
Assessing Microbial Monitoring Methods for Challenging Environmental Strains and Cultures



Abstract
:1. Introduction
2. Materials and Methods
2.1. Cultures and Media
2.2. Sampling and Testing
2.3. Quantitative PCR
2.4. Cell Count Calculations
2.5. Mixed Culture Testing
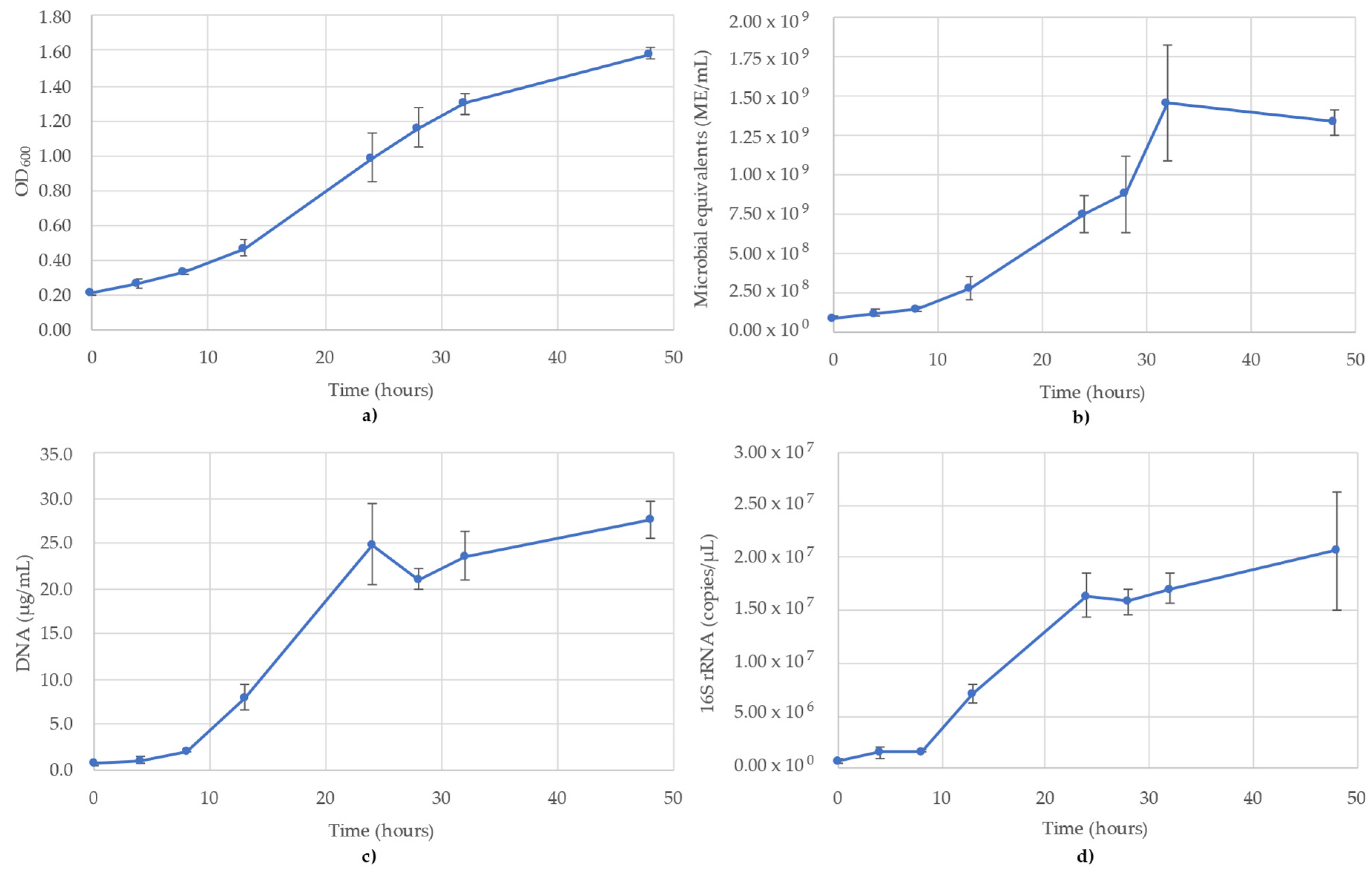
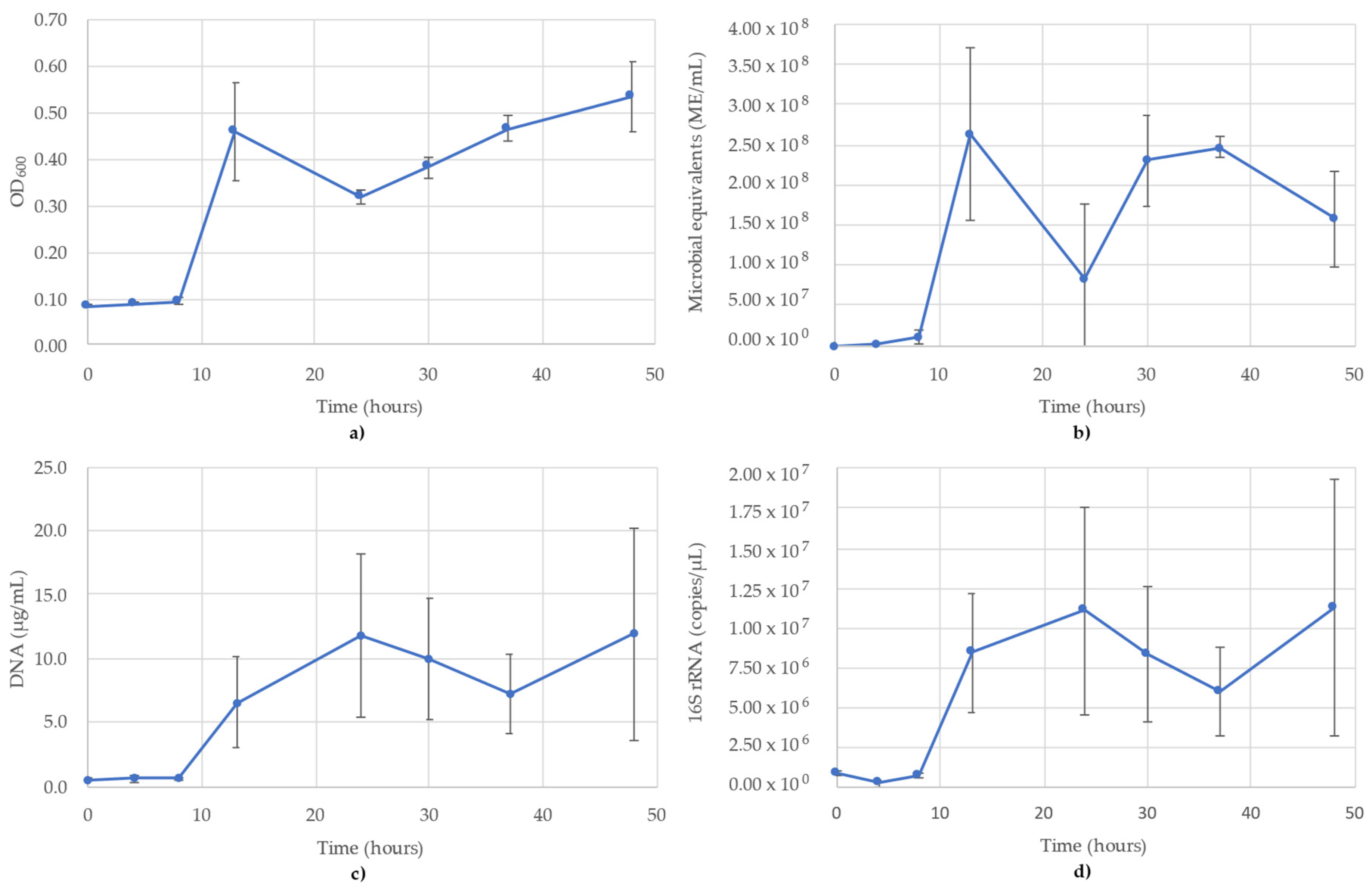
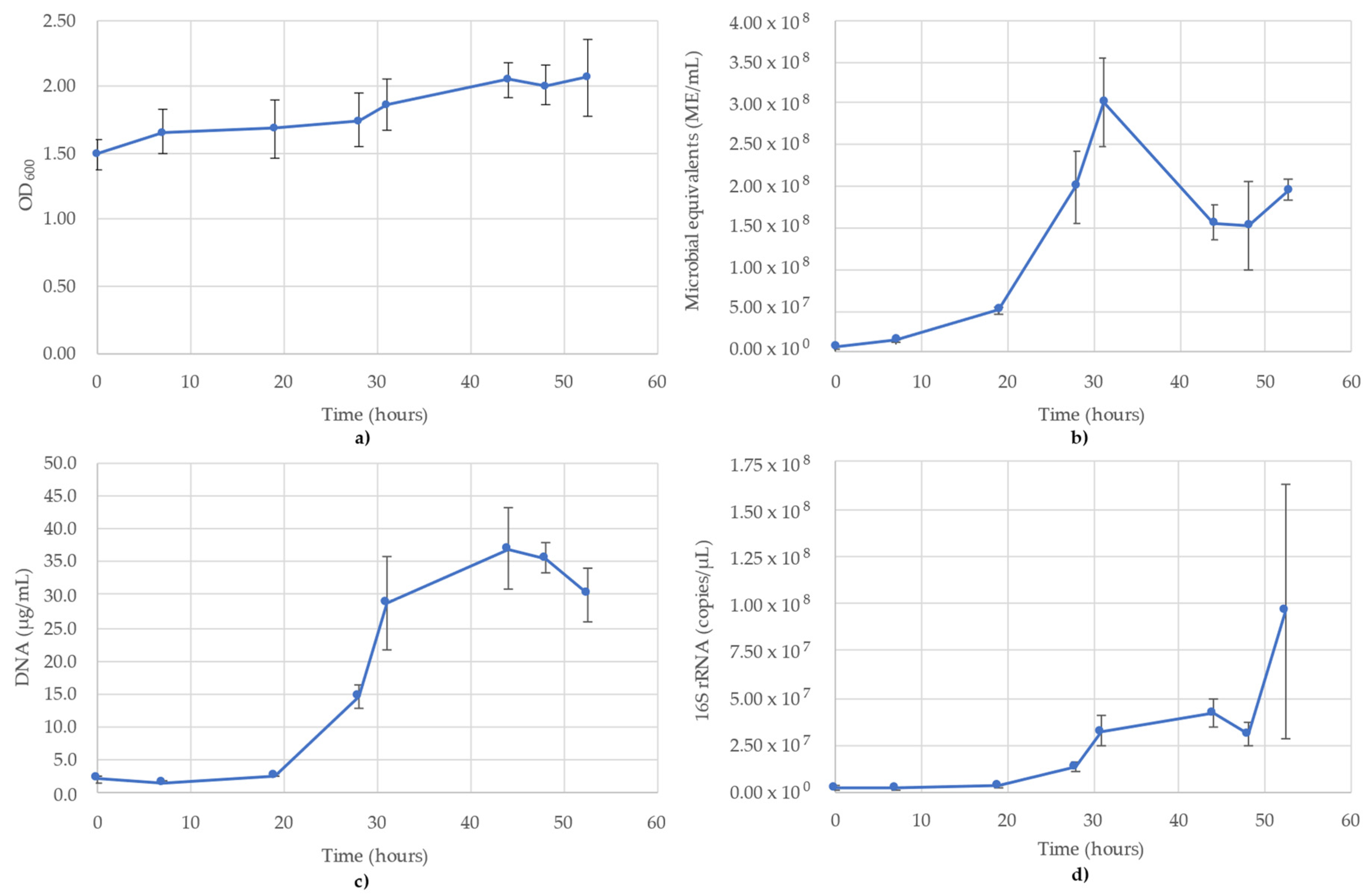
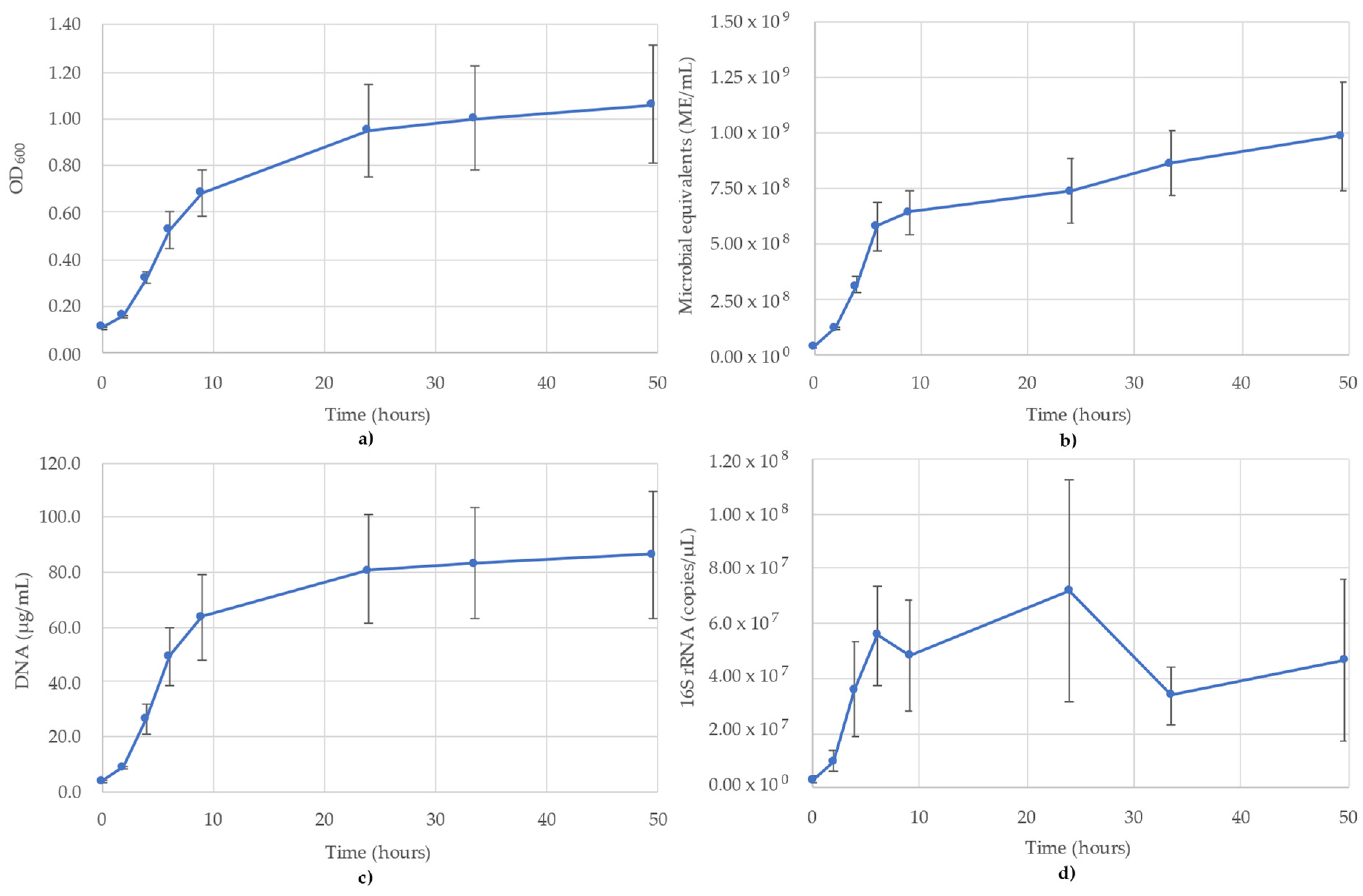
3. Results
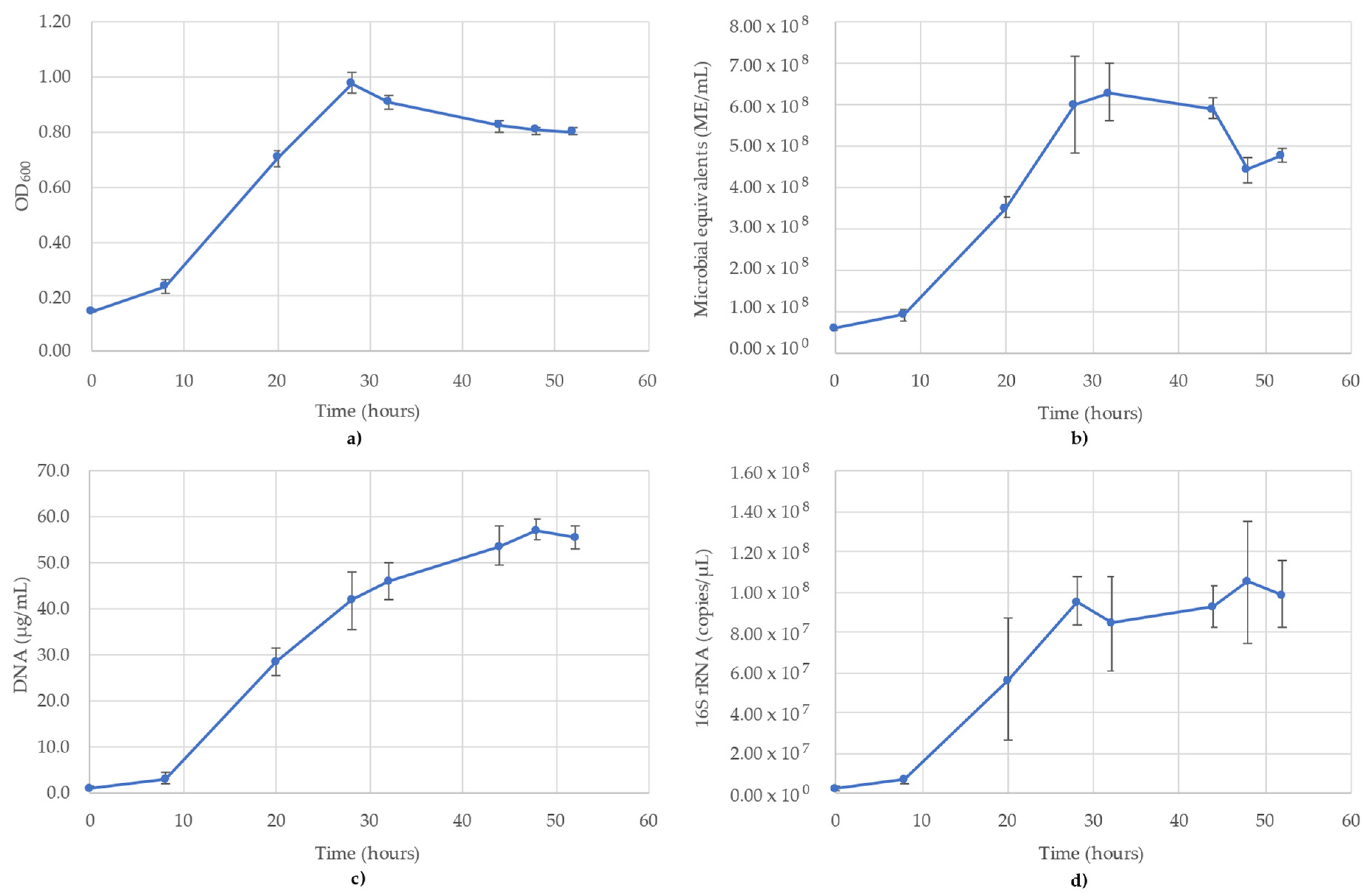
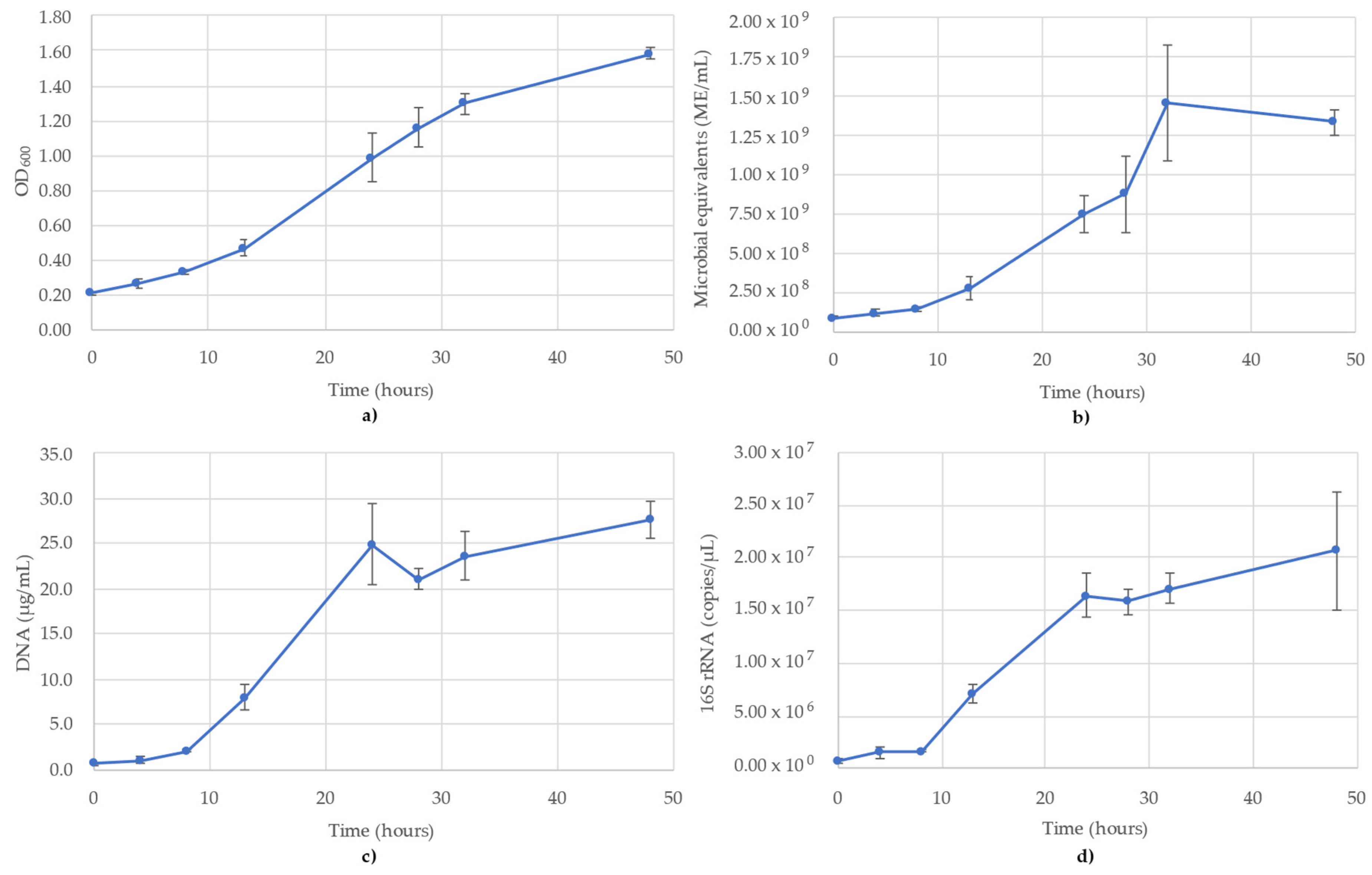
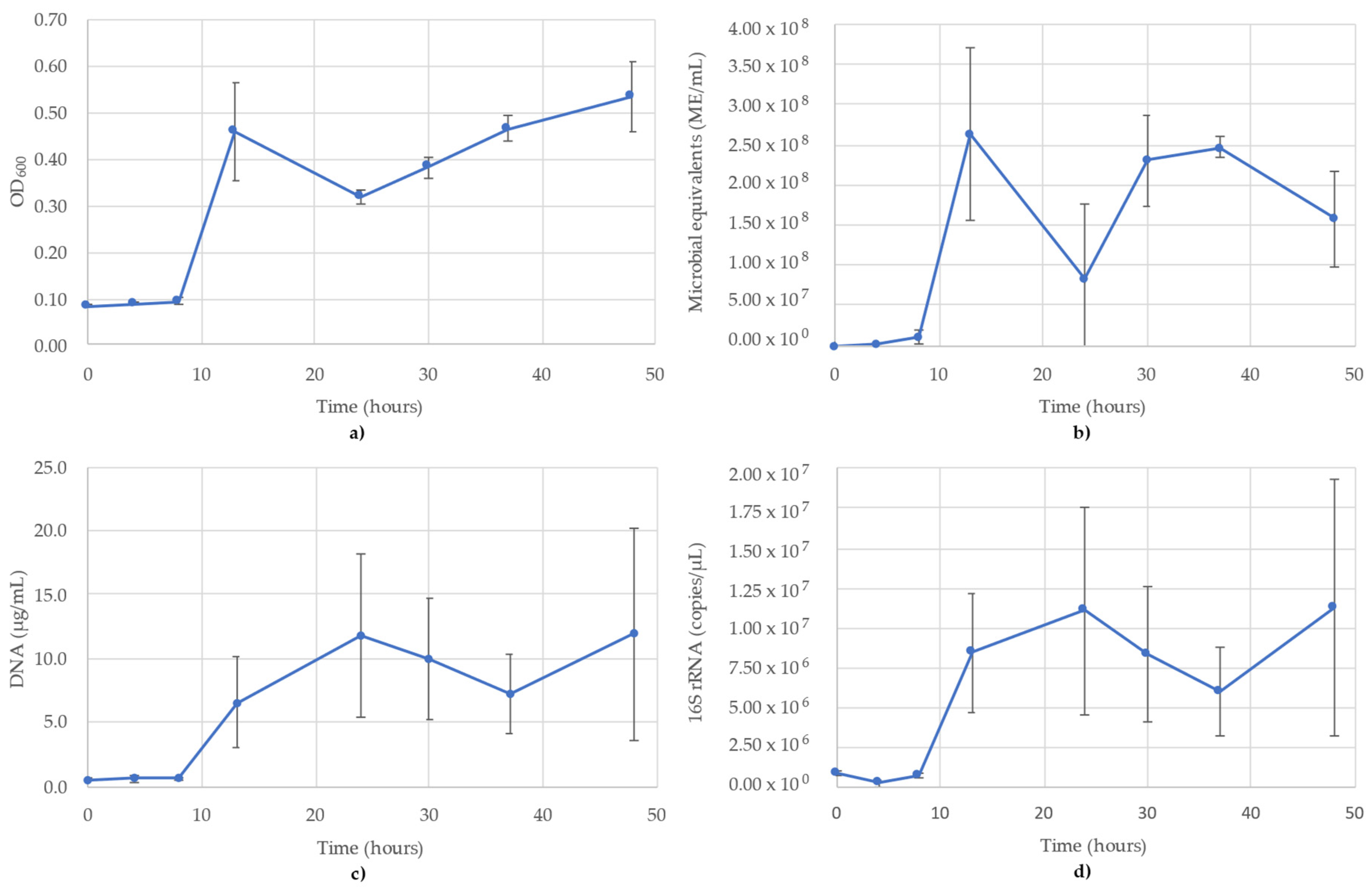
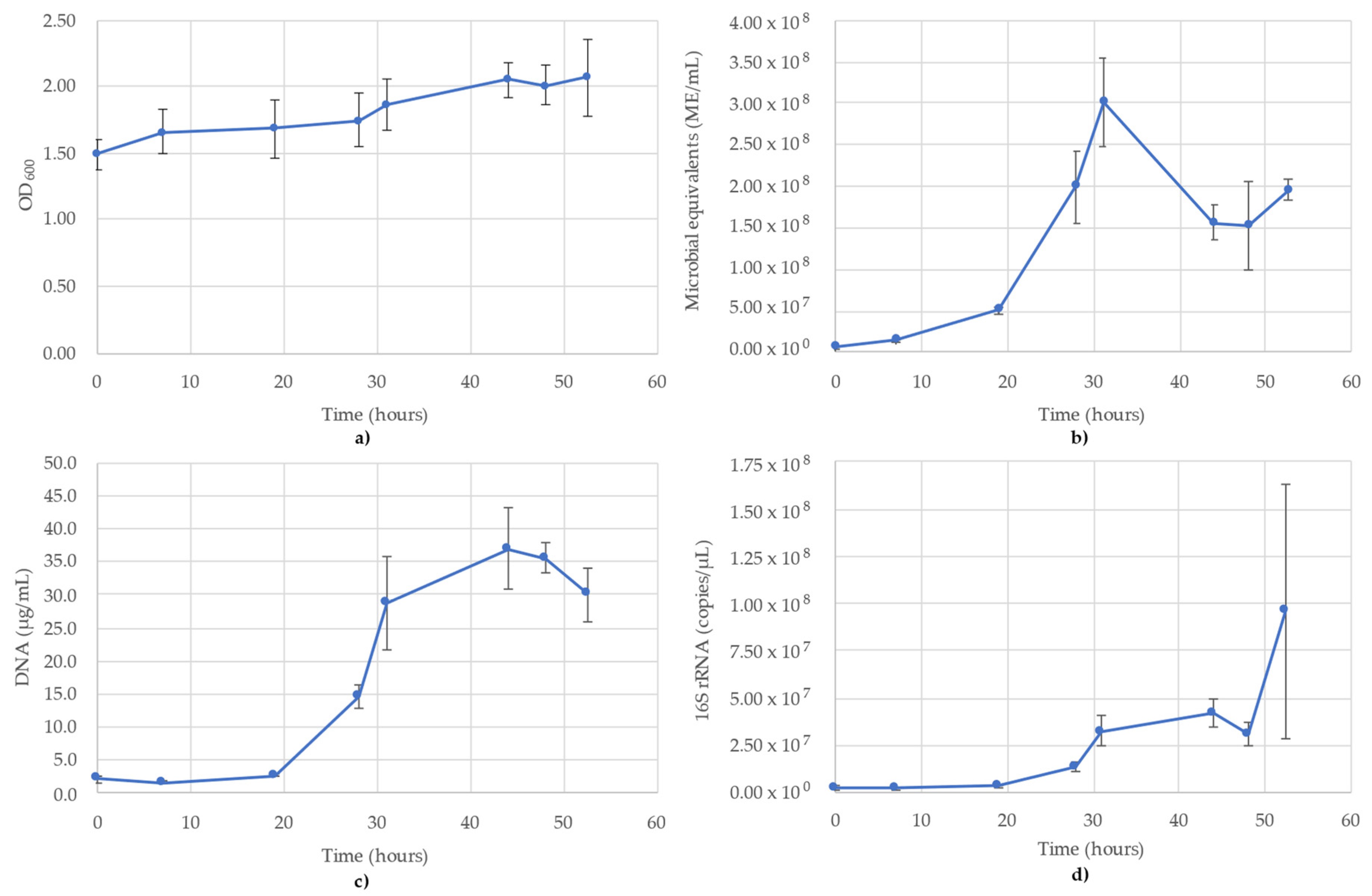
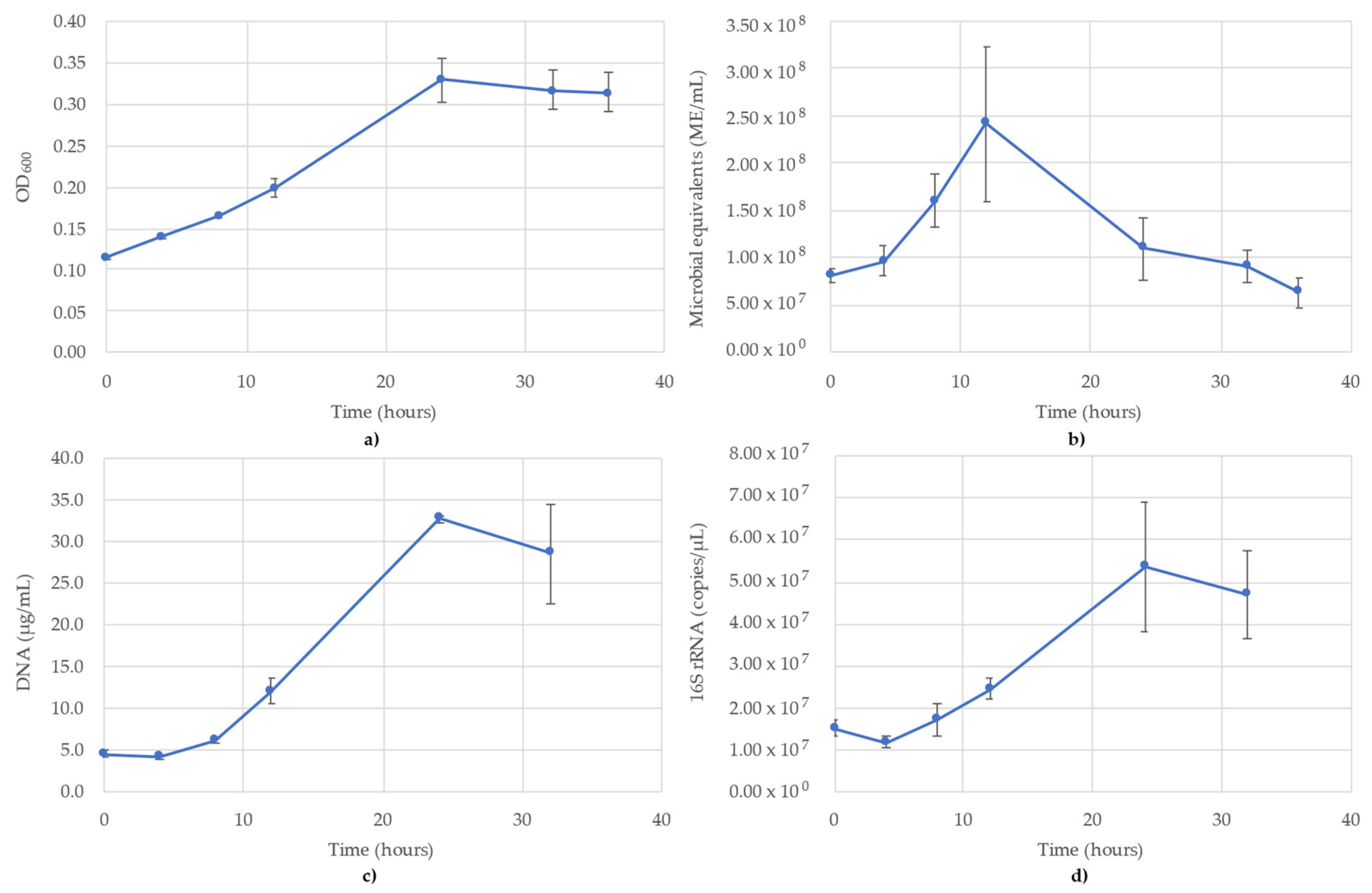
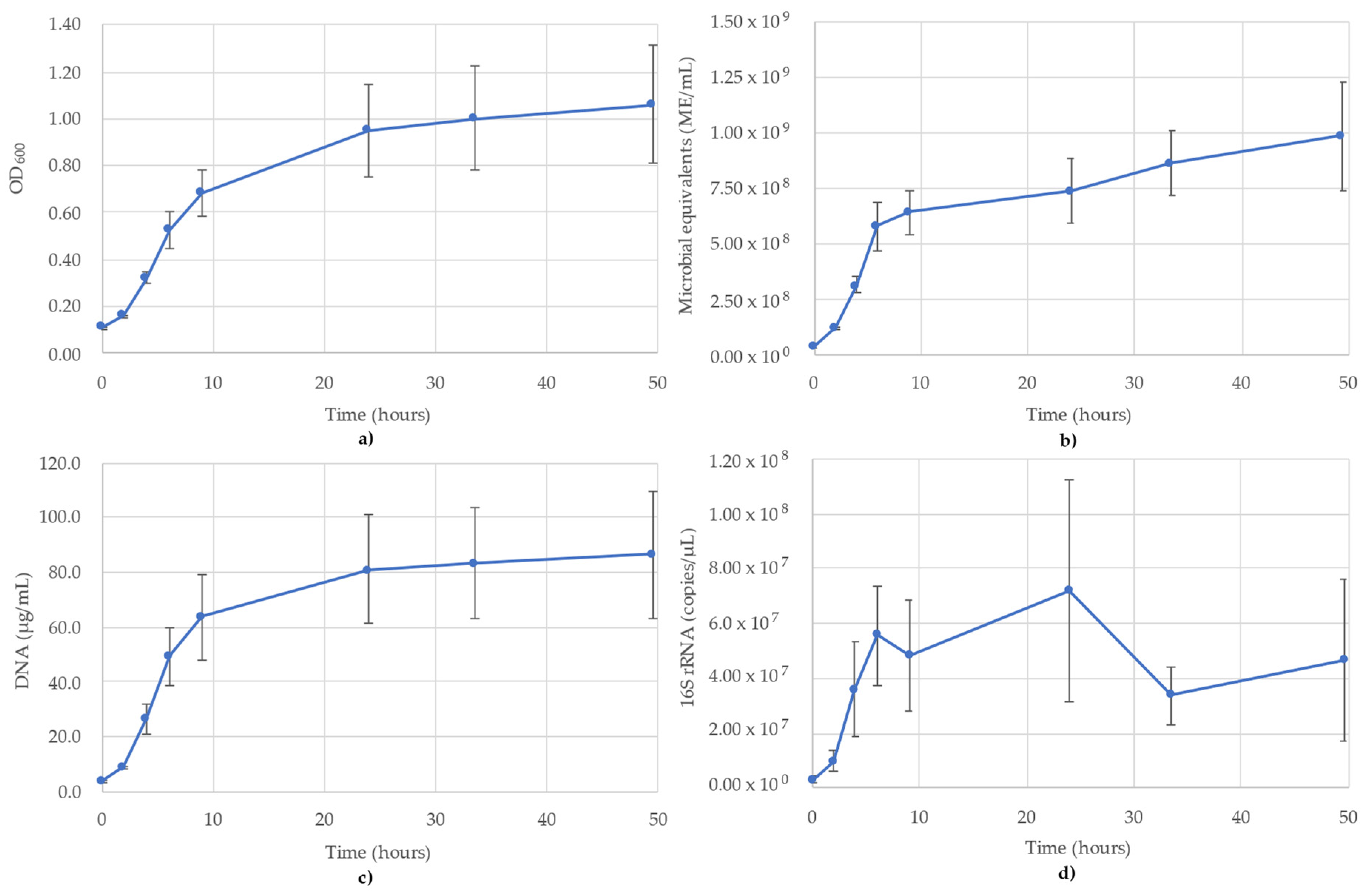
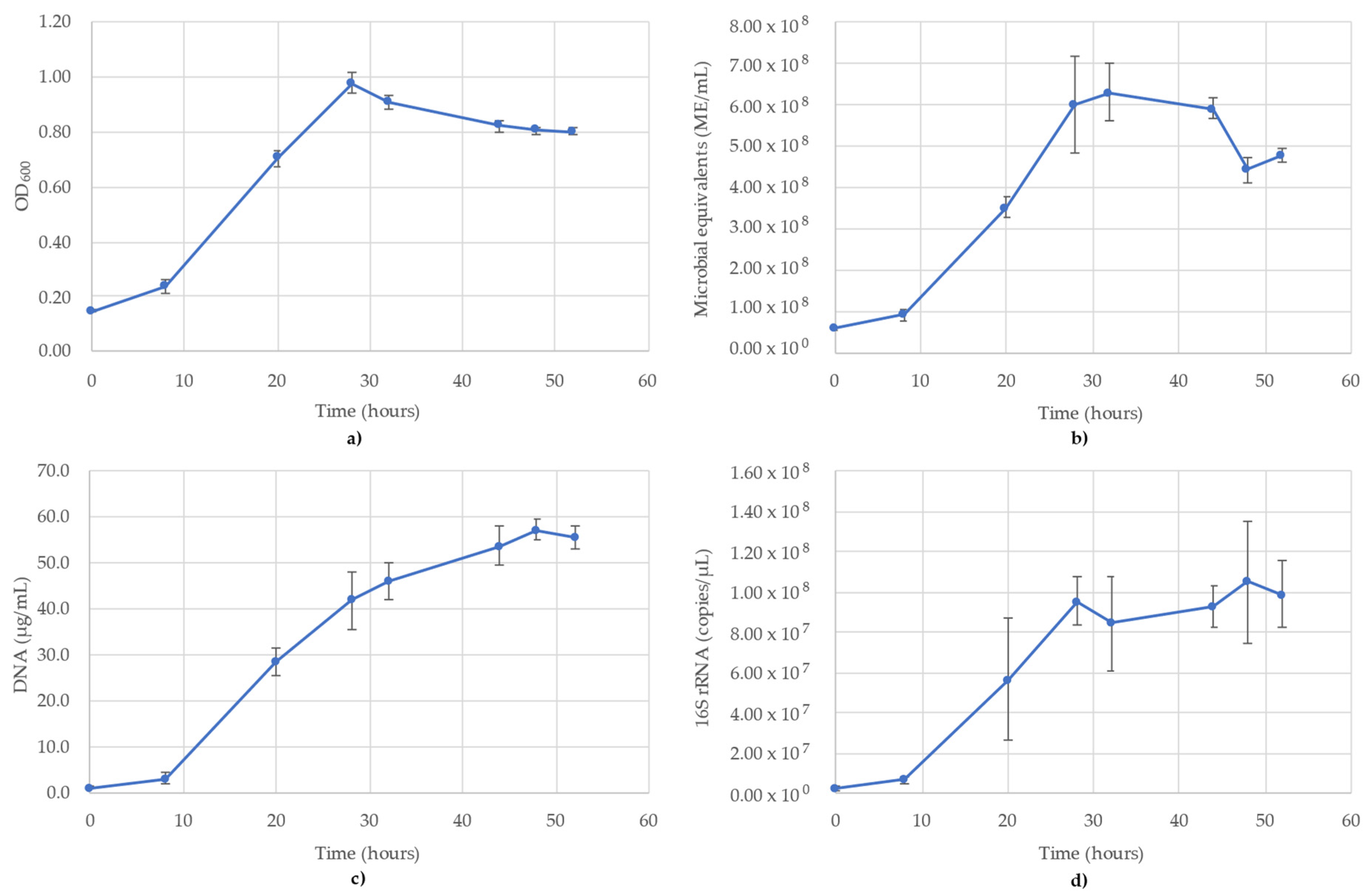
3.1. Acetobacterium Woodii Monitoring
3.2. Bacillus Subtilis Monitoring
3.3. Desulfovibrio Vulgaris Monitoring
3.4. Geoalkalibacter Subterraneus Monitoring
3.5. Pseudomonas Putida Monitoring
3.6. Thauera Aromatica Monitoring
3.7. Comparison of Cell Count Equivalents
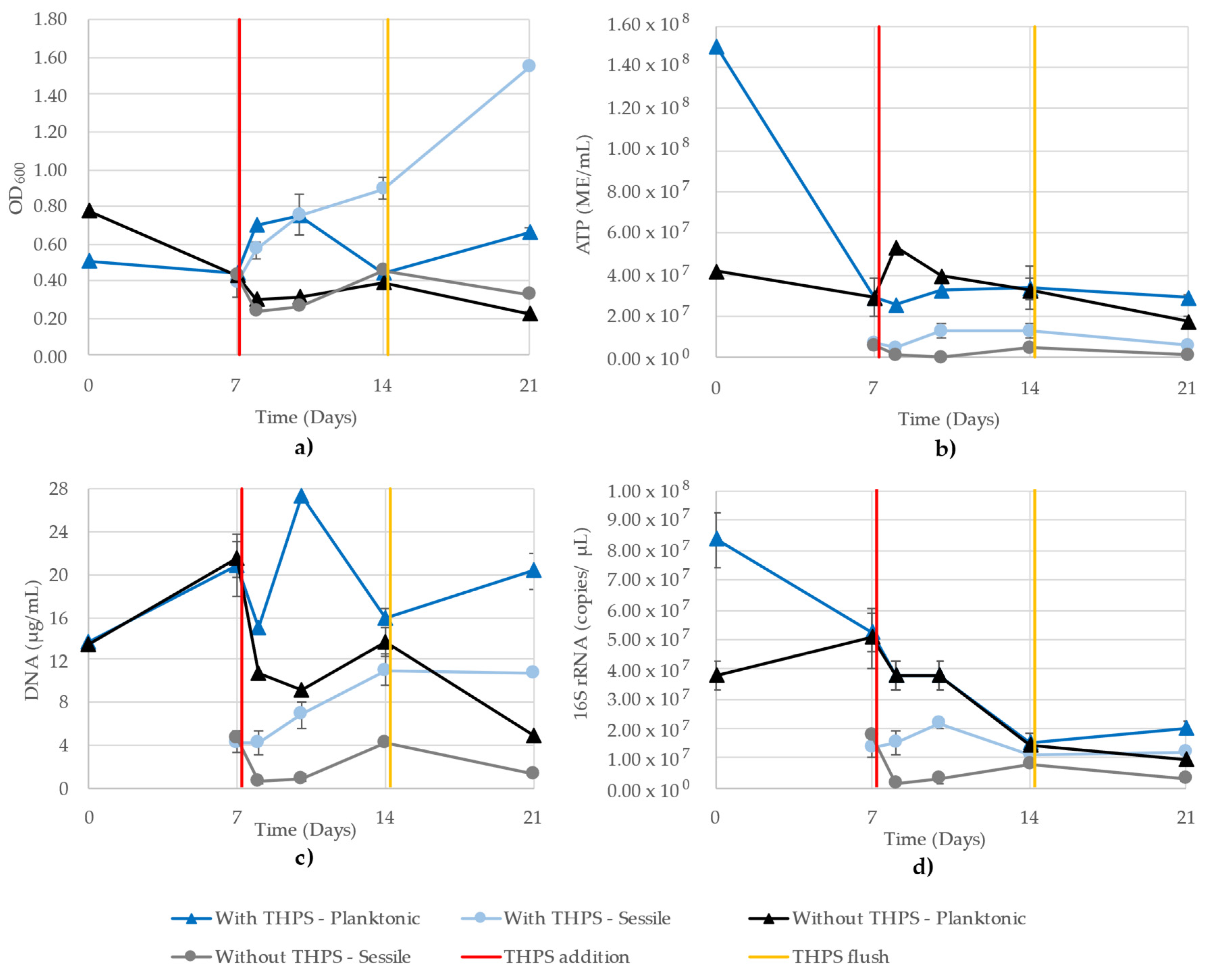
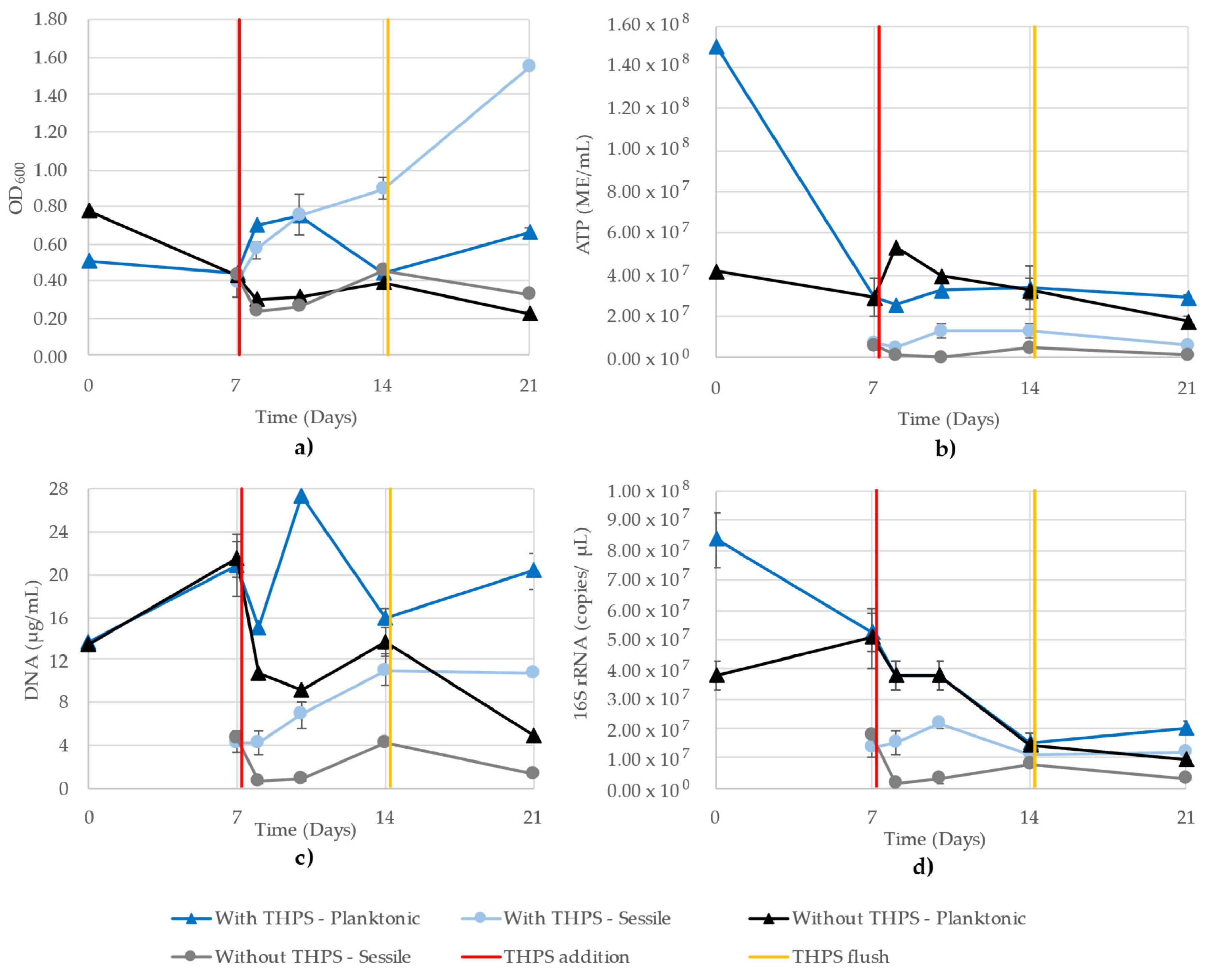
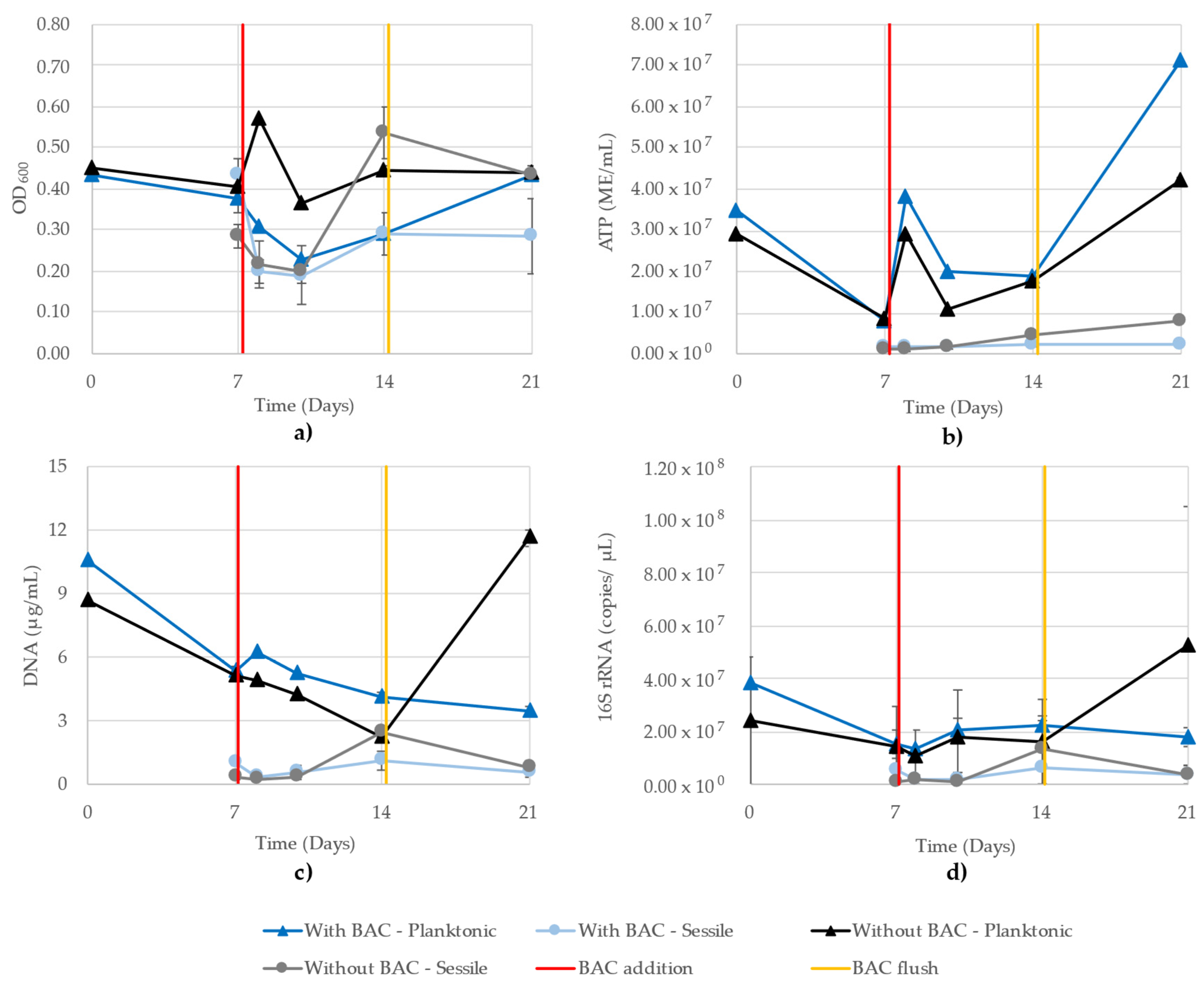
3.8. Mixed-Community Monitoring
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Madigan, M.T.; Bender, K.S.; Buckley, D.H.; Sattley, W.M.; Stahl, D.A. Brock Biology of Microorganisms, 15th ed.; Pearson Education: Harlow, UK, 2019. [Google Scholar]
- Colwell, R.R.; Brayton, P.R.; Grimes, D.J.; Roszak, D.B.; Huq, S.A.; Palmer, L.M. Viable but Non-Culturable Vibrio Cholerae and Related Pathogens in the Environment: Implications for Release of Genetically Engineered Microorganisms. Nat. Biotechnol. 1985, 3, 817–820. [Google Scholar] [CrossRef]
- Byrd, J.J.; Xu, H.S.; Colwell, R.R. Viable but Nonculturable Bacteria in Drinking Water. Appl. Environ. Microbiol. 1991, 57, 875–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Roberts, N.; Singleton, F.; Attwell, R.; Grimes, D.; Colwell, R. Survival and Viability of Nonculturable Escherichia coli and Vibrio cholerae in the Estuarine and Marine Environment. Microb. Ecol. 1982, 8, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Snellen, J.E.; Marr, A.G.; Starr, M.P. A Membrane Filter Direct Count Technique for Enumerating Bdellovibrio. Curr. Microbiol. 1978, 1, 117–122. [Google Scholar] [CrossRef]
- Duncan, C.L. Time of Enterotoxin Formation and Release during Sporulation of Clostridium Perfringens Type A. J. Bacteriol. 1973, 113, 932–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, J.A.; Curtis, B.S.; Curtis, W.R. Improving Accuracy of Cell and Chromophore Concentration Measurements Using Optical Density. BMC Biophys. 2013, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Bracho, M.; Araujo, I.; de Romero, M.; Ocando, L.; García, M.; Sarró, M.I.; le Gall, S.L.B. Comparison Of Bacterial Growth Of Sulfate Reducing Bacteria Evaluated By Serial Dilution, Pure Plate And Epifluorescence Techniques. In CORROSION 2007; OnePetro: Nashville, TN, USA, 2007; p. NACE-07530. [Google Scholar]
- Razban, B.; Nelson, K.Y.; Cullimore, D.R.; Cullimore, J.; McMartin, D.W. Quantitative Bacteriological Assessment of Aerobic Wastewater Treatment Quality and Plant Performance. J. Environ. Sci. Health Part A 2012, 47, 727–733. [Google Scholar] [CrossRef]
- Sharpe, A.N.; Michaud, G.L. Enumeration of High Numbers of Bacteria Using Hydrophobic Grid-Membrane Filters. Appl. Microbiol. 1975, 30, 519–524. [Google Scholar] [CrossRef]
- Tawakoli, P.N.; Al-Ahmad, A.; Hoth-Hannig, W.; Hannig, M.; Hannig, C. Comparison of Different Live/Dead Stainings for Detection and Quantification of Adherent Microorganisms in the Initial Oral Biofilm. Clin. Oral Investig. 2012, 17, 841–850. [Google Scholar] [CrossRef]
- Hannig, C.; Hannig, M.; Rehmer, O.; Braun, G.; Hellwig, E.; Al-Ahmad, A. Fluorescence Microscopic Visualization and Quantification of Initial Bacterial Colonization on Enamel in Situ. Arch. Oral Biol. 2007, 52, 1048–1056. [Google Scholar] [CrossRef]
- Xu, Z.; Liang, Y.; Lin, S.; Chen, D.; Li, B.; Li, L.; Deng, Y. Crystal Violet and XTT Assays on Staphylococcus Aureus Biofilm Quantification. Curr. Microbiol. 2016, 73, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Ommen, P.; Zobek, N.; Meyer, R.L. Quantification of Biofilm Biomass by Staining: Non-Toxic Safranin Can Replace the Popular Crystal Violet. J. Microbiol. Methods 2017, 141, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, D.; Wiedmann, M.; McLandsborough, L.A. Microtiter Plate Assay for Assessment of Listeria Monocytogenes Biofilm Formation. Appl. Environ. Microbiol. 2002, 68, 2950–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhupathiraju, V.K.; Hernandez, M.; Krauter, P.; Alvarez-Cohen, L. A New Direct Microscopy Based Method for Evaluating In-Situ Bioremediation. J. Hazard. Mater. 1999, 67, 299–312. [Google Scholar] [CrossRef]
- Ou, F.; McGoverin, C.; Swift, S.; Vanholsbeeck, F. Absolute Bacterial Cell Enumeration Using Flow Cytometry. J. Appl. Microbiol. 2017, 123, 464–477. [Google Scholar] [CrossRef] [Green Version]
- Pinder, A.C.; Purdy, P.W.; Poulter, S.A.G.; Clark, D.C. Validation of Flow Cytometry for Rapid Enumeration of Bacterial Concentrations in Pure Cultures. J. Appl. Bacteriol. 1990, 69, 92–100. [Google Scholar] [CrossRef]
- Holm, C.; Mathiasen, T.; Jespersen, L. A Flow Cytometric Technique for Quantification and Differentiation of Bacteria in Bulk Tank Milk. J. Appl. Microbiol. 2004, 97, 935–941. [Google Scholar] [CrossRef]
- Yang, S.Y.; Hsiung, S.K.; Hung, Y.C.; Chang, C.M.; Liao, T.L.; Lee, G.B. A Cell Counting/Sorting System Incorporated with a Microfabricated Flow Cytometer Chip. Meas. Sci. Technol. 2006, 17, 2001. [Google Scholar] [CrossRef]
- Harmsen, H.J.M.; Wildeboer-Veloo, A.C.M.; Grijpstra, J.; Knol, J.; Degener, J.E.; Welling, G.W. Development of 16S RRNA-Based Probes for the Coriobacterium Group and the Atopobium Cluster and Their Application for Enumeration of Coriobacteriaceae in Human Feces from Volunteers of Different Age Groups. Appl. Environ. Microbiol. 2000, 66, 4523–4527. [Google Scholar] [CrossRef] [Green Version]
- Langendijk, P.S.; Schut, F.; Jansen, G.J.; Raangs, G.C.; Kamphuis, G.R.; Wilkinson, M.H.F.; Welling, G.W. Quantitative Fluorescence in Situ Hybridization of Bifidobacterium spp. with Genus-Specific 16S RRNA-Targeted Probes and Its Application in Fecal Samples. Appl. Environ. Microbiol. 1995, 61, 3069–3075. [Google Scholar] [CrossRef] [Green Version]
- Jian, C.; Luukkonen, P.; Yki-Järvinen, H.; Salonen, A.; Korpela, K. Quantitative PCR Provides a Simple and Accessible Method for Quantitative Microbiota Profiling. PLoS ONE 2020, 15, e0227285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.J.; Nedwell, D.B.; Dong, L.F.; Osborn, A.M. Evaluation of Quantitative Polymerase Chain Reaction-Based Approaches for Determining Gene Copy and Gene Transcript Numbers in Environmental Samples. Environ. Microbiol. 2006, 8, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.T.; Taylor, L.T.; DeLong, E.F. Quantitative Analysis of Small-Subunit RRNA Genes in Mixed Microbial Populations via 5’-Nuclease Assays. Appl. Environ. Microbiol. 2000, 66, 4605–4614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breznak, J.A.; Costilow, R.N. Physicochemical Factors in Growth. In Methods for General and Molecular Microbiology; ASM Press: Washington, DC, USA, 2014; pp. 309–329. [Google Scholar] [CrossRef]
- Hungate, R.E. Chapter IV A Roll Tube Method for Cultivation of Strict Anaerobes. Methods Microbiol. 1969, 3, 117–132. [Google Scholar] [CrossRef]
- Hungate, R.E. The Anaerobic Mesophilic Cellulolytic Bacteria. Bacteriol. Rev. 1950, 14, 1–49. [Google Scholar] [CrossRef]
- Balch, W.; Wolfe, R. New Approach to the Cultivation of Methanogenic Bacteria: 2-Mercaptoethanesulfonic Acid (HS-CoM)-Dependent Growth of Methanobacterium Ruminantium in a Pressureized Atmosphere. Appl. Environ. Microbiol. 1976, 32, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Chan, E.C.S.; Siboo, R.; Touyz, L.Z.G.; Qiu, Y.-S.; Klitorinos, A. A Successful Method for Quantifying Viable Oral Anaerobic Spirochetes. Oral Microbiol. Immunol. 1993, 8, 80–83. [Google Scholar] [CrossRef]
- Martins, C.H.G.; Carvalho, T.C.; Souza, M.G.M.; Ravagnani, C.; Peitl, O.; Zanotto, E.D.; Panzeri, H.; Casemiro, L.A. Assessment of Antimicrobial Effect of Biosilicate® against Anaerobic, Microaerophilic and Facultative Anaerobic Microorganisms. J. Mater. Sci. Mater. Med. 2011, 22, 1439–1446. [Google Scholar] [CrossRef]
- Salanitro, J.P.; Fairchilds, I.G.; Zgornicki, Y.D. Isolation, Culture Characteristics, and Identification of Anaerobic Bacteria from the Chicken Cecum. Appl. Microbiol. 1974, 27, 678–687. [Google Scholar] [CrossRef]
- Montero, B.; Garcia-Morales, J.L.; Sales, D.; Solera, R. Evolution of Microorganisms in Thermophilic-Dry Anaerobic Digestion. Bioresour. Technol. 2008, 99, 3233–3243. [Google Scholar] [CrossRef]
- Himmelberg, A.M.; Brüls, T.; Farmani, Z.; Weyrauch, P.; Barthel, G.; Schrader, W.; Meckenstock, R.U. Anaerobic Degradation of Phenanthrene by a Sulfate-Reducing Enrichment Culture. Environ. Microbiol. 2018, 20, 3589–3600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pronk, J.T.; de Bruyn, J.C.; Bos, P.; Kuenen, J.G. Anaerobic Growth of Thiobacillus ferrooxidans. Appl. Environ. Microbiol. 1992, 58, 2227–2230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauerhofer, L.M.; Pappenreiter, P.; Paulik, C.; Seifert, A.H.; Bernacchi, S.; Rittmann, S.K.M.R. Methods for Quantification of Growth and Productivity in Anaerobic Microbiology and Biotechnology. Folia Microbiol. 2019, 64, 321–360. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Padrón, E.; Bordenave, S.; Lin, S.; Bhaskar, I.M.; Dong, X.; Sensen, C.W.; Fournier, J.; Voordouw, G.; Gieg, L.M. Carbon and Sulfur Cycling by Microbial Communities in a Gypsum-Treated Oil Sands Tailings Pond. Environ. Sci. Technol. 2010, 45, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Stams, A.J.M.; Sousa, D.Z.; Kleerebezem, R.; Plugge, C.M. Role of Syntrophic Microbial Communities in High-Rate Methanogenic Bioreactors. Water Sci. Technol. 2012, 66, 352–362. [Google Scholar] [CrossRef]
- Monod, J. The Growth of Bacterial Cultures. Ann. Rev. Microbiol. 1949, 3, 371–394. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.; Marcotte, E.M. Insights into the Regulation of Protein Abundance from Proteomic and Transcriptomic Analyses. Nat. Rev. Genet. 2012, 13, 227. [Google Scholar] [CrossRef]
- Frostegård, Å.; Tunlid, A.; Bååth, E. Microbial Biomass Measured as Total Lipid Phosphate in Soils of Different Organic Content. J. Microbiol. Methods 1991, 14, 151–163. [Google Scholar] [CrossRef]
- Nguyen, T.D.P.; Nguyen, D.H.; Lim, J.W.; Chang, C.-K.; Leong, H.Y.; Tran, T.N.T.; Vu, T.B.H.; Nguyen, T.T.C.; Show, P.L. Investigation of the Relationship between Bacteria Growth and Lipid Production Cultivating of Microalgae Chlorella Vulgaris in Seafood Wastewater. Energies 2019, 12, 2282. [Google Scholar] [CrossRef] [Green Version]
- Neubauer, C.; Kasi, A.S.; Grahl, N.; Sessions, A.L.; Kopf, S.H.; Kato, R.; Hogan, D.A.; Newman, D.K. Refining the Application of Microbial Lipids as Tracers of Staphylococcus Aureus Growth Rates in Cystic Fibrosis Sputum. J. Bacteriol. 2018, 200. [Google Scholar] [CrossRef] [Green Version]
- Hattori, N.; Sakakibara, T.; Kajiyama, N.; Igarashi, T.; Maeda, M.; Murakami, S. Enhanced Microbial Biomass Assay Using Mutant Luciferase Resistant to Benzalkonium Chloride. Anal. Biochem. 2003, 319, 287–295. [Google Scholar] [CrossRef]
- Schneider, D.A.; Gourse, R.L. Relationship between Growth Rate and ATP Concentration in Escherichia coli: A Bioassay for Available Cellular ATP. J. Biol. Chem. 2004, 279, 8262–8268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soini, J.; Falschlehner, C.; Mayer, C.; Böhm, D.; Weinel, S.; Panula, J.; Vasala, A.; Neubauer, P. Transient Increase of ATP as a Response to Temperature Up-Shift in Escherichia coli. Microb. Cell Factories 2005, 4, 9. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Ye, L.; Tong, A.H.Y.; Lok, S.; Zhang, T. Biased Diversity Metrics Revealed by Bacterial 16S Pyrotags Derived from Different Primer Sets. PLoS ONE 2013, 8, e53649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.W.; Martin, J.C.; Scott, P.; Parkhill, J.; Flint, H.J.; Scott, K.P. 16S RRNA Gene-Based Profiling of the Human Infant Gut Microbiota Is Strongly Influenced by Sample Processing and PCR Primer Choice. Microbiome 2015, 3, 26. [Google Scholar] [CrossRef] [Green Version]
- Baker, G.C.; Smith, J.J.; Cowan, D.A. Review and Re-Analysis of Domain-Specific 16S Primers. J. Microbiol. Methods 2003, 55, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Edgcomb, V.; McDonald, J.; Devereux, R.; Smith, D. Estimation of Bacterial Cell Numbers in Humic Acid-Rich Salt Marsh Sediments with Probes Directed to 16S Ribosomal DNA. Appl. Environ. Microbiol. 1999, 65, 1516–1523. [Google Scholar] [CrossRef] [Green Version]
- Müller, A.L.; Kjeldsen, K.U.; Rattei, T.; Pester, M.; Loy, A. Phylogenetic and Environmental Diversity of DsrAB-Type Dissimilatory (Bi)Sulfite Reductases. ISME J. 2015, 9, 1152–1165. [Google Scholar] [CrossRef] [Green Version]
- Grüntzig, V.; Nold, S.C.; Zhou, J.; Tiedje, J.M. Pseudomonas Stutzeri Nitrite Reductase Gene Abundance in Environmental Samples Measured by Real-Time PCR. Appl. Environ. Microbiol. 2001, 67, 760–768. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, B.R.; Nakatsu, C.H.; Nies, L. Detection and Enumeration of Aromatic Oxygenase Genes by Multiplex and Real-Time PCR. Appl. Environ. Microbiol. 2003, 69, 3350–3358. [Google Scholar] [CrossRef] [Green Version]
- McKew, B.A.; Coulon, F.; Yakimov, M.M.; Denaro, R.; Genovese, M.; Smith, C.J.; Osborn, A.M.; Timmis, K.N.; McGenity, T.J. Efficacy of Intervention Strategies for Bioremediation of Crude Oil in Marine Systems and Effects on Indigenous Hydrocarbonoclastic Bacteria. Environ. Microbiol. 2007, 9, 1562–1571. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.L.; Jiang, X.; Wu, Q.L.; Zhou, N.Y. Intragenomic Heterogeneity of 16S RRNA Genes Causes Overestimation of Prokaryotic Diversity. Appl. Environ. Microbiol. 2013, 79, 5962–5969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuks, G.; Elgart, M.; Amir, A.; Zeisel, A.; Turnbaugh, P.J.; Soen, Y.; Shental, N. Combining 16S RRNA Gene Variable Regions Enables High-Resolution Microbial Community Profiling. Microbiome 2018, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampelotto, P.H.; Sereia, A.F.R.; de Oliveira, L.F.V.; Margis, R. Exploring the Hospital Microbiome by High-Resolution 16S RRNA Profiling. Int. J. Mol. Sci. 2019, 20, 3099. [Google Scholar] [CrossRef] [Green Version]
- Teng, F.; Darveekaran Nair, S.S.; Zhu, P.; Li, S.; Huang, S.; Li, X.; Xu, J.; Yang, F. Impact of DNA Extraction Method and Targeted 16S-RRNA Hypervariable Region on Oral Microbiota Profiling. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Douterelo, I.; Calero-Preciado, C.; Soria-Carrasco, V.; Boxall, J.B. Whole Metagenome Sequencing of Chlorinated Drinking Water Distribution Systems. Environ. Sci. Water Res. Technol. 2018, 4, 2080–2091. [Google Scholar] [CrossRef] [Green Version]
- Dada, N.; Sheth, M.; Liebman, K.; Pinto, J.; Lenhart, A. Whole Metagenome Sequencing Reveals Links between Mosquito Microbiota and Insecticide Resistance in Malaria Vectors. Sci. Rep. 2018, 8, 2084. [Google Scholar] [CrossRef]
- Al-Hebshi, N.N.; Baraniya, D.; Chen, T.; Hill, J.; Puri, S.; Tellez, M.; Hassan, N.A.; Colwell, R.R.; Ismail, A. Metagenome Sequencing-Based Strain-Level and Functional Characterization of Supragingival Microbiome Associated with Dental Caries in Children. J. Oral Microbiol. 2018, 11, 1557986. [Google Scholar] [CrossRef]
- Jamieson, R.; Gordon, R.; Sharples, K.; Stratton, G.; Madani, A. Movement and Persistence of Fecal Bacteria in Agricultural Soils and Subsurface Drainage Water: A Review. Can. Biosyst. Eng. 2002, 44. [Google Scholar]
- Mathew, R.A.; Abraham, M. Bioremediation of Diesel Oil in Marine Environment. Oil Gas Sci. Technol. 2020, 75, 60. [Google Scholar] [CrossRef]
- Brukner, I.; Longtin, Y.; Oughton, M.; Forgetta, V.; Dascal, A. Assay for Estimating Total Bacterial Load: Relative QPCR Normalisation of Bacterial Load with Associated Clinical Implications. Diagn. Microbiol. Infect. Dis. 2015, 83, 1–6. [Google Scholar] [CrossRef]
- Manti, A.; Boi, P.; Falcioni, T.; Canonico, B.; Ventura, A.; Sisti, D.; Pianetti, A.; Balsamo, M.; Papa, S. Bacterial Cell Monitoring in Wastewater Treatment Plants by Flow Cytometry. Water Environ. Res. 2008, 80, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Maloney, L.C.; Nelson, Y.M.; Kitts, C.L. Characterization of Aerobic and Anaerobic Microbial Activity in Hydrocarbon-Contaminated Soil. In Proceedings of the Fourth International Conference on Remediation of Chlorinated and Recalcitrant Compounds, Monterey, CA, USA, 1 May 2004. [Google Scholar]
- Campana, R.; Scesa, C.; Patrone, V.; Vittoria, E.; Baffone, W. Microbiological Study of Cosmetic Products during Their Use by Consumers: Health Risk and Efficacy of Preservative Systems. Lett. Appl. Microbiol. 2006, 43, 301–306. [Google Scholar] [CrossRef]
- Pezzuto, A.; Losasso, C.; Mancin, M.; Gallocchio, F.; Piovesana, A.; Binato, G.; Gallina, A.; Marangon, A.; Mioni, R.; Favretti, M.; et al. Food Safety Concerns Deriving from the Use of Silver Based Food Packaging Materials. Front. Microbiol. 2015, 6, 1109. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global Patterns of 16S RRNA Diversity at a Depth of Millions of Sequences per Sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Begot, C.; Desnier, I.; Daudin, J.D.; Labadie, J.C.; Lebert, A. Recommendations for Calculating Growth Parameters by Optical Density Measurements. J. Microbiol. Methods 1996, 25, 225–232. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Guo, J.; Yu, Y.; Li, J.; Guo, Y.; Liu, C. Comparing Two Functions for Optical Density and Cell Numbers in Bacterial Exponential Growth Phase. J. Pure Appl. Microbiol. 2015, 9, 299–305. [Google Scholar]
- Beal, J.; Farny, N.G.; Haddock-Angelli, T.; Selvarajah, V.; Baldwin, G.S.; Buckley-Taylor, R.; Gershater, M.; Kiga, D.; Marken, J.; Sanchania, V.; et al. Robust Estimation of Bacterial Cell Count from Optical Density. Commun. Biol. 2020, 3, 512. [Google Scholar] [CrossRef]
- Kim, D.; Chung, S.; Lee, S.; Choi, J. Relation of Microbial Biomass to Counting Units for Pseudomonas Aeruginosa. Afr. J. Microbiol. Res. 2012, 6, 4620–4622. [Google Scholar] [CrossRef]
- Bakken, L.R.; Olsen, R.A. DNA-Content of Soil Bacteria of Different Cell Size. Soil Biol. Biochem. 1989, 21, 789–793. [Google Scholar] [CrossRef]
- Berlanga, M.; Guerrero, R. Living Together in Biofilms: The Microbial Cell Factory and Its Biotechnological Implications. Microb. Cell Factories 2016, 15, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolter, R. Biofilms in Lab and Nature: A Molecular Geneticist’s Voyage to Microbial Ecology. Int. Microbiol. 2010, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hobley, L.; Harkins, C.; MacPhee, C.E.; Stanley-Wall, N.R. Giving Structure to the Biofilm Matrix: An Overview of Individual Strategies and Emerging Common Themes. FEMS Microbiol. Rev. 2015, 39, 649–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Advantages | Disadvantages/Limitations | Relative Cost a |
|---|---|---|---|
| Culturing | -Easy to quantify -Easy to determine presence on contamination | -Unable to quantify complex communities -Very limited number of species can be grown -Time required depends on species’ doubling time | $10 |
| Optical density | -Fast -Sample is recoverable | -True quantification is limited by transmission -Counts should be verified by another method -Precipitants in liquid interfere | $1 |
| Staining and microscopy | -Combining stains allows differential counting | -Biased by field of view in microscope -Dense growth prevents accurate counting | $100 |
| ATP assay | -Rapid test able to be used in lab or in the field -Measures ATP from all living cells | -One-time equipment is expensive, reagents are consumable and expensive -Microbial equivalent calculations do not provide true cell count | $1000 |
| DNA concentration | -DNA can be used for downstream applications -Once extracted, DNA is stable for long time frames, allowing flexibility in measuring | -Minimum volume/cell mass of sample required -Variance in DNA concentrations between species skews cell count conversions -DNA must be extracted then measured | $100 |
| Flow cytometry | -Very accurate enumeration -Combining different stains enables counting of multiple types of cells | -Requires expensive equipment -Not applicable to environmental samples | $1000 |
| Quantitative PCR | -Enumeration can be as broad or specific as desired based on primers used -Multiplexing can count multiple targets in single runs | -Expensive equipment and consumable reagents -Technically more challenging than other methods | $1000 |
| Metagenomic sequencing | -Detects all species present in complex environments | -Provides relative cell counts, not true cell counts -Equipment is very expensive | $1000–10,000 b |
| Species and Strain | NCBI Accession Number | Genome Length (bp) | 16S rRNA Copy Number * |
|---|---|---|---|
| Acetobacterium woodii DSM 1030 | CP002987.1 | 4,044,777 | 5 |
| Bacillus subtilis subtilis 168 | CP010052 | 4,215,619 | 10 |
| Desulfovibrio vulgaris Miyazaki F | CP001197.1 | 4,040,304 | 4 |
| Geoalkalibacter subterraneus Red1 | CP010311 | 3,475,523 | 4 |
| Pseudomonas putida KT2440 | AE015451.2 | 6,181,873 | 7 |
| Thauera aromatica MZ1T | CP001281.2 | 4,496,212 | 4 |
| Species and Strain | Medium | Temperature (°C) |
|---|---|---|
| Acetobacterium woodii DSM 1030 | DSM 135 | 30 |
| Bacillus subtilis subtilis 168 | DSM 1 | 30 |
| Desulfovibrio vulgaris Miyazaki F | DSM 63 | 37 |
| Geoalkalibacter subterraneus Red1 | DSM 1249 | 37 |
| Pseudomonas putida KT2440 | DSM 1a | 26 |
| Thauera aromatica MZ1T | DSM 586 | 30 |
| Primer Name | Sequence (5′-3′) | Melting Temperature (°C) |
|---|---|---|
| 519_F | CAG CMG CCG CGG TAA | 57.6 |
| 806_R | GGA CTA CHV GGG TWT CTA AT | 50.7 |
| gBlock DNA Sequence (5′-3′) (Multidrug Resistance Efflux Pump Gene A–(Thymine Spacer)–Universal 16S rRNA–(Thymine Spacer)–Multidrug Resistance Efflux Pump Gene A) |
|---|
| Multidrug resistance gene A amplicon TTTTTTTTTTT GTG CCA GCA GCC GCG GTA ATA CAG AGG GTG CAA GCG TTA ATC GGA ATT ACT GGG CGT AAA GCG CGC GTA GGT GGT TTG TTA AGT TGG ATG TGA AAG CCC CGG GCT CAA CCT GGG AAC TGC ATC CAA AAC TGG CAA GCT AGA GTA CGG TAG AGG GTG GTG GAA TTT CCT GTG TAG CGG TGA AAT GCG TAG ATA TAG GAA GGA ACA CCA GTG GCG AAG GCG ACC ACC TGG ACT GAT ACT GAC ACT GAG GTG CGA AAG CGT GGG GAG CAA ACA GGA TTA GAT ACC CTG GTA GTC C TTTTTTTTTT Multidrug resistance gene B amplicon |
| Species | OD600 vs. ATP | OD600 vs. DNA | OD600 vs. 16S | ATP vs. DNA | ATP vs. 16S | DNA vs. 16S |
|---|---|---|---|---|---|---|
| A. woodii | 0.97 | 0.96 | 0.98 | 0.92 | 0.93 | 1.00 |
| B. subtilis | 0.89 | 0.84 | 0.86 | 0.64 | 0.66 | 0.98 |
| D. vulgaris | 0.65 | 0.93 | 0.74 | 0.74 | 0.50 | 0.62 |
| G. subterraneus | 0.24 | 0.99 | 0.98 | 0.16 | 0.15 | 1.00 |
| P. putida | 0.98 | 0.99 | 0.71 | 0.99 | 0.73 | 0.77 |
| T. aromatica | 0.97 | 0.90 | 0.95 | 0.89 | 0.92 | 0.98 |
| Average | 0.78 | 0.94 | 0.87 | 0.72 | 0.65 | 0.89 |
| Sample | OD600 vs. ATP | OD600 vs. DNA | OD600 vs. 16S | ATP vs. DNA | ATP vs. 16S | DNA vs. 16S |
|---|---|---|---|---|---|---|
| Planktonic growth with THPS | 0.27 | 0.45 | 0.20 | 0.48 | 0.82 | 0.27 |
| Planktonic growth without THPS | 0.27 | 0.45 | 0.38 | 0.11 | 0.50 | 0.66 |
| Sessile growth with THPS | 0.04 | 0.83 | 0.27 | 0.40 | 0.33 | 0.39 |
| Sessile growth without THPS | 0.93 | 0.96 | 0.80 | 0.96 | 0.88 | 0.91 |
| Average | 0.38 | 0.67 | 0.41 | 0.49 | 0.63 | 0.56 |
| Sample | OD600 vs. ATP | OD600 vs. DNA | OD600 vs. 16S | ATP vs. DNA | ATP vs. 16S | DNA vs. 16S |
|---|---|---|---|---|---|---|
| Planktonic growth with BAC | 0.52 | 0.33 | 0.36 | 0.12 | 0.02 | 0.76 |
| Planktonic growth without BAC | 0.51 | 0.03 | 0.17 | 0.79 | 0.73 | 0.86 |
| Sessile growth with BAC | 0.20 | 0.70 | 0.77 | 0.34 | 0.51 | 0.96 |
| Sessile growth without BAC | 0.77 | 0.89 | 0.88 | 0.48 | 0.46 | 0.76 |
| Average | 0.50 | 0.49 | 0.55 | 0.43 | 0.43 | 0.84 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, D.C.; Turner, R.J. Assessing Microbial Monitoring Methods for Challenging Environmental Strains and Cultures. Microbiol. Res. 2022, 13, 235-257. https://doi.org/10.3390/microbiolres13020020
Brown DC, Turner RJ. Assessing Microbial Monitoring Methods for Challenging Environmental Strains and Cultures. Microbiology Research. 2022; 13(2):235-257. https://doi.org/10.3390/microbiolres13020020
Chicago/Turabian StyleBrown, Damon C., and Raymond J. Turner. 2022. "Assessing Microbial Monitoring Methods for Challenging Environmental Strains and Cultures" Microbiology Research 13, no. 2: 235-257. https://doi.org/10.3390/microbiolres13020020
APA StyleBrown, D. C., & Turner, R. J. (2022). Assessing Microbial Monitoring Methods for Challenging Environmental Strains and Cultures. Microbiology Research, 13(2), 235-257. https://doi.org/10.3390/microbiolres13020020