
Diversity Profiling of Seed Associated Endophytic Microbiome in Important Species of Caricaceae Family


Abstract
:1. Introduction
2. Materials and Methods
2.1. Source of the Seeds
2.2. Isolation of DNA from the Seeds
2.3. Preparation of MiSeq Library in Illumina, Quality Check and Sequencing
2.4. Pipelines and Metagenomic Analysis
2.5. Fungal ITS Metagenomic Analysis
2.6. Data Availability and Graphs
3. Results
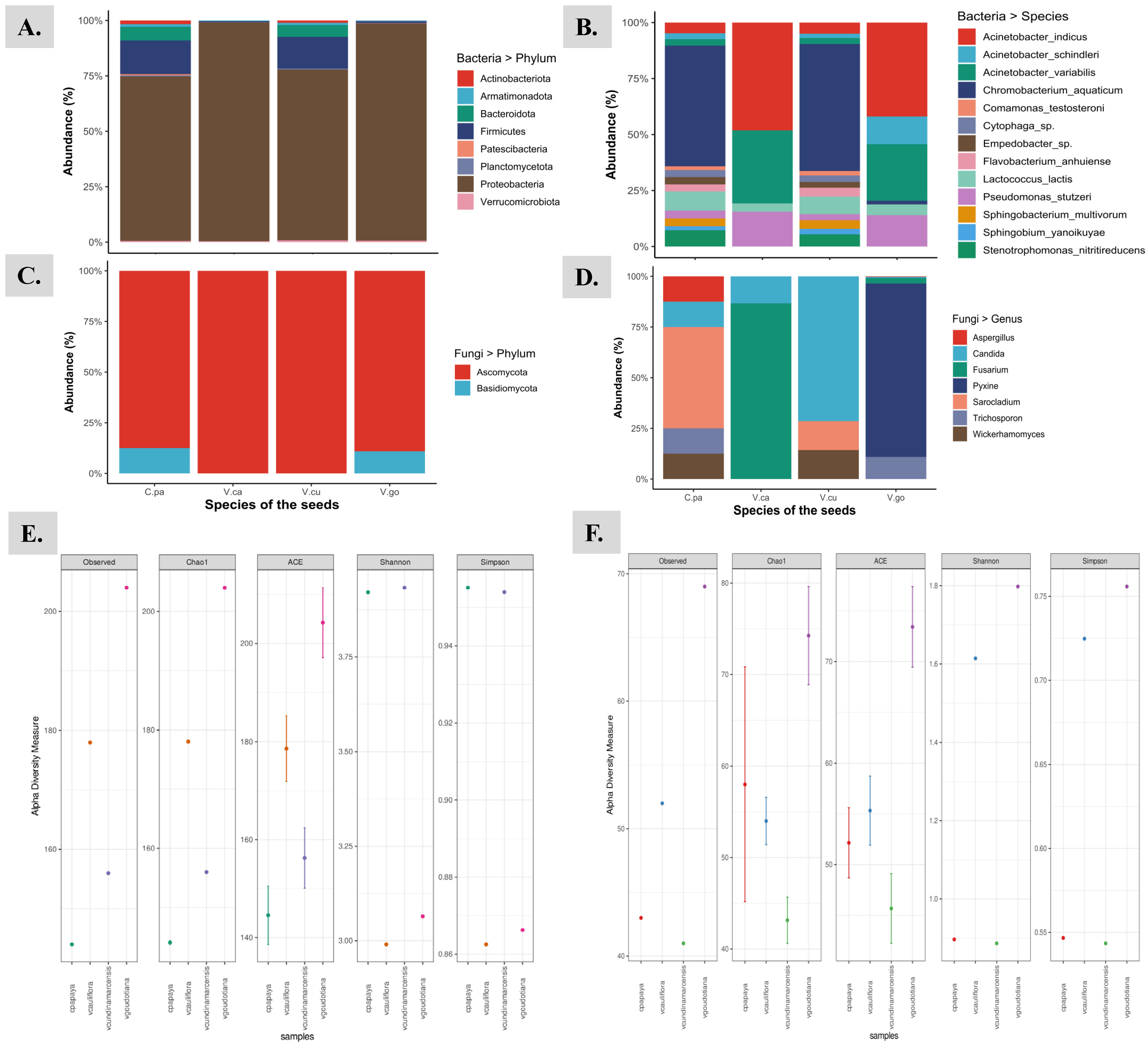
3.1. Structural Composition and Alpha Diversity of the Seed Associated Endophytic Microbes
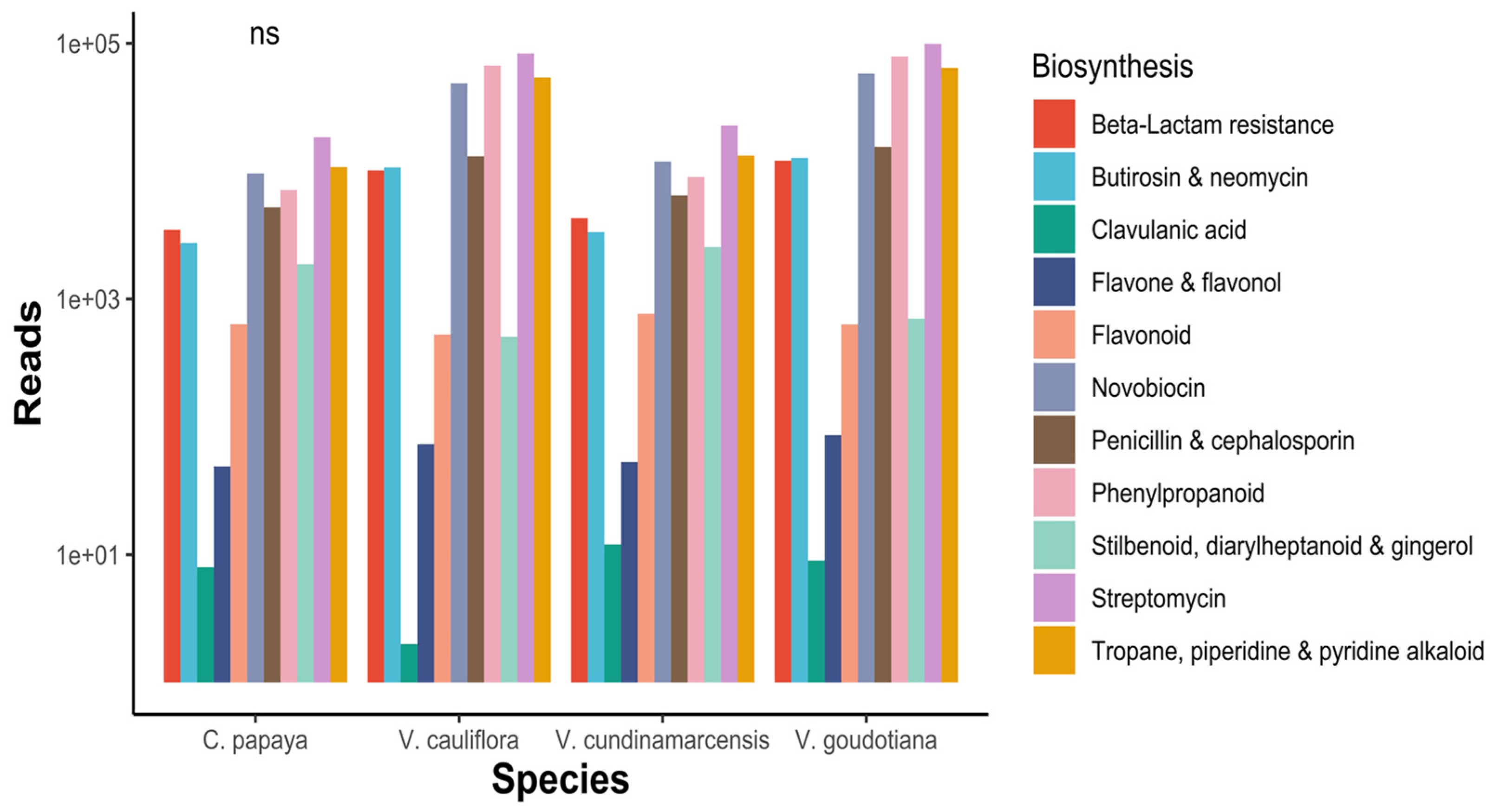
3.2. Functional Annotations
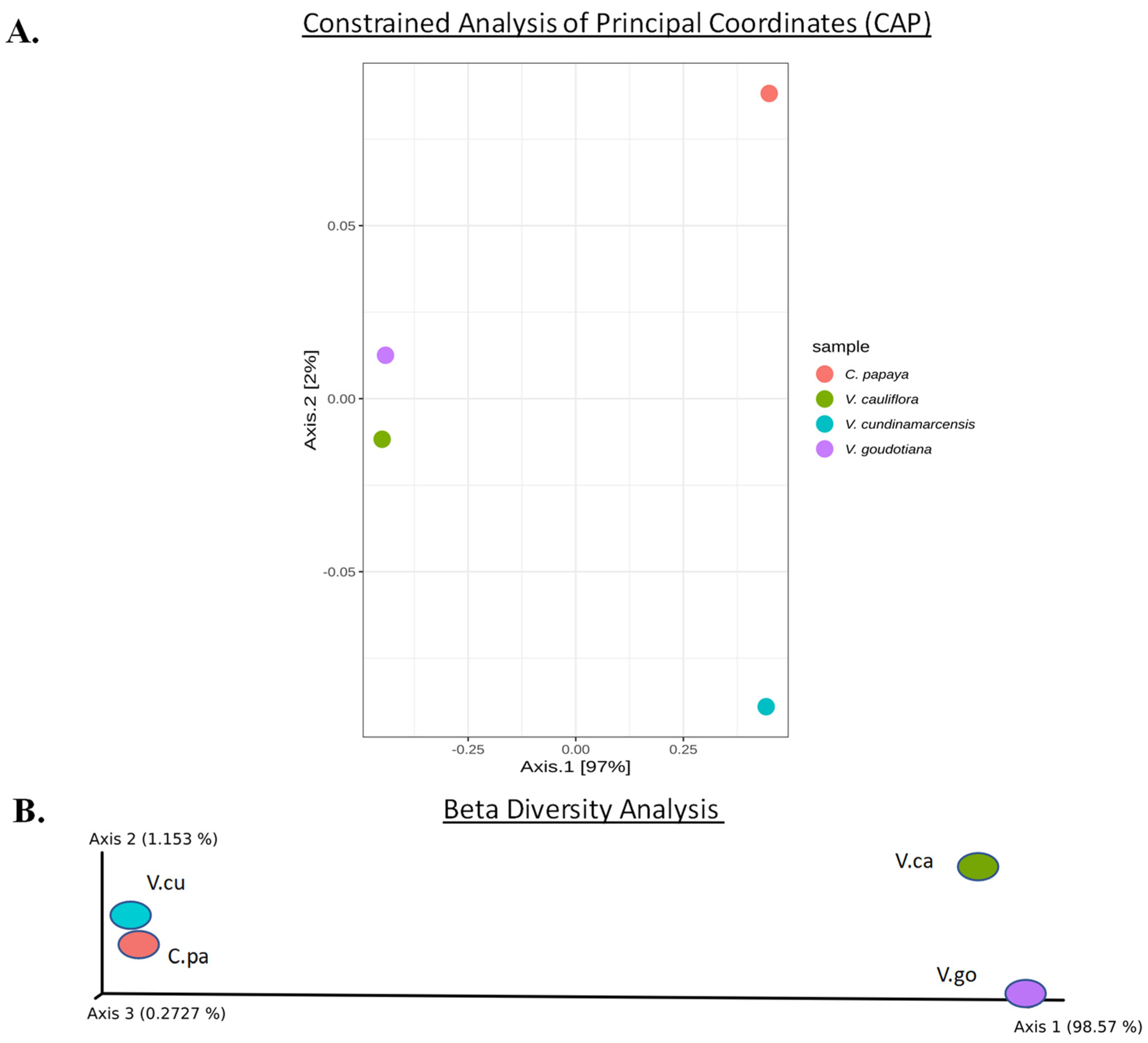
3.3. Statistical Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saran, P.L. Papaya; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Altendorf, S. Major Tropical Fruits Review 2017; FAO: Rome, Italy, 2019; Available online: http://www.fao.org/fileadmin/templates/est/COMM_MARKETS_MONITORING/Tropical_Fruits/Documents/CA2895EN.pdf (accessed on 29 July 2021).
- Mishra, R.; Gaur, R.K.; Patil, B.L. Current knowledge of viruses infecting papaya and their transgenic management. In Plant Viruses: Evolution and Management; Springer: Berlin/Heidelberg, Germany, 2016; pp. 189–203. [Google Scholar]
- Yamamoto, H.Y. Comparison of the Carotenoids in Yellow-and Red-Fleshed Carica papaya. Nature 1964, 201, 1049–1050. [Google Scholar] [CrossRef]
- Antunes Carvalho, F.; Renner, S.S. A Dated Phylogeny of the Papaya Family (Caricaceae) Reveals the Crop’s Closest Relatives and the Family’s Biogeographic History. Mol. Phylogenet. Evol. 2012, 65, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Liebman, B. Nutritional Aspects of Fruit. Nut Action Newslett. 1992, 1, 10–11. [Google Scholar]
- Chandrika, U.G.; Jansz, E.R.; Wickramasinghe, S.M.D.N.; Warnasuriya, N.D. Carotenoids in Yellow- and Red-fleshed Papaya (Carica papaya L.). J. Sci. Food Agric. 2003, 83, 1279–1282. [Google Scholar] [CrossRef]
- Jiménez-Coello, M.; Guzman-Marín, E.; Ortega-Pacheco, A.; Perez-Gutiérrez, S.; Acosta-Viana, K.Y. Assessment of the Anti-Protozoal Activity of Crude Carica papaya Seed Extract against Trypanosoma Cruzi. Molecules 2013, 18, 12621–12632. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Lucas, J.; Castañeda, D.; Hormigo, D. New Trends for a Classical Enzyme: Papain, a Biotechnological Success Story in the Food Industry. Trends Food Sci. Technol. 2017, 68, 91–101. [Google Scholar] [CrossRef]
- Scheldeman, X.; Willemen, L.; Coppens d’Eeckenbrugge, G.; Romeijn-Peeters, E.; Restrepo, M.T.; Romero Motoche, J.; Jiménez, D.; Lobo, M.; Medina, C.I.; Reyes, C.; et al. Distribution, diversity and environmental adaptation of highland papayas (Vasconcellea spp.) in tropical and subtropical America. In Plant Conservation and Biodiversity; Hawksworth, D.L., Bull, A.T., Eds.; Springer: Dordrecht, The Netherlands, 2007; pp. 293–310. ISBN 978-1-4020-6444-9. [Google Scholar]
- Sawant, A.C. Crossing Relationships in the Genus Carica. Evolution 1958, 12, 263–266. [Google Scholar] [CrossRef]
- Dinesh, M.R.; Rekha, A.; Ravishankar, K.V.; Praveen, K.S.; Santosh, L.C. Breaking the Intergeneric Crossing Barrier in Papaya Using Sucrose Treatment. Sci. Hortic. 2007, 114, 33–36. [Google Scholar] [CrossRef]
- Siar, S.V.; Beligan, G.A.; Sajise, A.J.C.; Villegas, V.N.; Drew, R.A. Papaya Ringspot Virus Resistance in Carica Papaya via Introgression from Vasconcellea quercifolia. Euphytica 2011, 181, 159–168. [Google Scholar] [CrossRef]
- O’Brien, C.M.; Drew, R.A. Marker-Assisted Hybridisation and Backcrossing between Vasconcellea Species and Carica Papaya for PRSV-P Resistance. In Proceedings of the International Symposium on Molecular Markers in Horticulture 859, Corvallis, OR, USA, 25–27 September 2010. [Google Scholar]
- Gouda, S.; Das, G.; Sen, S.K.; Shin, H.S.; Patra, J.K. Endophytes: A Treasure House of Bioactive Compounds of Medicinal Importance. Front. Microbiol. 2016, 7, 1538. [Google Scholar] [CrossRef] [Green Version]
- Krings, M.; Taylor, T.N.; Hass, H.; Kerp, H.; Dotzler, N.; Hermsen, E.J. Fungal Endophytes in a 400-Million-Yr-Old Land Plant: Infection Pathways, Spatial Distribution, and Host Responses. New Phytol. 2007, 174, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Redecker, D.; Kodner, R.; Graham, L.E. Glomalean Fungi from the Ordovician. Science 2000, 289, 1920–1921. [Google Scholar] [CrossRef] [Green Version]
- Clay, K. Fungal Endophytes of Grasses: A Defensive Mutualism between Plants and Fungi. Ecology 1988, 69, 10–16. [Google Scholar] [CrossRef]
- Clay, K. Fungal Endophytes of Plants: Biological and Chemical Diversity. Nat. Toxins 1993, 1, 147–149. [Google Scholar] [CrossRef]
- Breen, J.P. Acremonium Endophyte Interactions with Enhanced Plant Resistance to Insects. Annu. Rev. Entomol. 1994, 39, 401–423. [Google Scholar] [CrossRef]
- Jaber, L.R.; Vidal, S. Fungal Endophyte Negative Effects on Herbivory Are Enhanced on Intact Plants and Maintained in a Subsequent Generation. Ecol. Entomol. 2010, 35, 25–36. [Google Scholar] [CrossRef]
- Leuchtmann, A.; Schmidt, D.; Bush, L.P. Different Levels of Protective Alkaloids in Grasses with Stroma-Forming and Seed-Transmitted Epichloe/Neotyphodium Endophytes. J. Chem. Ecol. 2000, 26, 1025–1036. [Google Scholar] [CrossRef]
- Faeth, S.H.; Saari, S. Fungal Grass Endophytes and Arthropod Communities: Lessons from Plant Defence Theory and Multitrophic Interactions. Fungal Ecol. 2012, 5, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Nair, D.N.; Padmavathy, S. Impact of Endophytic Microorganisms on Plants, Environment and Humans. Sci. World J. 2014, 2014, 250693. [Google Scholar] [CrossRef] [Green Version]
- Mendes, R.; Kruijt, M.; De Bruijn, I.; Dekkers, E.; Van Der Voort, M.; Schneider, J.H.M.; Piceno, Y.M.; DeSantis, T.Z.; Andersen, G.L.; Bakker, P.A.H.M.; et al. Deciphering the Rhizosphere Microbiome for Disease-Suppressive Bacteria. Science 2011, 332, 1097–1100. [Google Scholar] [CrossRef]
- Raaijmakers, J.M.; Weller, D.M. Natural Plant Protection by 2,4-Diacetylphloroglucinol-Producing Pseudomonas Spp. in Take-All Decline Soils. Mol. Plant-Microbe Interact. 1998, 11, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Kloepper, J.W.; Leong, J.; Teintze, M.; Schroth, M.N. Enhanced Plant Growth by Siderophores Produced by Plant Growth-Promoting Rhizobacteria. Nature 1980, 286, 885–886. [Google Scholar] [CrossRef]
- Cha, J.Y.; Han, S.; Hong, H.J.; Cho, H.; Kim, D.; Kwon, Y.; Kwon, S.K.; Crusemann, M.; Bok Lee, Y.; Kim, J.F.; et al. Microbial and Biochemical Basis of a Fusarium Wilt-Suppressive Soil. ISME J. 2016, 10, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Cazorla, F.M.; Romero, D.; Pérez-García, A.; Lugtenberg, B.J.J.; De Vicente, A.; Bloemberg, G. Isolation and Characterization of Antagonistic Bacillus Subtilis Strains from the Avocado Rhizoplane Displaying Biocontrol Activity. J. Appl. Microbiol. 2007, 103, 1950–1959. [Google Scholar] [CrossRef] [PubMed]
- Haggag, W.M. Colonization of Exopolysaccharide-Producing Paenibacillus Polymyxa on Peanut Roots for Enhancing Resistance against Crown Rot Disease. Afr. J. Biotechnol. 2007, 6, 13. [Google Scholar] [CrossRef]
- Prasannakumar, M.K.; Mahesh, H.B.; Desai, R.U.; Kunduru, B.; Narayan, K.S.; Teli, K.; Puneeth, M.E.; Rajadurai, R.C.; Parivallal, B.; Babu, G.V. Metagenome Sequencing of Fingermillet-Associated Microbial Consortia Provides Insights into Structural and Functional Diversity of Endophytes. 3 Biotech 2020, 10, 15. [Google Scholar] [CrossRef]
- Liu, D.; Anderson, N.A.; Kinkel, L.L. Selection and Characterization of Strains of Streptomyces Suppressive to the Potato Scab Pathogen. Can. J. Microbiol. 1996, 42, 487–502. [Google Scholar] [CrossRef]
- Vujanovic, V.; Germida, J.J. Seed Endosymbiosis: A Vital Relationship in Providing Prenatal Care to Plants. Can. J. Plant Sci. 2017, 97, 972–998. [Google Scholar] [CrossRef] [Green Version]
- Handelsman, J. Metagenomics: Application of Genomics to Uncultured Microorganisms. Microbiol. Mol. Biol. Rev. 2005, 69, 195. [Google Scholar] [CrossRef] [Green Version]
- Hongoh, Y. Diversity and Genomes of Uncultured Microbial Symbionts in the Termite Gut. Biosci. Biotechnol. Biochem. 2010, 74, 1145–1151. [Google Scholar] [CrossRef]
- Adu-Oppong, B.; Gasparrini, A.J.; Dantas, G. Genomic and Functional Techniques to Mine the Microbiome for Novel Antimicrobials and Antimicrobial Resistance Genes. Ann. N. Y. Acad. Sci. 2017, 1388, 42–58. [Google Scholar] [CrossRef] [Green Version]
- Anantharaman, K.; Brown, C.T.; Hug, L.A.; Sharon, I.; Castelle, C.J.; Probst, A.J.; Thomas, B.C.; Singh, A.; Wilkins, M.J.; Karaoz, U.; et al. Thousands of Microbial Genomes Shed Light on Interconnected Biogeochemical Processes in an Aquifer System. Nat. Commun. 2016, 7, 13219. [Google Scholar] [CrossRef]
- Zaheer, R.; Noyes, N.; Ortega Polo, R.; Cook, S.R.; Marinier, E.; Van Domselaar, G.; Belk, K.E.; Morley, P.S.; McAllister, T.A. Impact of Sequencing Depth on the Characterization of the Microbiome and Resistome. Sci. Rep. 2018, 8, 5890. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Marques, J.; Hout, A.; Ferreira, R.M.; Weber, M.; Pinto-Ribeiro, I.; Van Doorn, L.J.; Knetsch, C.W.; Figueiredo, C. Impact of Host DNA and Sequencing Depth on the Taxonomic Resolution of Whole Metagenome Sequencing for Microbiome Analysis. Front. Microbiol. 2019, 10, 1277. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a Reference Resource for Gene and Protein Annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded Microbial Genome Coverage and Improved Protein Family Annotation in the COG Database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef] [PubMed]
- Sankar Narayan, K.; Esack, E.R.; Radhapriya, P.; Gopal, V.B.; Muthu, S.; Perumal, P. Impact of Geography on Adaptation of Phyllanthus Amarus Seeds. 3 Biotech 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Hijmans, R.J.; van Etten, J.; Cheng, J.; Mattiuzzi, M.; Sumner, M.; Greenberg, J.A.; Lamigueiro, O.P.; Bevan, A.; Racine, E.B.; Shortridge, A. Package ‘Raster’. R Package 2015, 269, 734. [Google Scholar]
- Thomas, P.; Agrawal, M.; Bharathkumar, C.B. Diverse Cellular Colonizing Endophytic Bacteria in Field Shoots and in Vitro Cultured Papaya with Physiological and Functional Implications. Physiol. Plant. 2019, 166, 729–747. [Google Scholar] [CrossRef] [PubMed]
- Padmanaban, A. End to End Sample Quality Control for Next Generation Sequencing Library Preparation and SureSelect Target Enrichment on the Agilent 2200 TapeStation System Application Note; Agilent Technologies: Bangalore, India, 2015. [Google Scholar]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Ewels, P.A.; Peltzer, A.; Fillinger, S.; Patel, H.; Alneberg, J.; Wilm, A.; Garcia, M.U.; di Tommaso, P.; Nahnsen, S. The Nf-Core Framework for Community-Curated Bioinformatics Pipelines. Nat. Biotechnol. 2020, 38, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; da Silva Santos, L.B.; Bourne, P.E. The FAIR Guiding Principles for Scientific Data Management and Stewardship. Sci. Data 2016, 3, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data 2010; Babraham Bioinformatics: Cambridge, UK, 2017. [Google Scholar]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Optimizing Taxonomic Classification of Marker-Gene Amplicon Sequences with QIIME 2’s Q2-Feature-Classifier Plugin. Microbiome 2018, 6, 1–17. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, K.S.; Kirkegaard, R.H.; Karst, S.M.; Albertsen, M. Ampvis2: An R Package to Analyse and Visualise 16S RRNA Amplicon Data. BioRxiv 2018. [Google Scholar] [CrossRef] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2: An Improved and Customizable Approach for Metagenome Inference. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Gweon, H.S.; Oliver, A.; Taylor, J.; Booth, T.; Gibbs, M.; Read, D.S.; Griffiths, R.I.; Schonrogge, K. PIPITS: An Automated Pipeline for Analyses of Fungal Internal Transcribed Spacer Sequences from the I Llumina Sequencing Platform. Methods Ecol. Evol. 2015, 6, 973–980. [Google Scholar] [CrossRef]
- Anslan, S.; Nilsson, R.H.; Wurzbacher, C.; Baldrian, P.; Tedersoo, L.; Bahram, M. Great Differences in Performance and Outcome of High-Throughput Sequencing Data Analysis Platforms for Fungal Metabarcoding. MycoKeys 2018, 39, 29–40. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Bengtsson-Palme, J.; Ryberg, M.; Hartmann, M.; Branco, S.; Wang, Z.; Godhe, A.; de Wit, P.; Sánchez-García, M.; Ebersberger, I.; de Sousa, F. Improved Software Detection and Extraction of ITS1 and ITS 2 from Ribosomal ITS Sequences of Fungi and Other Eukaryotes for Analysis of Environmental Sequencing Data. Methods Ecol. Evol. 2013, 4, 914–919. [Google Scholar] [CrossRef]
- Magdalita, P.M.; Persley, D.M.; Godwin, I.D.; Drew, R.A.; Adkins, S.W. Screening Carica Papaya× C. Cauliflora Hybrids for Resistance to Papaya Ringspot Virus-type P. Plant Pathol. 1997, 46, 837–841. [Google Scholar] [CrossRef] [Green Version]
- Rajamanickam, S.; Karthikeyan, G.; Kavino, M.; Manoranjitham, S.K. Biohardening of Micropropagated Banana Using Endophytic Bacteria to Induce Plant Growth Promotion and Restrain Rhizome Rot Disease Caused by Pectobacterium Carotovorum Subsp. Carotovorum. Sci. Hortic. 2018, 231, 179–187. [Google Scholar] [CrossRef]
- Ghosh, S.; Penterman, J.N.; Little, R.D.; Chavez, R.; Glick, B.R. Three Newly Isolated Plant Growth-Promoting Bacilli Facilitate the Seedling Growth of Canola, Brassica Campestris. Plant Physiol. Biochem. 2003, 41, 277–281. [Google Scholar] [CrossRef]
- Rivarez, M.P.S.; Parac, E.P.; Dimasingkil, S.F.M.; Magdalita, P.M. Defense Biopriming and Antimicrobial Activity of Endophytic Bacteria and Associated Bacillus Species Contribute to Bacterial Crown Rot Tolerance in Papaya. BioxRiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Kaspar, F.; Neubauer, P.; Gimpel, M. Bioactive Secondary Metabolites from Bacillus Subtilis: A Comprehensive Review. J. Nat. Prod. 2019, 82, 2038–2053. [Google Scholar] [CrossRef] [PubMed]
- Kakinuma, A.; Sugino, H.; Isono, M.; Tamura, G.; Arima, K. Determination of Fatty Acid in Surfactin and Elucidation of the Total Structure of Surfactin. Agric. Biol. Chem. 1969, 33, 973–976. [Google Scholar] [CrossRef]
- Caulier, S.; Nannan, C.; Gillis, A.; Licciardi, F.; Bragard, C.; Mahillon, J. Overview of the Antimicrobial Compounds Produced by Members of the Bacillus Subtilis Group. Front. Microbiol. 2019, 10, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denner, H.W.B. The Legislative Aspects of the Use of Industrial Enzymes in the Manufacture of Food and Food Ingredients; Nature Press: New York, NY, USA, 1983; Volume 111. [Google Scholar]
- Amruta, N.; Prasanna Kumar, M.K.; Puneeth, M.E.; Sarika, G.; Kandikattu, H.K.; Vishwanath, K.; Narayanaswamy, S. Exploring the Potentiality of Novel Rhizospheric Bacterial Strains against the Rice Blast Fungus Magnaporthe Oryzae. Plant Pathol. J. 2018, 34, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Cawoy, H.; Bettiol, W.; Fickers, P.; Onge, M. Bacillus-Based Biological Control of Plant Diseases. In Pesticides in the Modern World—Pesticides Use and Management; Stoytcheva, M., Ed.; Books on Demand: Norderstedt, Germany, 2011. [Google Scholar]
- El-Maraghy, S.S.; Tohamy, T.A.; Hussein, K.A. Expression of SidD Gene and Physiological Characterization of the Rhizosphere Plant Growth-Promoting Yeasts. Heliyon 2020, 6, e04384. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial 16S rRNA V3–4 Regions | ||||||
|---|---|---|---|---|---|---|
| Sr. No | Plant Species | Number of Reads | Total Bases | Data (Mb) | Filtered Reads | OTUs |
| 1 | Carica papaya | 106,801 | 53,472,228 | ~53 | 5443 | 144 |
| 2 | Vasconcellea cauliflora | 146,299 | 73,737,062 | ~74 | 12,429 | 177 |
| 3 | Vasconcellea cundinamarcensis | 138,844 | 69,421,623 | ~69 | 6429 | 156 |
| 4 | Vasconcellea goudotiana | 176,230 | 88,649,395 | ~89 | 15,043 | 204 |
| Fungal ITS | ||||||
| 5 | Carica papaya | 164,957 | 79,392,901 | ~79 | 154,546 | 43 |
| 6 | Vasconcellea cauliflora | 178,790 | 87,637,325 | ~88 | 175,852 | 52 |
| 7 | Vasconcellea cundinamarcensis | 186,838 | 90,803,894 | ~91 | 169,084 | 41 |
| 8 | Vasconcellea goudotiana | 181,128 | 88,669,669 | ~89 | 168,084 | 69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, B.L.; Narayan, K.S.; Gopalkrishna, A.M. Diversity Profiling of Seed Associated Endophytic Microbiome in Important Species of Caricaceae Family. Microbiol. Res. 2021, 12, 779-792. https://doi.org/10.3390/microbiolres12040057
Patil BL, Narayan KS, Gopalkrishna AM. Diversity Profiling of Seed Associated Endophytic Microbiome in Important Species of Caricaceae Family. Microbiology Research. 2021; 12(4):779-792. https://doi.org/10.3390/microbiolres12040057
Chicago/Turabian StylePatil, Basavaprabhu L., Karthik S. Narayan, and Amulya M. Gopalkrishna. 2021. "Diversity Profiling of Seed Associated Endophytic Microbiome in Important Species of Caricaceae Family" Microbiology Research 12, no. 4: 779-792. https://doi.org/10.3390/microbiolres12040057
APA StylePatil, B. L., Narayan, K. S., & Gopalkrishna, A. M. (2021). Diversity Profiling of Seed Associated Endophytic Microbiome in Important Species of Caricaceae Family. Microbiology Research, 12(4), 779-792. https://doi.org/10.3390/microbiolres12040057