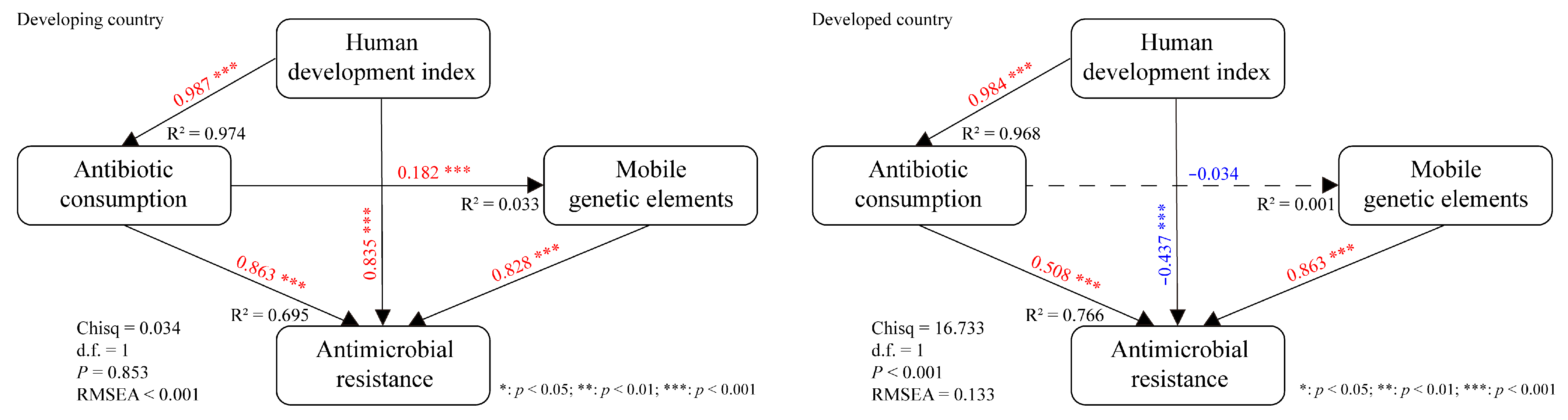
3.5. Interpreting the Mediation Effects of Various Factors on S. aureus AMR
3.6. Global Risk Assessment of S. aureus AMR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lowy, F.D. Staphylococcus aureus infections. N. Engl. J. Med. 1998, 339, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Wang, H.; Zhu, C.; Zhang, M.; Shang, F.; Xue, T. Effect of biofilm on the survival of Staphylococcus aureus isolated from raw milk in high temperature and drying environment. Food Res. Int. 2021, 149, 110672. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, S.; Lee, H.; Ha, J.; Lee, J.; Choi, Y.; Oh, H.; Yoon, Y.; Choi, K.H. icaA Gene of Staphylococcus aureus Responds to NaCl, Leading to Increased Biofilm Formation. J. Food Prot. 2018, 81, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.G.; Mills, K.B.; Crosby, H.A.; Horswill, A.R. The Staphylococcus aureus regulatory program in a human skin-like environment. mBio 2024, 15, e0045324. [Google Scholar] [CrossRef]
- Bassetti, M.; Trecarichi, E.M.; Mesini, A.; Spanu, T.; Giacobbe, D.R.; Rossi, M.; Shenone, E.; Pascale, G.D.; Molinari, M.P.; Cauda, R.; et al. Risk factors and mortality of healthcare-associated and community-acquired Staphylococcus aureus bacteraemia. Clin. Microbiol. Infect. 2012, 18, 862–869. [Google Scholar] [CrossRef]
- Kourtis, A.P.; Hatfield, K.; Baggs, J.; Mu, Y.; See, I.; Epson, E.; Nadle, J.; Kainer, M.A.; Dumyati, G.; Petit, S.; et al. Vital Signs: Epidemiology and Recent Trends in Methicillin-Resistant and in Methicillin-Susceptible Staphylococcus aureus Bloodstream Infections-United States. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 214–219. [Google Scholar] [CrossRef]
- Qin, X.; Ding, L.; Hao, M.; Li, P.; Hu, F.; Wang, M. Antimicrobial resistance of clinical bacterial isolates in China: Current status and trends. JAC Antimicrob. Resist. 2024, 6, dlae052. [Google Scholar] [CrossRef]
- von Hoven, G.; Husmann, M. Staphylococcus aureus α-Toxin’s Close Contacts Ensure the Kill. Trends Microbiol. 2019, 27, 89–90. [Google Scholar] [CrossRef]
- Zhu, Z.; Hu, Z.; Li, S.; Fang, R.; Ono, H.K.; Hu, D.L. Molecular Characteristics and Pathogenicity of Staphylococcus aureus Exotoxins. Int. J. Mol. Sci. 2023, 25, 395. [Google Scholar] [CrossRef]
- Gajdács, M. The Continuing Threat of Methicillin-Resistant Staphylococcus aureus. Antibiotics 2019, 8, 52. [Google Scholar] [CrossRef]
- Speziale, P.; Rindi, S.; Pietrocola, G. Antibody-Based Agents in the Management of Antibiotic-Resistant Staphylococcus aureus Diseases. Microorganisms 2018, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, G.; Midiri, A.; Gerace, E.; Biondo, C. Bacterial Antibiotic Resistance: The Most Critical Pathogens. Pathogens 2021, 10, 1310. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, J.A.; Holden, M.T. Staphylococcus aureus: Superbug, super genome? Trends Microbiol. 2004, 12, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Xu, J.; Wang, Y.; Shi, L.; Dong, Y.; Liu, F.; Long, J.; Duan, G.; Jin, Y.; Chen, S.; et al. The longitudinal trend and influential factors exploring of global antimicrobial resistance in Klebsiella pneumoniae. Sci. Total Environ. 2024, 950, 175357. [Google Scholar] [CrossRef]
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial Resistance in ESKAPE Pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef]
- Hutchings, M.I.; Truman, A.W.; Wilkinson, B. Antibiotics: Past, present and future. Curr. Opin. Microbiol. 2019, 51, 72–80. [Google Scholar] [CrossRef]
- Allison, S. Glutamine mitigates antimicrobial resistance. Nat. Rev. Nephrol. 2022, 18, 137. [Google Scholar] [CrossRef]
- DelaFuente, J.; Toribio-Celestino, L.; Santos-Lopez, A.; León-Sampedro, R.; Alonso-Del Valle, A.; Costas, C.; Hernández-García, M.; Cui, L.; Rodríguez-Beltrán, J.; Bikard, D.; et al. Within-patient evolution of plasmid-mediated antimicrobial resistance. Nat. Ecol. Evol. 2022, 6, 1980–1991. [Google Scholar] [CrossRef]
- Larsen, J.; Raisen, C.L.; Ba, X.; Sadgrove, N.J.; Padilla-González, G.F.; Simmonds, M.S.J.; Loncaric, I.; Kerschner, H.; Apfalter, P.; Hartl, R.; et al. Emergence of methicillin resistance predates the clinical use of antibiotics. Nature 2022, 602, 135–141. [Google Scholar] [CrossRef]
- Klein, E.Y.; Van Boeckel, T.P.; Martinez, E.M.; Pant, S.; Gandra, S.; Levin, S.A.; Goossens, H.; Laxminarayan, R. Global increase and geographic convergence in antibiotic consumption between 2000 and 2015. Proc. Natl. Acad. Sci. USA 2018, 115, E3463–E3470. [Google Scholar] [CrossRef]
- Sands, K.; Carvalho, M.J.; Portal, E.; Thomson, K.; Dyer, C.; Akpulu, C.; Andrews, R.; Ferreira, A.; Gillespie, D.; Hender, T.; et al. Characterization of antimicrobial-resistant Gram-negative bacteria that cause neonatal sepsis in seven low- and middle-income countries. Nat. Microbiol. 2021, 6, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Browne, A.J.; Chipeta, M.G.; Haines-Woodhouse, G.; Kumaran, E.P.A.; Hamadani, B.H.K.; Zaraa, S.; Henry, N.J.; Deshpande, A.; Reiner, R.C., Jr.; Day, N.P.J.; et al. Global antibiotic consumption and usage in humans, 2000–18: A spatial modelling study. Lancet Planet. Health 2021, 5, e893–e904. [Google Scholar] [CrossRef] [PubMed]
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Malhotra-Kumar, S.; Lammens, C.; Coenen, S.; Van Herck, K.; Goossens, H. Effect of azithromycin and clarithromycin therapy on pharyngeal carriage of macrolide-resistant streptococci in healthy volunteers: A randomised, double-blind, placebo-controlled study. Lancet 2007, 369, 482–490. [Google Scholar] [CrossRef]
- Hou, J.; Long, X.; Wang, X.; Li, L.; Mao, D.; Luo, Y.; Ren, H. Global trend of antimicrobial resistance in common bacterial pathogens in response to antibiotic consumption. J. Hazard. Mater. 2023, 442, 130042. [Google Scholar] [CrossRef]
- Harkins, C.P.; Pichon, B.; Doumith, M.; Parkhill, J.; Westh, H.; Tomasz, A.; de Lencastre, H.; Bentley, S.D.; Kearns, A.M.; Holden, M.T.G. Methicillin-resistant Staphylococcus aureus emerged long before the introduction of methicillin into clinical practice. Genome Biol. 2017, 18, 130. [Google Scholar] [CrossRef]
- Lee, A.S.; de Lencastre, H.; Garau, J.; Kluytmans, J.; Malhotra-Kumar, S.; Peschel, A.; Harbarth, S. Methicillin-resistant Staphylococcus aureus. Nat. Rev. Dis. Primers 2018, 4, 18033. [Google Scholar] [CrossRef]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef]
- Rice, L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef]
- Pennone, V.; Prieto, M.; Álvarez-Ordóñez, A.; Cobo-Diaz, J.F. Antimicrobial Resistance Genes Analysis of Publicly Available Staphylococcus aureus Genomes. Antibiotics 2022, 11, 1632. [Google Scholar] [CrossRef]
- Durrant, M.G.; Li, M.M.; Siranosian, B.A.; Montgomery, S.B.; Bhatt, A.S. A Bioinformatic Analysis of Integrative Mobile Genetic Elements Highlights Their Role in Bacterial Adaptation. Cell Host Microbe 2020, 28, 767. [Google Scholar] [CrossRef] [PubMed]
- Forster, S.C.; Liu, J.; Kumar, N.; Gulliver, E.L.; Gould, J.A.; Escobar-Zepeda, A.; Mkandawire, T.; Pike, L.J.; Shao, Y.; Stares, M.D.; et al. Strain-level characterization of broad host range mobile genetic elements transferring antibiotic resistance from the human microbiome. Nat. Commun. 2022, 13, 1445. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Maddamsetti, R.; Weiss, A.; Ha, Y.; Wang, T.; Wang, S.; You, L. Intra- and interpopulation transposition of mobile genetic elements driven by antibiotic selection. Nat. Ecol. Evol. 2022, 6, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Sotgiu, G.; Pontali, E.; Migliori, G.B. Linezolid to treat MDR-/XDR-tuberculosis: Available evidence and future scenarios. Eur. Respir. J. 2015, 45, 25–29. [Google Scholar] [CrossRef]
- Cervera, C.; van Delden, C.; Gavaldà, J.; Welte, T.; Akova, M.; Carratalà, J. Multidrug-resistant bacteria in solid organ transplant recipients. Clin. Microbiol. Infect. 2014, 20 (Suppl. S7), 49–73. [Google Scholar] [CrossRef]
- Lakhundi, S.; Zhang, K. Methicillin-Resistant Staphylococcus aureus: Molecular Characterization, Evolution, and Epidemiology. Clin. Microbiol. Rev. 2018, 31, e00020-18. [Google Scholar] [CrossRef]
- Zhou, Z.; Shuai, X.; Lin, Z.; Yu, X.; Ba, X.; Holmes, M.A.; Xiao, Y.; Gu, B.; Chen, H. Association between particulate matter (PM)2.5 air pollution and clinical antibiotic resistance: A global analysis. Lancet Planet. Health 2023, 7, E649–E659. [Google Scholar] [CrossRef]
- Li, W.; Liu, C.; Ho, H.C.; Shi, L.; Zeng, Y.; Yang, X.; Xia, H.; Zhang, W.; Huang, C.; Yang, L. Estimating the effect of increasing ambient temperature on antimicrobial resistance in China: A nationwide ecological study with the difference-in-differences approach. Sci. Total Environ. 2023, 882, 163518. [Google Scholar] [CrossRef]
- Hendriksen, R.S.; Munk, P.; Njage, P.; van Bunnik, B.; McNally, L.; Lukjancenko, O.; Röder, T.; Nieuwenhuijse, D.; Pedersen, S.K.; Kjeldgaard, J.; et al. Global monitoring of antimicrobial resistance based on metagenomics analyses of urban sewage. Nat. Commun. 2019, 10, 1124. [Google Scholar] [CrossRef]
- Van Bavel, J.J.; Cichocka, A.; Capraro, V.; Sjåstad, H.; Nezlek, J.B.; Pavlović, T.; Alfano, M.; Gelfand, M.J.; Azevedo, F.; Birtel, M.D.; et al. National identity predicts public health support during a global pandemic. Nat. Commun. 2022, 13, 517. [Google Scholar] [CrossRef]
- Gandra, S.; Alvarez-Uria, G.; Turner, P.; Joshi, J.; Limmathurotsakul, D.; van Doorn, H.R. Antimicrobial Resistance Surveillance in Low- and Middle-Income Countries: Progress and Challenges in Eight South Asian and Southeast Asian Countries. Clin. Microbiol. Rev. 2020, 33, e00048-19. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef]
- Sim, E.M.; Kim, R.; Gall, M.; Arnott, A.; Howard, P.; Valcanis, M.; Howden, B.P.; Sintchenko, V. Added Value of Genomic Surveillance of Virulence Factors in Shiga Toxin-Producing Escherichia coli in New South Wales, Australia. Front. Microbiol. 2021, 12, 713724. [Google Scholar] [CrossRef]
- Pärnänen, K.; Karkman, A.; Hultman, J.; Lyra, C.; Bengtsson-Palme, J.; Larsson, D.G.J.; Rautava, S.; Isolauri, E.; Salminen, S.; Kumar, H.; et al. Maternal gut and breast milk microbiota affect infant gut antibiotic resistome and mobile genetic elements. Nat. Commun. 2018, 9, 3891. [Google Scholar] [CrossRef]
- Zhou, Z.; Alikhan, N.F.; Sergeant, M.J.; Luhmann, N.; Vaz, C.; Francisco, A.P.; Carriço, J.A.; Achtman, M. GrapeTree: Visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res. 2018, 28, 1395–1404. [Google Scholar] [CrossRef]
- Norsigian, C.J.; Danhof, H.A.; Brand, C.K.; Oezguen, N.; Midani, F.S.; Palsson, B.O.; Savidge, T.C.; Britton, R.A.; Spinler, J.K.; Monk, J.M. Systems biology analysis of the Clostridioides difficile core-genome contextualizes microenvironmental evolutionary pressures leading to genotypic and phenotypic divergence. NPJ Syst. Biol. Appl. 2020, 6, 31. [Google Scholar] [CrossRef]
- Tokuda, M.; Shintani, M. Microbial evolution through horizontal gene transfer by mobile genetic elements. Microb. Biotechnol. 2024, 17, e14408. [Google Scholar] [CrossRef] [PubMed]
- Arnold, B.J.; Huang, I.T.; Hanage, W.P. Horizontal gene transfer and adaptive evolution in bacteria. Nat. Rev. Microbiol. 2022, 20, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Muzzi, A.; Donati, C. Population genetics and evolution of the pan-genome of Streptococcus pneumoniae. Int. J. Med. Microbiol. 2011, 301, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C. Where will new antibiotics come from? Nat. Rev. Microbiol. 2003, 1, 65–70. [Google Scholar] [CrossRef]
- Alirol, E.; Getaz, L.; Stoll, B.; Chappuis, F.; Loutan, L. Urbanisation and infectious diseases in a globalised world. Lancet Infect. Dis. 2011, 11, 131–141. [Google Scholar] [CrossRef]
- Ajulo, S.; Awosile, B. Global antimicrobial resistance and use surveillance system (GLASS 2022): Investigating the relationship between antimicrobial resistance and antimicrobial consumption data across the participating countries. PLoS ONE 2024, 19, e0297921. [Google Scholar] [CrossRef]
- Contarin, R.; Drapeau, A.; François, P.; Madec, J.Y.; Haenni, M.; Dordet-Frisoni, E. The interplay between mobilome and resistome in Staphylococcus aureus. mBio 2024, 15, e0242824. [Google Scholar] [CrossRef]
- Zhou, L.; Xu, P.; Gong, J.; Huang, S.; Chen, W.; Fu, B.; Zhao, Z.; Huang, X. Metagenomic profiles of the resistome in subtropical estuaries: Co-occurrence patterns, indicative genes, and driving factors. Sci. Total Environ. 2022, 810, 152263. [Google Scholar] [CrossRef]
- Blair, J.M.A. A climate for antibiotic resistance. Nat. Clim. Change 2018, 8, 460–461. [Google Scholar] [CrossRef]
- MacFadden, D.R.; McGough, S.F.; Fisman, D.; Santillana, M.; Brownstein, J.S. Antibiotic Resistance Increases with Local Temperature. Nat. Clim. Change 2018, 8, 510–514. [Google Scholar] [CrossRef]
- van Bavel, B.; Berrang-Ford, L.; Moon, K.; Gudda, F.; Thornton, A.J.; Robinson, R.F.S.; King, R. Intersections between climate change and antimicrobial resistance: A systematic scoping review. Lancet Planet. Health 2024, 8, e1118–e1128. [Google Scholar] [CrossRef] [PubMed]
- Rzymski, P.; Gwenzi, W.; Poniedziałek, B.; Mangul, S.; Fal, A. Climate warming, environmental degradation and pollution as drivers of antibiotic resistance. Environ. Pollut. 2024, 346, 123649. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Hasegawa, H.; Maeda, S. High temperatures promote cell-to-cell plasmid transformation in Escherichia coli. Biochem. Biophys. Res. Commun. 2019, 515, 196–200. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, Q.; Zhang, J.; Xu, H.; Miao, L.; Zhang, T.; Liu, D.; Zhu, Q.; Yan, H.; Yan, D. Climate warming promotes collateral antibiotic resistance development in cyanobacteria. Water Res. 2024, 256, 121642. [Google Scholar] [CrossRef]
- Li, W.; Liu, C.; Ho, H.C.; Shi, L.; Zeng, Y.; Yang, X.; Huang, Q.; Pei, Y.; Huang, C.; Yang, L. Association between antibiotic resistance and increasing ambient temperature in China: An ecological study with nationwide panel data. Lancet Reg. Health West. Pac. 2023, 30, 100628. [Google Scholar] [CrossRef]
- Carney, R.L.; Labbate, M.; Siboni, N.; Tagg, K.A.; Mitrovic, S.M.; Seymour, J.R. Urban beaches are environmental hotspots for antibiotic resistance following rainfall. Water Res. 2019, 167, 115081. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fu, X.; Ou, Z.; Li, J.; Lin, S.; Wu, Y.; Wang, X.; Deng, Y.; Sun, Y. Environmental determinants and demographic influences on global urban microbiomes, antimicrobial resistance and pathogenicity. NPJ Biofilms Microbiomes 2023, 9, 94. [Google Scholar] [CrossRef]
- Meinen, A.; Tomczyk, S.; Wiegand, F.N.; Abu Sin, M.; Eckmanns, T.; Haller, S. Antimicrobial resistance in Germany and Europe—A systematic review on the increasing threat accelerated by climate change. J. Health Monit. 2023, 8, 93–108. [Google Scholar] [CrossRef]
- Fagunwa, O.E.; Ashiru-Oredope, D.; Gilmore, B.F.; Doherty, S.; Oyama, L.B.; Huws, S.A. Climate change as a challenge for pharmaceutical storage and tackling antimicrobial resistance. Sci. Total Environ. 2024, 956, 177367. [Google Scholar] [CrossRef]
- Xie, J.; Jin, L.; He, T.; Chen, B.; Luo, X.; Feng, B.; Huang, W.; Li, J.; Fu, P.; Li, X. Bacteria and Antibiotic Resistance Genes (ARGs) in PM2.5 from China: Implications for Human Exposure. Environ. Sci. Technol. 2019, 53, 963–972. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Results of Generalized Linear Model a | ||||
|---|---|---|---|---|
| Variables | Estimates | SE | t Value | p Value |
| rcs(BIO3,3) | 1.082 | 0.469 | 2.309 | 0.021 |
| rcs(BIO4,3) | 0.015 | 0.006 | 2.562 | 0.011 |
| rcs(BIO5,3) | 0.354 | 0.122 | 2.894 | 0.004 |
| rcs(BIO6,3) | 0.527 | 0.188 | 2.810 | 0.005 |
| rcs(BIO10,3) | −0.260 | 0.129 | −2.016 | 0.044 |
| rcs(BIO11,3) | −0.501 | 0.251 | −2.001 | 0.046 |
| rcs(BIO12,3) | 0.008 | 0.002 | 3.616 | <0.001 |
| rcs(BIO13,3) | 0.035 | 0.011 | 3.025 | 0.003 |
| rcs(BIO14,3) | −0.062 | 0.020 | −3.015 | 0.003 |
| PM2.5 | 0.033 | 0.009 | 3.737 | <0.001 |
| MGEs | 0.138 | 0.006 | 23.339 | <0.001 |
| Antibiotic consumption | 0.063 | 0.020 | 3.177 | 0.002 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, H.; Xu, J.; Wang, Y.; Li, Y.; Hao, Y.; Long, J.; Liu, F.; Zhu, J.; Yang, H. The Global Antimicrobial Resistance Trends of Staphylococcus aureus and Influencing Factors. Microbiol. Res. 2025, 16, 118. https://doi.org/10.3390/microbiolres16060118
Yuan H, Xu J, Wang Y, Li Y, Hao Y, Long J, Liu F, Zhu J, Yang H. The Global Antimicrobial Resistance Trends of Staphylococcus aureus and Influencing Factors. Microbiology Research. 2025; 16(6):118. https://doi.org/10.3390/microbiolres16060118
Chicago/Turabian StyleYuan, Haitao, Jie Xu, Ying Wang, Yuan Li, Yuqing Hao, Jinzhao Long, Fang Liu, Jingyuan Zhu, and Haiyan Yang. 2025. "The Global Antimicrobial Resistance Trends of Staphylococcus aureus and Influencing Factors" Microbiology Research 16, no. 6: 118. https://doi.org/10.3390/microbiolres16060118
APA StyleYuan, H., Xu, J., Wang, Y., Li, Y., Hao, Y., Long, J., Liu, F., Zhu, J., & Yang, H. (2025). The Global Antimicrobial Resistance Trends of Staphylococcus aureus and Influencing Factors. Microbiology Research, 16(6), 118. https://doi.org/10.3390/microbiolres16060118