
Comparative Analysis of Healthy Gut Microbiota in German and Korean Populations: Insights from Large-Scale Cohort Studies


Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Microbiome Analysis and Statistical Analysis
3. Results
3.1. Data Preprocessing Summary
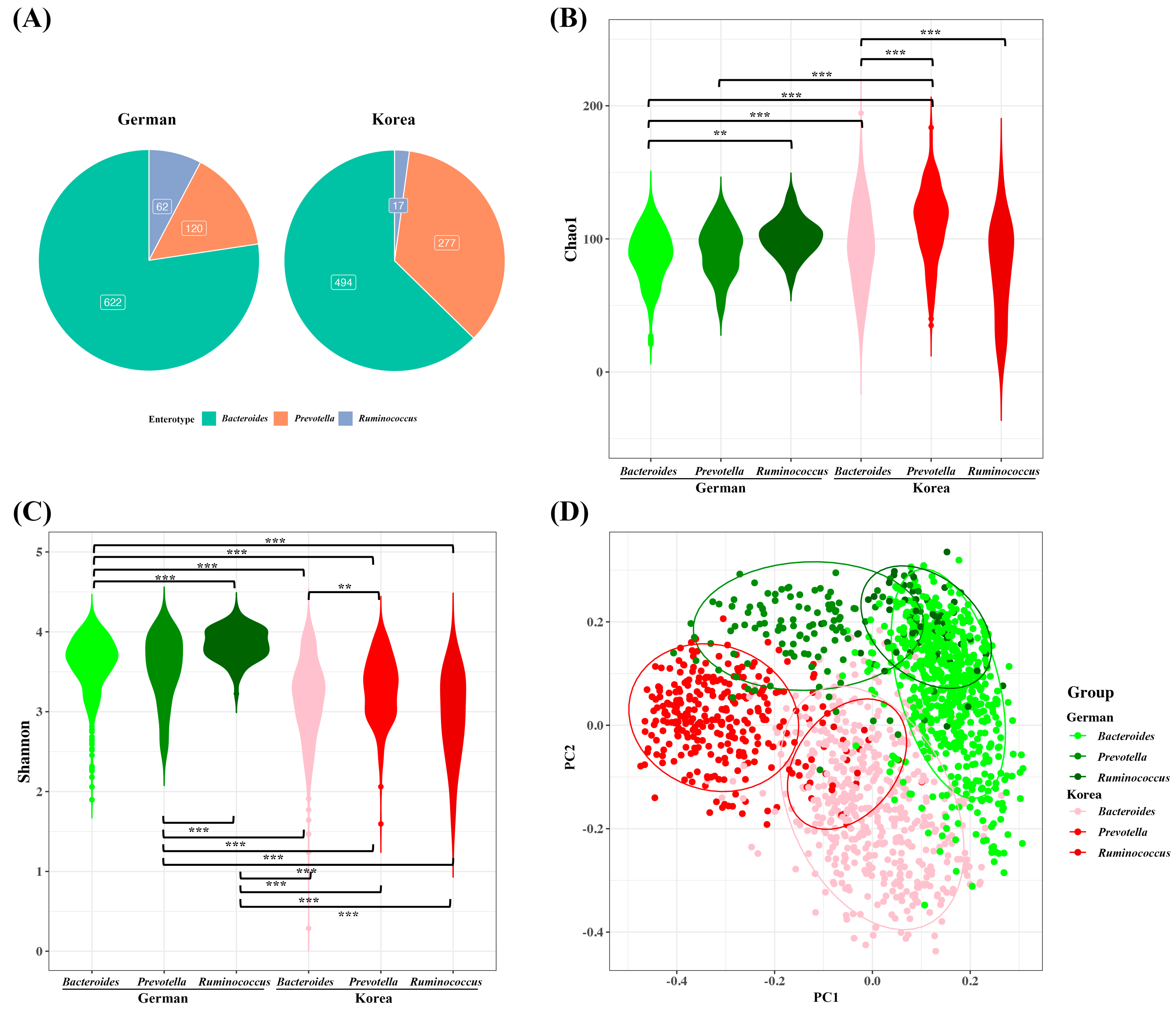
3.2. Diversity and Abundance of Microbiota
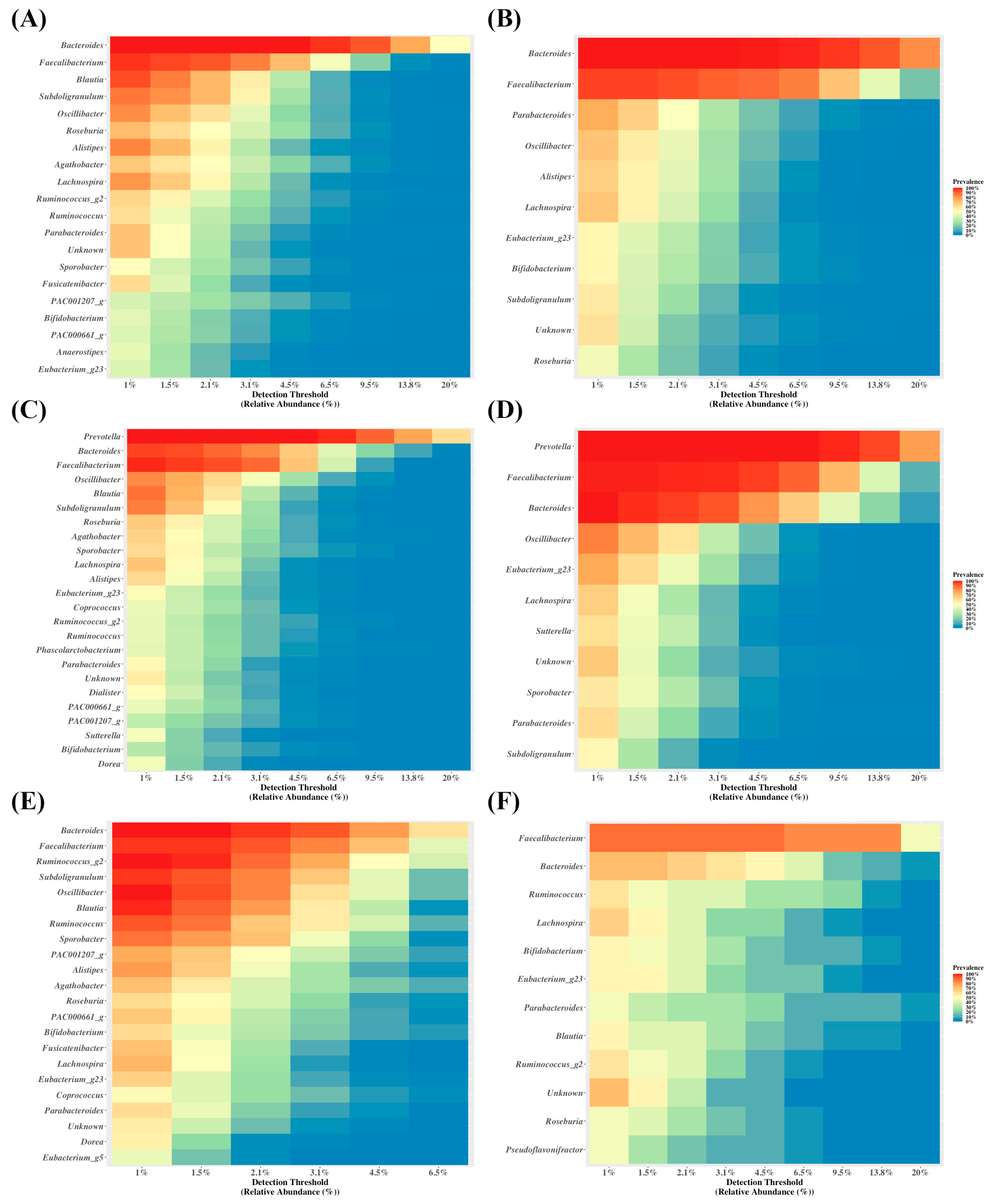
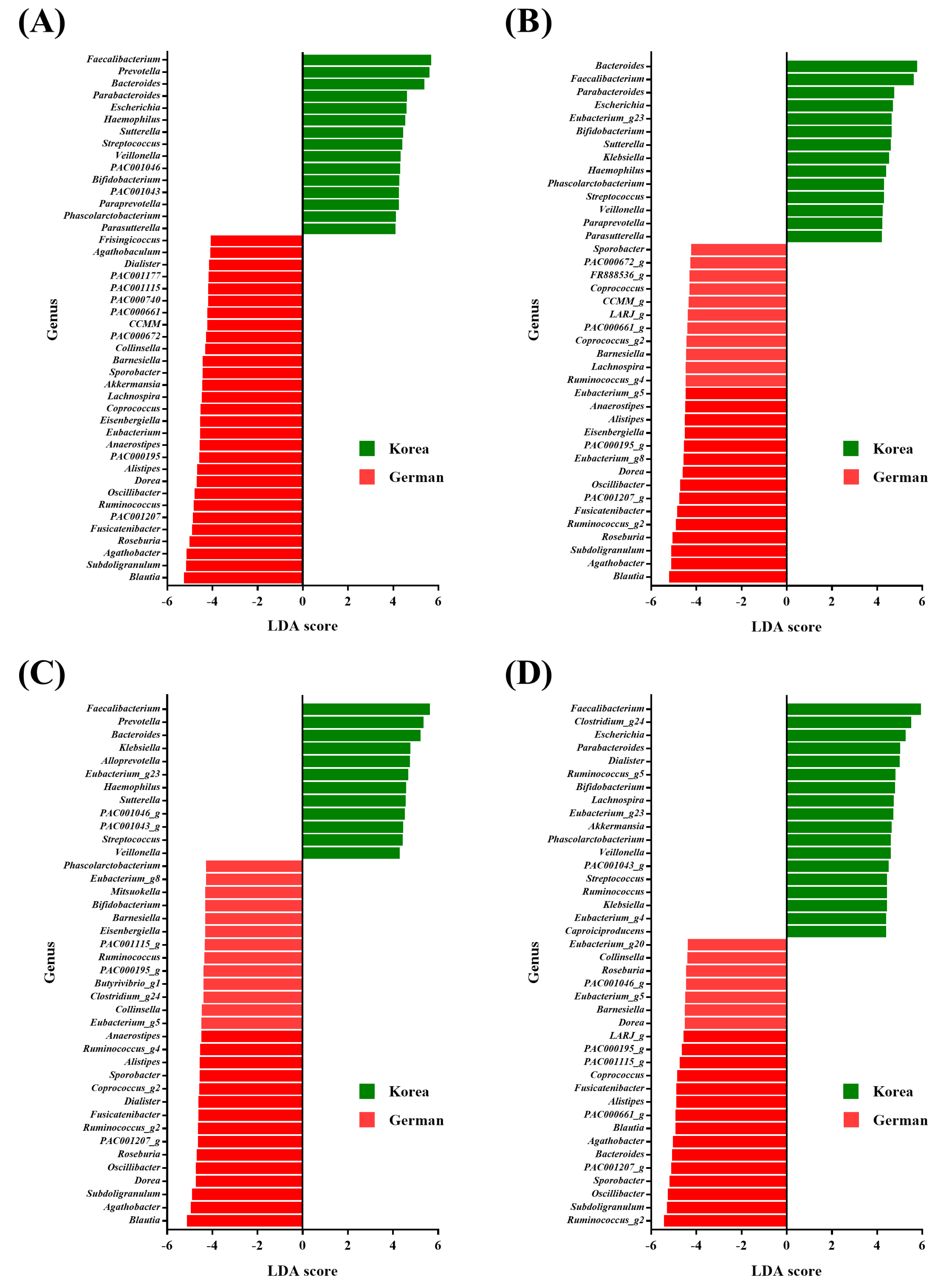
3.3. Species Taxa Comparison
3.4. Network Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Frank, D.N.; Amand, A.L.S.; Feldman, R.F.; Boedeker, E.C.; Harpaz, H.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed]
- Leviatan, S.; Shoer, S.; Rothschild, D.; Gorodetski, M.; Segal, E. An expanded reference map of the human gut microbiome reveals hundreds of previously unknown species. Nat. Commun. 2022, 13, 3863. [Google Scholar] [CrossRef] [PubMed]
- Tuniyazi, M.; Li, S.; Hu, X.; Fu, Y.; Zhang, N. The Role of Early Life Microbiota Composition in the Development of Allergic Diseases. Microorganisms 2022, 10, 1190. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Bai, Y.; Zha, L.; Ullah, N.; Ullah, H.; Shah, S.R.H.; Sun, H.; Zhang, C. Mechanism of the Gut Microbiota Colonization Resistance and Enteric Pathogen Infection. Front. Cell. Infect. Microbiol. 2021, 11, 716299. [Google Scholar] [CrossRef] [PubMed]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Haneishi, Y.; Furuya, Y.; Hasegawa, M.; Picarelli, A.; Rossi, M.; Miyamoto, J. Inflammatory Bowel Diseases and Gut Microbiota. Int. J. Mol. Sci. 2023, 24, 3817. [Google Scholar] [CrossRef] [PubMed]
- Igudesman, D.; Crandell, J.L.; Corbin, K.D.; Hoorper, J.; Thomas, J.M.; Bulik, C.M.; Pence, B.W.; Pratley, R.E.; Kosorok, M.R.; Maahs, D.M.; et al. Associations of Dietary Intake with the Intestinal Microbiota and Short-Chain Fatty Acids among Young Adults with Type 1 Diabetes and Overweight or Obesity. J. Nutr. 2022, 153, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Stadlbauer, V. Liver-Gut-Interaction: Role of Microbiome Transplantation in the Future Treatment of Metabolic Disease. J. Pers. Med. 2023, 13, 220. [Google Scholar] [CrossRef]
- Clemente, J.C.; Ursell, L.K.; Laura, W.P.; Rob, K. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef]
- Lim, M.Y.; Hong, S.; Bang, S.J.; Chung, W.H.; Shin, J.H.; Kim, J.H.; Nam, Y.D. Gut Microbiome Structure and Association with Host Factors in a Korean Population. Msystems 2021, 6, e0017921. [Google Scholar] [CrossRef]
- Brandl, B.; Rennekamp, R.; Reitmeier, S.; Pietrynik, K.; Dirndorfer, S.; Haller, D.; Hofmann, T.; Skurk, T.; Hauner, H. Offering Fiber-Enriched Foods Increases Fiber Intake in Adults with or without Cardiometabolic Risk: A Randomized Controlled Trial. Front. Nutr. 2022, 9, 816229. [Google Scholar] [CrossRef] [PubMed]
- Takagi, T.; Inoue, R.; Oshima, A.; Sakazume, H.; Ogawa, K.; Tominaga, T.; Mihara, Y.; Sugaya, T.; Mizushima, K.; Uchiyama, K.; et al. Typing of the Gut Microbiota Community in Japanese Subjects. Microorganisms 2022, 10, 664. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Ma, T.; Sakandar, H.A.; Menghe, B.; Sun, Z. Gut microbiome and aging nexus and underlying mechanism. Appl. Microbiol. Biotechnol. 2022, 106, 5349–5358. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.K.; Paul, S.; Dutta, C. Geography, Ethnicity or Subsistence-Specific Variations in Human Microbiome Composition and Diversity. Front. Microbiol. 2017, 8, 1162. [Google Scholar] [CrossRef] [PubMed]
- Gaulke, C.A.; Sharpton, T.J. The influence of ethnicity and geography on human gut microbiome composition. Nat. Med. 2018, 24, 1495–1496. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Maier, L.; Pruteanu, M.; Kuhn, M.; Zeller, G.; Telzerow, A.; Anderson, E.E.; Brochado, A.R.; Fernandez, K.C.; Dose, H.; Mori, H.; et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 2018, 555, 623–628. [Google Scholar] [CrossRef]
- Nogacka, A.M.; Salazar, N.; Arboleya, S.; Suarez, M.; Frenandez, N.; Solís, G.; de los Reyes-Gavilán, C.G.; Gueimonde, M. Early microbiota, antibiotics and health. Cell. Mol. Life Sci. 2018, 75, 83–91. [Google Scholar] [CrossRef]
- Peterson, J.; Garges, S.; Giovanni, M.; Mclnnes, P.; Wang, L.; Schloss, J.A.; Bonazzi, V.; McEwen, J.E.; Wetterstrand, K.A.; Deal, C.; et al. The NIH Human Microbiome Project. Genome Res. 2009, 19, 2317–2323. [Google Scholar]
- Wei, L.Q.; Cheong, I.H.; Yang, G.H.; Li, X.G.; Kozlakidis, Z.; Ding, L.; Liu, N.N.; Wang, H. The Application of High-Throughput Technologies for the Study of Microbiome and Cancer. Front. Genet. 2021, 12, 699793. [Google Scholar] [CrossRef]
- Song, E.J.; Lee, E.S.; Nam, Y.D. Progress of analytical tools and techniques for human gut microbiome research. J. Microbiol. 2018, 56, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Allali, I.; Arnold, J.W.; Roach, J.; Cadenas, M.B.; Butz, N.; Hassan, H.M.; Koci, M.; Ballou, A.; Mendoza, M.; Ali, R.; et al. A comparion of sequencing platforms and bioinformatics popelines for compositional analysis of the gut microbiome. BMC Microbiol. 2017, 17, 194. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.; Beiko, R.G. 16S rRNA Gene Analysis with QIIME2. Methods Mol. Biol. 2018, 1849, 113–129. [Google Scholar] [PubMed]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chum, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Salonen, A.; Salojarvi, J.; Lahti, L.; de Vos, W.M. The adult intestinal core microbiota is determined by analysis depth and health status. Clin. Microbiol. Infect. 2012, 18, 16–20. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Subelj, L.; Bajec, M. Unfolding communities in large complex networks: Combining defensive and offensive label propagation for core extraction. Phys. Rev. E 2011, 83, 036103. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.-M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Pielou, E.C. Shannon’s formula as a measure of specific diversity: Its use and misuse. Am. Nat. 1966, 100, 463–465. [Google Scholar] [CrossRef]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Wrzosek, L.; Ciocan, D.; Borentain, P.; Spatz, M.; Puchois, V.; Hugot, C.; Ferrere, G.; Mayeur, C.; Perlemuter, G.; Cassard, A.-M. Transplantation of human microbiota into conventional mice durably reshapes the gut microbiota. Sci. Rep. 2018, 8, 6854. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Tang, H.; Li, M.; Pang, X.; Wang, L.; Zhang, M.; Zhao, Y.; Zhang, X.; Shen, J. The abundance of fecal Faecalibacterium prausnitzii in relation to obesity and gender in Chinese adults. Arch. Microbiol. 2014, 196, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Aprile, F.; Bruno, G.; Palma, R.; Mascellino, M.T.; Panetta, C.; Scalese, G.; Oliva, A.; Severi, C.; Pontone, S. Microbiota Alterations in Precancerous Colon Lesions: A Systematic Review. Cancers 2021, 13, 3061. [Google Scholar] [CrossRef] [PubMed]
- Andoh, A.; Nishida, A. Alteration of the Gut Microbiome in Inflammatory Bowel Disease. Digestion 2023, 104, 16–23. [Google Scholar] [CrossRef]
- Shetty, S.A.; Hugenholtz, H.; Lahti, L.; Smidt, H.; de Vos, W.M. Intestinal microbiome landscaping: Insight in community assemblage and implications for microbial modulation strategies. FEMS Microbiol. Rev. 2017, 41, 182–199. [Google Scholar] [CrossRef]
- Neu, A.T.; Allen, E.E.; Roy, K. Defining and quantifying the core microbiome: Challenges and prospects. Proc. Natl. Acad. Sci. USA 2021, 118, e2104429118. [Google Scholar] [CrossRef]
- Pan, M.; Barua, N.; Ip, M. Mucin-degrading gut commensals isolated from healthy faecal donor suppress intestinal epithelial inflammation and regulate tight junction barrier function. Front. Immunol. 2022, 13, 1021094. [Google Scholar] [CrossRef]
- Vital, M.; Karch, A.; Pieper, D.H. Colonic Butyrate-Producing Communities in Humans: An Overview Using Omics Data. Msystems 2017, 2, e00130-17. [Google Scholar] [CrossRef]
- Canani, R.B.; Costanzo, M.D.; Leone, L.; Pedata, M.; Meli, R.; Calignano, A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J. Gastroenterol. 2011, 17, 1519–1528. [Google Scholar] [CrossRef]
- Odamaki, T.; Kato, K.; Sugahara, H.; Hashikura, N.; Takahashi, S.; Xiao, J.Z.; Abe, F.; Osawa, R. Age-related changes in gut microbiota composition from newborn to centenarian: A cross-sectional study. BMC Microbiol. 2016, 16, 90. [Google Scholar] [CrossRef] [PubMed]
- Zmora, N.; Suez, J.; Elinav, E. You are what you eat: Diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 35–56. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.M.; Mailing, L.J.; Niemiro, G.M.; Moore, R.; Cook, M.D.; White, B.A.; Holscher, H.D.; Woods, J.A. Exercise Alters Gut Microbiota Composition and Function in Lean and Obese Humans. Med. Sci. Sport. Exerc. 2018, 50, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Cabral, D.J.; Wurster, J.I.; Korry, B.J.; Penumutchu, S.; Belenky, P. Consumption of a Western-Style Diet Modulates the Response of the Murine Gut Microbiome to Ciprofloxacin. Msystems 2020, 5, e00317-20. [Google Scholar] [CrossRef] [PubMed]
- Fuczmarski, M.K.; Bodt, B.A.; Shupe, E.S.; Zonderman, A.B.; Evans, M.K. Dietary Patterns Associated with Lower 10-Year Atherosclerotic Cardiovascular Disease Risk among Urban African-American and White Adults Consuming Western Diets. Nutrients 2018, 10, 158. [Google Scholar] [CrossRef] [PubMed]
- Statovci, D.; Aguilera, M.; MacSharry, J.; Melgar, S. The Impact of Western Diet and Nutrients on the Microbiota and Immune Response at Mucosal Interfaces. Front. Immunol. 2017, 8, 838. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Paramithiotis, P.; Shin, H.S. Kimchi and Other Widely Consumed Traditional Fermented Foods of Korea: A Review. Front. Microbiol. 2016, 7, 1493. [Google Scholar] [CrossRef]
- Aleti, G.; Baker, J.L.; Tang, X.; Alvarez, R.; Dinis, M.; Tran, N.C.; Melnik, A.V.; Zhong, C.; Ernst, M.; Dorrestein, P.C.; et al. Identification of the Bacterial Biosynthetic Gene Clusters of the Oral Microbiome Illuminates the Unexplored Social Language of Bacteria during Health and Disease. mBio 2019, 10, e00321-e19. [Google Scholar] [CrossRef]
- Mark Welch, J.L.; Rossetti, B.J.; Rieken, C.W.; Dewhirst, F.E.; Borisy, G.G. Biogeography of a human oral microbiome at the micron scale. Proc. Natl. Acad. Sci. USA 2016, 113, E791–E800. [Google Scholar] [CrossRef]
- Bowen, W.H.; Burne, R.A.; Wu, H.; Koo, H. Oral Biofilms: Pathogens, Matrix, and Polymicrobial Interactions in Microenvironments. Trends Microbiol. 2018, 26, 229–242. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| German Cohort (PRJNA701859) | Korean Coort (PRJEB33905) | |
|---|---|---|
| Sample number | 804 | 788 |
| Input read count | 34,184 ± 10,067 (79,178–13,802) | 134,103 ± 111,787 (1,076,815–24,859) |
| Filtered read count | 24,448 ± 7640 | 94,568 ± 73,972 |
| Denoised read count | 23,548 ± 7449 | 92,642 ± 73,051 |
| Merged read count | 21,364 ± 6924 | 88,912 ± 71,105 |
| Non-chimeric read count | 16,783 ± 5229 (39,184–3873) | 60,943 ± 46,978 (460,695–8075) |
| Percentage of input non-chimeric (%) | 49.1 ± 5.5 | 66.3 ± 12.4 |
| Total OTU count | 24,182 | 62,472 |
| Paired read length | 432.1 ± 12.5 | 432.6 ± 17.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Son, M.K.; Song, Y.; Chung, J.; Na, H.S. Comparative Analysis of Healthy Gut Microbiota in German and Korean Populations: Insights from Large-Scale Cohort Studies. Microbiol. Res. 2024, 15, 109-119. https://doi.org/10.3390/microbiolres15010007
Son MK, Song Y, Chung J, Na HS. Comparative Analysis of Healthy Gut Microbiota in German and Korean Populations: Insights from Large-Scale Cohort Studies. Microbiology Research. 2024; 15(1):109-119. https://doi.org/10.3390/microbiolres15010007
Chicago/Turabian StyleSon, Min Kee, Yuri Song, Jin Chung, and Hee Sam Na. 2024. "Comparative Analysis of Healthy Gut Microbiota in German and Korean Populations: Insights from Large-Scale Cohort Studies" Microbiology Research 15, no. 1: 109-119. https://doi.org/10.3390/microbiolres15010007
APA StyleSon, M. K., Song, Y., Chung, J., & Na, H. S. (2024). Comparative Analysis of Healthy Gut Microbiota in German and Korean Populations: Insights from Large-Scale Cohort Studies. Microbiology Research, 15(1), 109-119. https://doi.org/10.3390/microbiolres15010007