
Microbiota of Punctuated Snake Eel Ophichthus remiger (Valenciennes, 1842) Reared in Recirculation System Is Dominated by Latilactobacillus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Snake Eel Rearing Conditions
2.2. Sampling, DNA Extraction, and PCR Amplicon Sequencing
2.3. Bioinformatics Analysis
3. Results
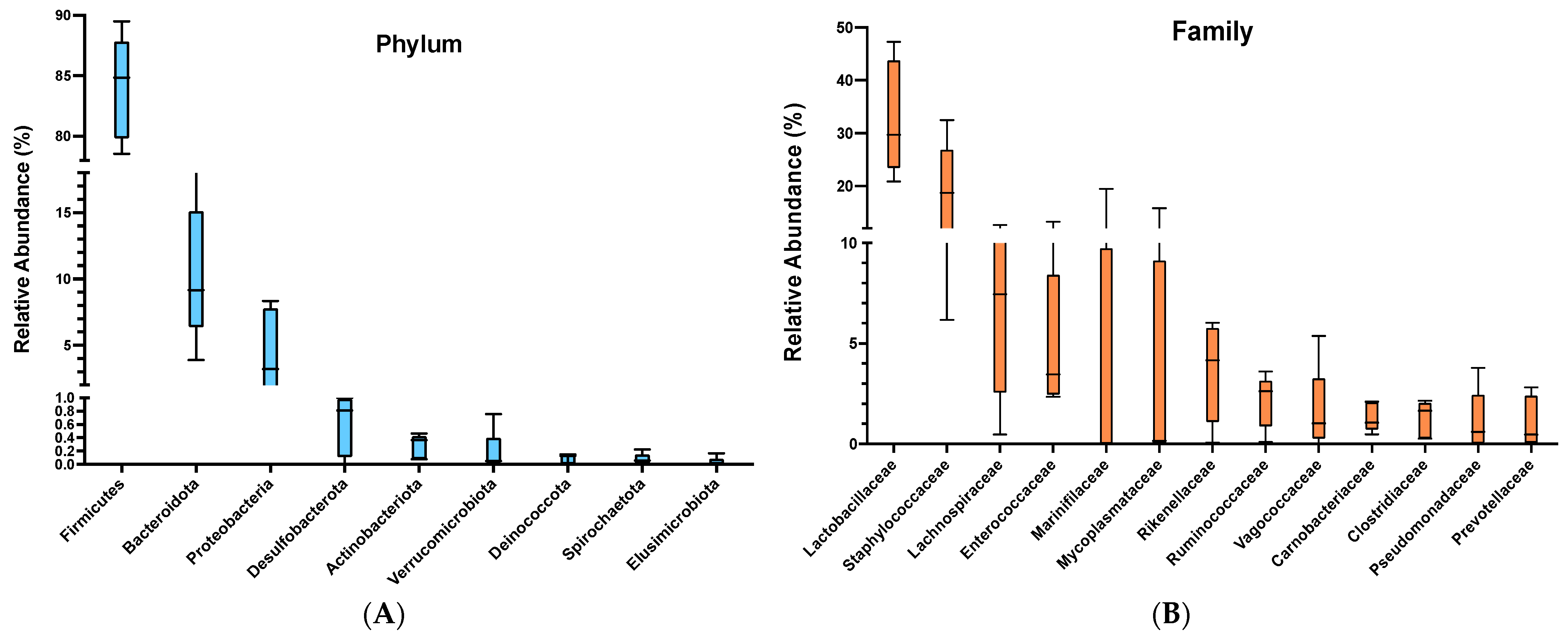
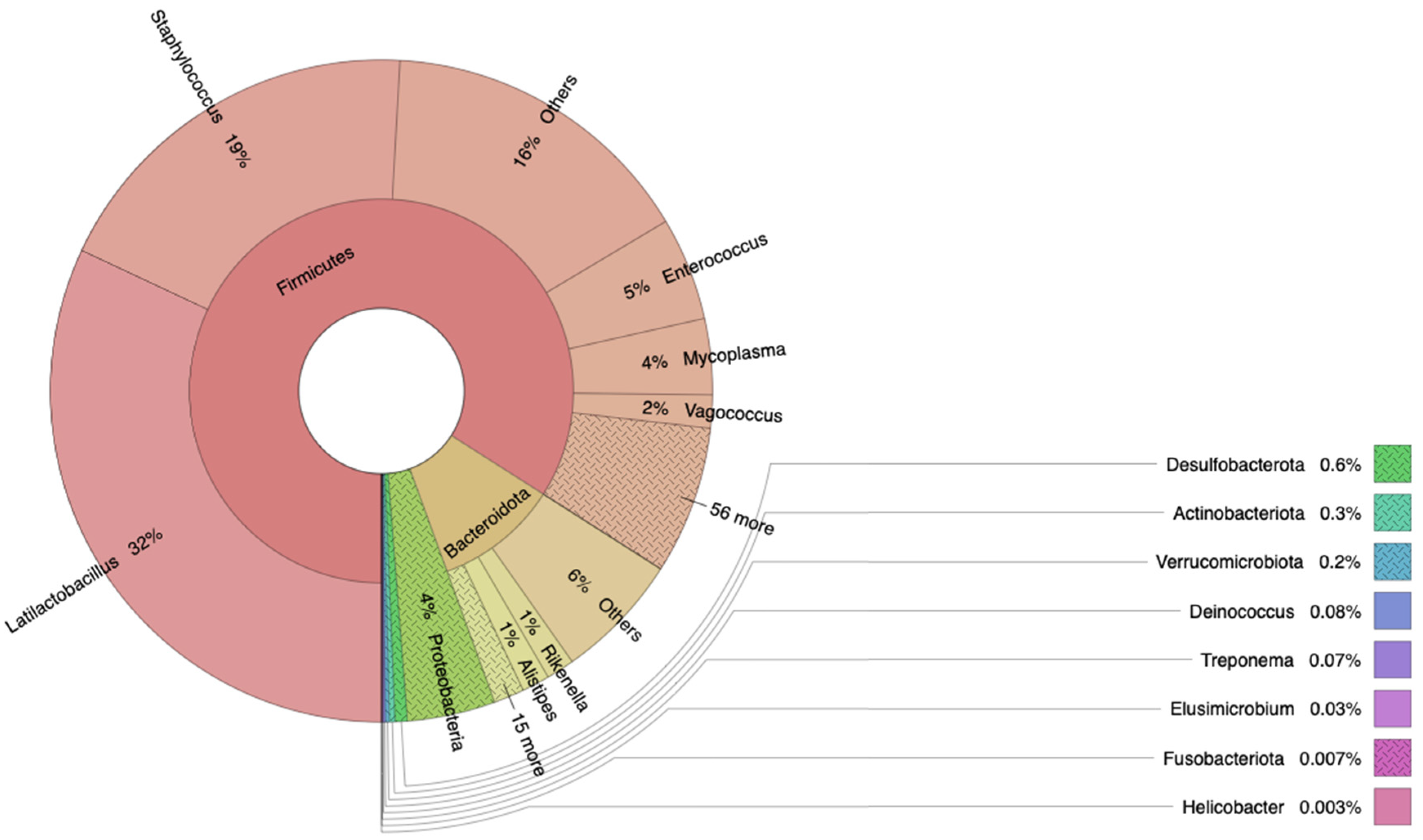
3.1. Bacterial Composition of Snake Eel Microbiota
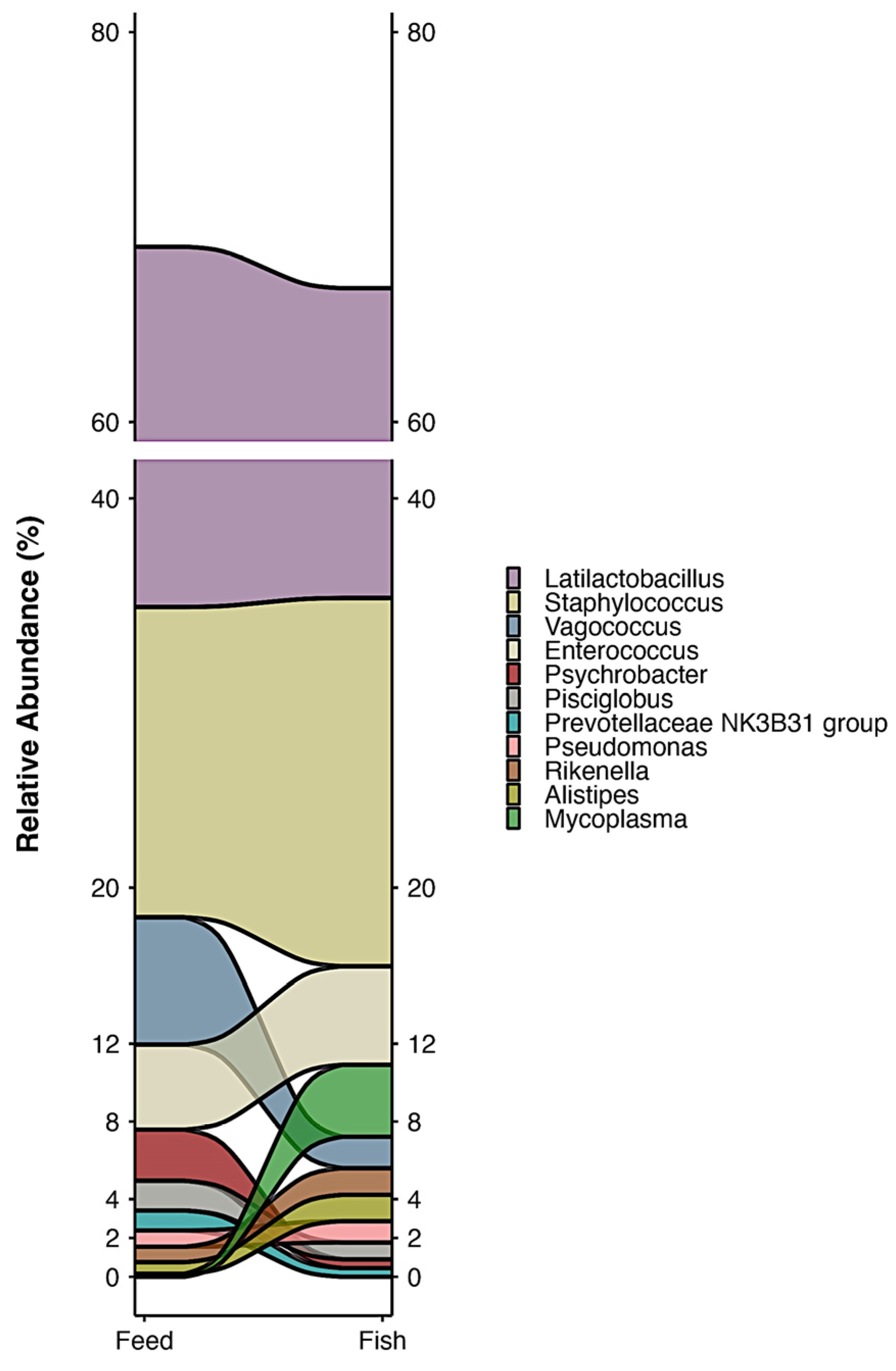
3.2. Bacterial Composition of Feed
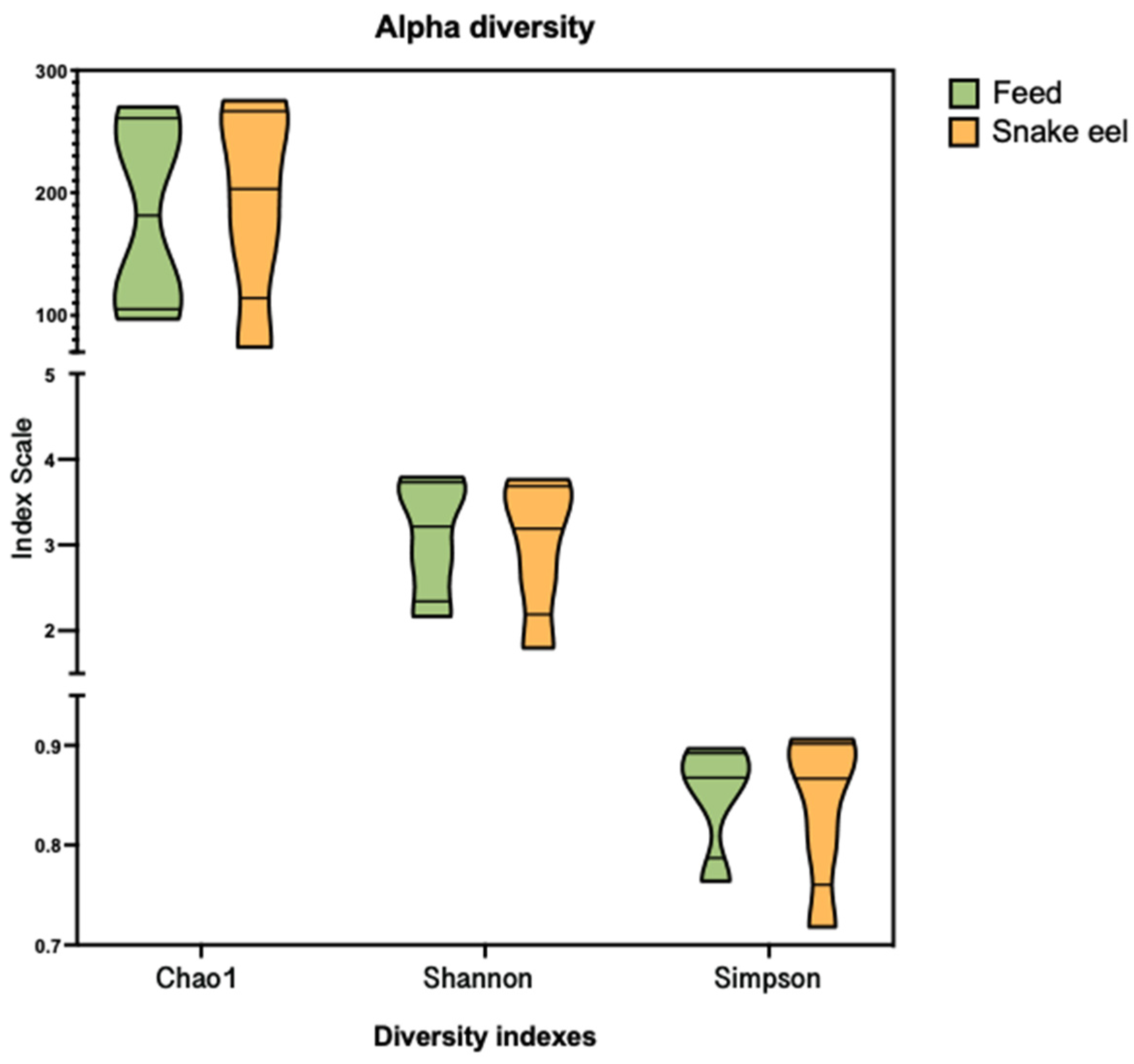
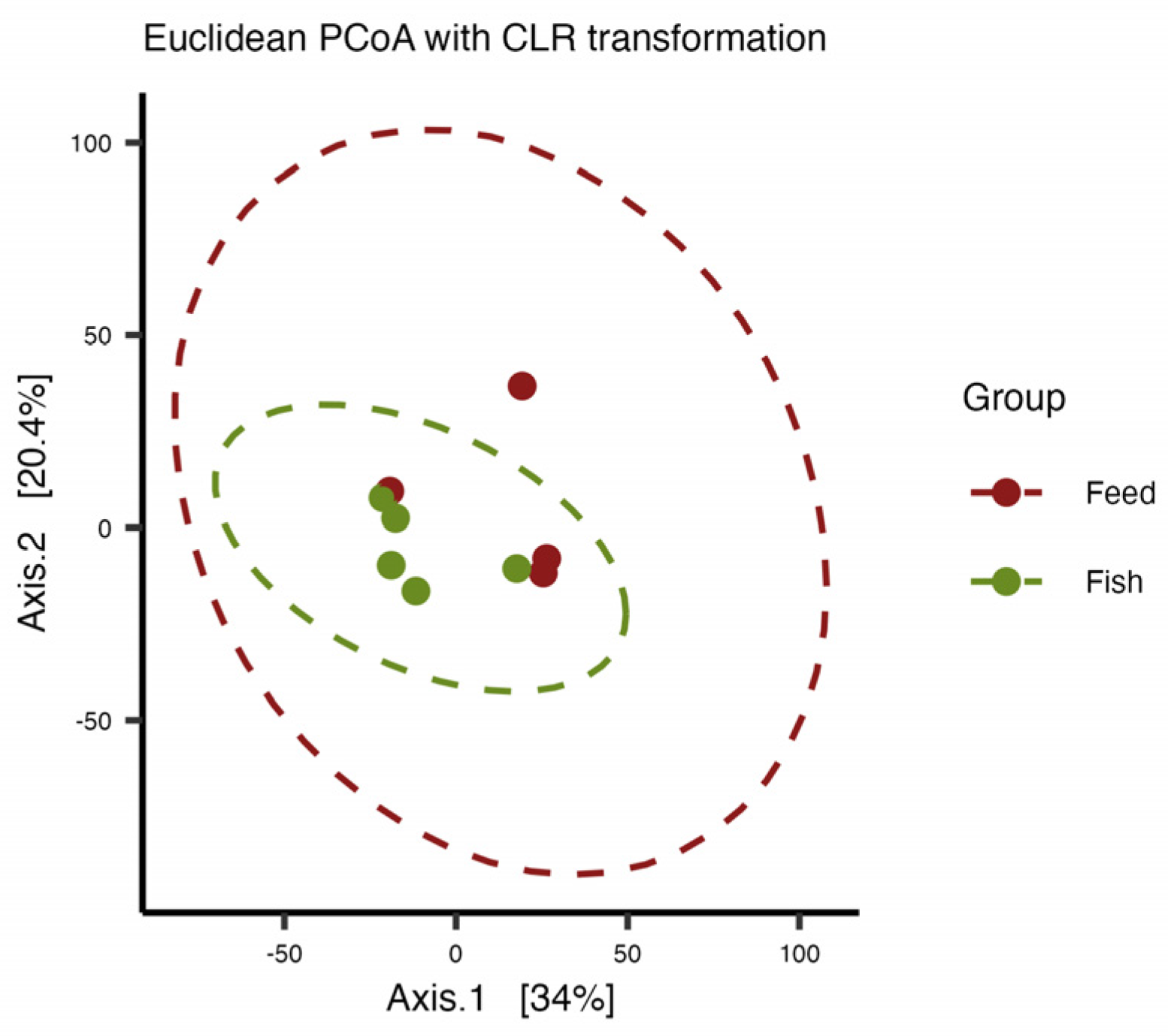
3.3. Alpha and Beta Diversity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ben-Hasan, A.; Walters, C.; Hordyk, A.; Christensen, V.; Al-Husaini, M. Alleviating Growth and Recruitment Overfishing Through Simple Management Changes: Insights from an Overexploited Long-Lived Fish. Mar. Coast. Fish. 2021, 13, 87–98. [Google Scholar] [CrossRef]
- Prince, J.; Hordyk, A. What to Do When You Have Almost Nothing: A Simple Quantitative Prescription for Managing Extremely Data-Poor Fisheries. Fish Fish. 2019, 20, 224–238. [Google Scholar] [CrossRef]
- Gozzer-Wuest, R.; Vinatea Chávez, R.A.; Olea Stranger, G.; Araya Goncalves, G.; Hiriart-Bertrand, L.; Labraña-Cornejo, R.; Alonso-Población, E. Identifying Priority Areas for Improvement in Chilean Fisheries. Front. Mar. Sci. 2023, 10, 1073397. [Google Scholar] [CrossRef]
- Subsecretaría de Pesca y Acuicultura. 2023. Available online: https://www.subpesca.cl/portal/618/articles-117812_recurso_1.pdf (accessed on 2 May 2024).
- Borges, L. The evolution of a discard policy in Europe. Fish Fish. 2015, 16, 534–540. [Google Scholar] [CrossRef]
- Wehrtmann, I.S.; Arana, P.M.; Barriga, E.; Gracia, A.; Pezzuto, P.R. Deep-water Shrimp Fisheries in Latin America: A Review. Lat. Am. J. Aquat. Res. 2012, 40, 497–535. [Google Scholar] [CrossRef]
- Romero, J.; Catalán, N.; Ramírez, C.; Miranda, C.D.; Oliva, M.; Flores, H.; Romero, M.S.; Rojas, R. High Abundance of Candidatus Arthromitus in Intestinal Microbiota of Seriolella violacea (Palm Ruff) under Reared Conditions. Fishes 2023, 8, 109. [Google Scholar] [CrossRef]
- McCord, A.I.; Chapman, C.A.; Weny, G.; Tumukunde, A.; Hyeroba, D.; Klotz, K.; Koblings, A.S.; Mbora, D.N.M.; Cregger, M.A.; White, B.A.; et al. Fecal microbiomes of non-human primates in Western Uganda reveal species-specific communities largely resistant to habitat perturbation. Am. Primatol. 2014, 76, 347–354. [Google Scholar] [CrossRef]
- Bletz, M.C.; Goedbloed, D.J.; Sanchez, E.; Reinhardt, T.; Tebbe, C.C.; Bhuju, S.; Geffers, R.; Jarek, M.; Vences, M.; Steinfartz, S. Amphibian gut microbiota shifts differentially in community structure but converges on habitat-specific predicted functions. Nat. Commun. 2016, 7, 13699. [Google Scholar] [CrossRef]
- Clark, R.I.; Walker, D.W. Role of gut microbiota in aging-related health decline: Insights from invertebrate models. Cell. Mol. Life Sci. 2018, 75, 93–101. [Google Scholar] [CrossRef]
- Grond, K.; Sandercock, B.K.; Jumpponen, A.; Zeglin, L.H. The avian gut microbiota: Community, physiology and function inwild birds. J. Avian Biol. 2018, 49, e01788. [Google Scholar] [CrossRef]
- Trevelline, B.K.; MacLeod, K.J.; Langkilde, T.; Kohl, K.D. Gestation alters the gut microbiota of an oviparous lizard. FEMS Microbiol. Ecol. 2019, 95, fiz086. [Google Scholar] [CrossRef]
- Liu, X.J.; Cao, Y.L.; Ouyang, S.; Wu, X.P. Comparative analysis of gut microbiota diversity in endangered, economical, and common freshwater mussels using 16S rRNA gene sequencing. Ecol. Evol. 2020, 21, 12015–12023. [Google Scholar] [CrossRef]
- Romero, J.; Ringø, E.; Merrifield, D.L. The gut microbiota of fish. In Aquaculture Nutrition: Gut Health, Probiotics and Prebiotics; Merrifield, D., Ringø, E., Eds.; Wiley-Blackwell Publishing: Oxford, UK, 2014; pp. 75–100. [Google Scholar]
- Hamidoghli, A.; Bae, J.; Won, S.; Lee, S.; Kim, D.J.; Bai, S.C. A Review on Japanese Eel (Anguilla japonica) Aquaculture, With Special Emphasis on Nutrition. Rev. Fish. Sci. Aquac. 2019, 27, 226–241. [Google Scholar] [CrossRef]
- Rasmussen, G.; Pedersen, M.I. Growth Rate and Biological Production of Yellow Eel Anguilla anguilla and Production of Silver Eel and the Number of Glass Eel to Fulfill the Danish EMP. Biol. Life Sci. 2023; preprint. [Google Scholar] [CrossRef]
- Available online: https://www.undercurrentnews.com/2024/04/12/glass-eel-prices-hit-all-time-high-at-15000-kg-in-japan-on-continuous-poor-harvest/ (accessed on 2 July 2024).
- Kusumawaty, D.; Augustine, S.M.N.; Aryani, A.; Effendi, Y.; Emran, T.B.; Tallei, T.E. Configuration of gut bacterial community profile and their potential functionality in the digestive tract of the wild and cultivated Indonesian shortfin elver-phase eels (Anguilla bicolor bicolor McClelland, 1844). 3 Biotech 2023, 13, 153. [Google Scholar] [CrossRef]
- Liu, X.; Fan, Y.; Mo, T.; Chen, Q.; Chen, W. Comparative Study of the Gut Microbiota Community between the Farmed and Wild Mastacembelus armatus (Zig-Zag Eel). Metabolites 2022, 12, 1193. [Google Scholar] [CrossRef]
- Roeselers, G.; Mittge, E.K.; Stephens, W.Z.; Parichy, D.M.; Cavanaugh, C.M.; Guillemin, K.; Rawls, J.F. Evidence for a core gut microbiota in the zebrafish. ISME J. 2011, 5, 1595–1608. [Google Scholar] [CrossRef]
- Villasante, A.; Ramírez, C.; Catalán, N.; Opazo, R.; Dantagnan, P.; Romero, J. Effect of dietary carbohydrate-to-protein ratio on gut microbiota in atlantic salmon (Salmo salar). Animals 2019, 9, 89. [Google Scholar] [CrossRef]
- Ramírez, C.; Romero, J. Fine flounder (Paralichthys adspersus) microbiome showed important differences between wild and reared specimens. Front. Microbiol. 2017, 8, 271. [Google Scholar] [CrossRef]
- Romero, J.; Díaz, O.; Miranda, C.D.; Rojas, R. Red Cusk-Eel (Genypterus chilensis) Gut Microbiota Description of Wild and Aquaculture Specimens. Microorganisms 2022, 10, 105. [Google Scholar] [CrossRef]
- Ramírez, C.; Romero, J. The microbiome of Seriola lalandi of wild and aquaculture origin reveals differences in composition and potential function. Front. Microbiol. 2017, 8, 1844. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, V.; Amaral-Zettler, L.; Davidson, J.; Summerfelt, S.; Good, C. The influence of fishmeal-free diets on microbial communities in Atlantic salmon Salmo salar recirculation aquaculture systems. Appl. Environ. Microbiol. 2016, 82, 4470–4481. [Google Scholar] [CrossRef] [PubMed]
- Hossain, S. European eel (Anguilla anguilla) GI tract conserves a unique metagenomics profile in the recirculation aquaculture system (RAS). Aquac. Int. 2021, 29, 1529–1544. [Google Scholar] [CrossRef]
- Gajardo, K.; Jaramillo-Torres, A.; Kortner, T.; Merrifield, D.; Tinsley, J.; Bakke, A.; Krogdahl, Å. Alternative protein sources in the diet modulate microbiota and functionality in the distal intestine of Atlantic salmon (Salmo salar). Appl. Environ. Microbiol. 2017, 83, e02615-16. [Google Scholar] [CrossRef]
- Froese, R.; Pauly, D. (Eds.) FishBase. World Wide Web Electronic Publication. Version (10/2024). 2024. Available online: www.fishbase.org (accessed on 2 February 2024).
- Goicochea, C.; Mostacero, J.; Moquillaza, P. Edad y Crecimiento de Ophichthus remiger (Valenciennes) en el Norte del Mar Peruano, 2004. Inf. Inst. Mar Perú 2012, 39, 9–17. [Google Scholar]
- Chirichigno, N.; Vélez, J. Claves Para Identificar los Peces Marinos del Perú, 2nd ed.; Publicación Especial del Instituto del Mar del Perú: Callao, Peru, 1998. [Google Scholar]
- Galán Galán, J.; Carbajal, W.; Castañeda Condori, J. La Anguila Ophichthus remiger en Aguas de las Islas Lobos de Afuera, Lambayeque, Durante el 2005. Inf. Inst. Mar Perú 2007, 34, 281–294. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romero, J.; Ramírez, C.; Hurtado, L.; Serrano, E.; Rojas, R. Microbiota of Punctuated Snake Eel Ophichthus remiger (Valenciennes, 1842) Reared in Recirculation System Is Dominated by Latilactobacillus. Microbiol. Res. 2025, 16, 38. https://doi.org/10.3390/microbiolres16020038
Romero J, Ramírez C, Hurtado L, Serrano E, Rojas R. Microbiota of Punctuated Snake Eel Ophichthus remiger (Valenciennes, 1842) Reared in Recirculation System Is Dominated by Latilactobacillus. Microbiology Research. 2025; 16(2):38. https://doi.org/10.3390/microbiolres16020038
Chicago/Turabian StyleRomero, Jaime, Carolina Ramírez, Luz Hurtado, Edison Serrano, and Rodrigo Rojas. 2025. "Microbiota of Punctuated Snake Eel Ophichthus remiger (Valenciennes, 1842) Reared in Recirculation System Is Dominated by Latilactobacillus" Microbiology Research 16, no. 2: 38. https://doi.org/10.3390/microbiolres16020038
APA StyleRomero, J., Ramírez, C., Hurtado, L., Serrano, E., & Rojas, R. (2025). Microbiota of Punctuated Snake Eel Ophichthus remiger (Valenciennes, 1842) Reared in Recirculation System Is Dominated by Latilactobacillus. Microbiology Research, 16(2), 38. https://doi.org/10.3390/microbiolres16020038