Abstract
The spliced human papillomavirus 16 (HPV16) E1C RNA is associated with high-grade precursor lesions and cervical cancer. This qualifies E1C as a biomarker for high-grade lesions in HPV-based cervical cancer precursor screening. Here, we aimed to characterize the biological activity of HPV16 E1C RNA. In HEK-293T cells overexpressing HPV16 E1C RNA, we detected 9 kDa E1C protein in the cytoplasm using immunological assays with a newly generated E1C-specific monoclonal antibody or in mass spectrometry only after proteasome inhibition with MG132, indicating instability of the E1C protein. In HPV16-transformed cervical cancer cell lines in which the level of endogenous E1C RNA is much lower, E1C protein was not detected even after proteasome inhibition. Transient E1C overexpression in HEK-293T cells, co-transfected with a firefly luciferase reporter gene under the control of the HPV16 upstream regulatory region (URR), activated the HPV16 URR by 38%. This activation was also present when E1C translation was abolished by mutation. However, a construct expressing a random RNA sequence with similar GC content and 45% homology to the E1C RNA sequence also stimulated URR activity, indicating that special E1C RNA motifs might be responsible for the activation. In HPV16-transformed cell lines W12-episomal (W12-epi), W12-integrated HPV (W12-int), CaSki and SiHa stably overexpressing E1C RNA from lentiviral transduction, levels of endogenous HPV16 RNAs E6*I and E7 remained unchanged, while E1^E4 levels were significantly reduced by 20–30% in W12-epi, W12-int and CaSki cells. Overall, our study shows that E1C RNA is active and might contribute to transformation independent of the E6*I or E7 pathways. However, E1C overexpression resulted in only subtle changes in HPV16 RNA expression and very low copies of endogenous E1C RNA were detected in cervical cancer cell lines. This could weigh towards a less prominent role of E1C RNA in natural HPV transformation.
1. Introduction
Persistent infection by high-risk HPV types, predominantly HPV16 [1], can cause anogenital and oropharyngeal cancers. Cancer of the uterine cervix (CxCa), the fourth most common cancer in women [2], with 570,000 new cases per year, has the highest worldwide incidence among HPV-associated malignancies [3].
CxCa develops through precursor lesions, which, based on increasing severity, are histologically classified as cervical intraepithelial neoplasia (CIN) grade 1, CIN2 and CIN3 and cytologically as low- (LSIL) and high-grade squamous intraepithelial lesions (HSIL) [4].
Regulation of HPV16 transcription with at least two promoters and multiple alternatively used splice donor and acceptor sites is complex and is tightly linked to the differentiation stage of the infected keratinocyte [5]. During productive infection, expression of the early genes E6 and E7 enhance proliferation and lateral expansion of the infected basal and parabasal cells, while E1 and E2 are essential for the initial amplification and maintenance of viral DNA. In further differentiated upper epidermal cell layers, viral DNA is replicated and the late genes L1 and L2, encoding the capsid proteins, are expressed together with E4, resulting in assembly and release of viral particles. Epigenetic regulation of HPV transcription is necessary for productive infection. Viral genome exists in an epigenetically repressed state in the undifferentiated basal cells by polycomb repressor complexes, which deposit repressive H3K27Me3 resulting in decreased viral gene expression sufficient for the episomes to replicate. Upon differentiation of the basal cells, epigenetic repression is decreased, leading to increased expression of viral replication proteins and capsid protein production. Studies in HPV16-episome containing W12 cells have shown that in poorly differentiated cells compared to differentiated cells, viral URR is enriched in methylated CpG dinucleotides [6]. In HPV-driven cell transformation, expression of E6 and E7, frequently addressed as viral oncogenes, is deregulated and the ordered viral gene expression events are disrupted, resulting in continuous cellular proliferation and neoplasia [7].
Quantitative analysis of spliced and unspliced HPV16 RNA species in HPV16-positive HSIL versus LSIL identified strong upregulation of E6*I and E6*II RNA (both encoding E7 protein) and a marked downregulation of E1^E4 (encoding E4 protein) and L1 RNA in HSIL [7,8]. Additionally, a spliced E1C (nucleotide (N) 880-N2582) RNA with coding potential for a 9 kDa protein) was present in a substantial fraction of HSIL, although in very low copy numbers (median 16 copies/PCR), and virtually absent in LSIL. These studies demonstrated the potential of HPV16 E1C RNA as a HSIL-specific diagnostic marker [8,9]. One explanation for the low copy number in HSIL could be a low number of E1C-positive cancer cells. Another explanation could be that E1C plays a role only in HSIL where E2 is still expressed. Thus, E1C could function in the maintenance of transformation when the HPV genome is present as a full-length integrate or episome.
The only study so far addressing a potential function of E1C [10] suggested that experimentally overexpressed HPV16 E1C could activate the HPV16 URR and counteract URR repression by HPV16 transcription regulator protein E2. In translation studies, the same group could not detect E1C protein and discussed low expression levels or protein instability as potential reasons [11].
The aim of this study was to further characterize the biological activity of HPV16 E1C RNA. Firstly, the presence of E1C protein was investigated in HEK-293T cells with experimental overexpression as well as in HPV16-positive cervical cancer cell lines with endogenous E1C RNA. Secondly, the effect of wild-type and mutated E1C on URR activity was analyzed in co-transfection studies in HEK-293T cells. Finally, we analyzed the effect of experimentally overexpressed E1C on endogenous HPV16 transcripts involved in cell cycle progression and transformation in cell lines where HPV genomes are present as episomes or integrants.
2. Materials and Methods
2.1. Generation of E1C Monoclonal Antibody (mAb)
To generate an HPV16 E1C-specific mAb, C57black6N mice (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) were immunized by 3 subcutaneous injections in both hind legs, 7–9 days apart (day 0 with Freund’s Adjuvant complete, day 6 with Freund’s Adjuvant incomplete, day 15 only PBS, day 16 test sera, day 17 fusion, with a decapeptide encompassing the splice-generated N-terminal E1C protein sequence (Figure 1A) conjugated to keyhole limpet hemocyanin (20 µg/100 µL in phosphate-buffered saline (PBS) per mouse/injection day) (Peptide Speciality Laboratories, Heidelberg, Germany). Lymph nodes of the popliteal fossa were isolated, and lymphocytes were fused with Sp2/0 cells as described previously [12]. Mother clones producing HPV16 E1C-specific antibodies were subcloned twice. E1C-specific antibody (Ab) reactivity was determined by a direct ELISA with Glutathione S-Transferase (GST)-E1C according to the method described by Sehr et al., 2001 [13] and in Western blot (see below) using lysates from E. coli overexpressing GST-HPV16 E1C or GST-HPV16 full length E1 [14] in parallel. GST-HPV16 E1C fusion protein was encoded from plasmid pGEXE1C-tag generated by insertion of a synthetic E1C-encoding sequence into pGEX-tag [13]. Ab from the hybridoma clone 61/1/2 specifically reacting with HPV16 E1C was purified using Protein A Sepharose® beads (Protein A Sepharose CL-4b, GE Healthcare, Chicago, IL, USA) to an IgG1 concentration of 0.1 mg/mL (determined as described in [15]).
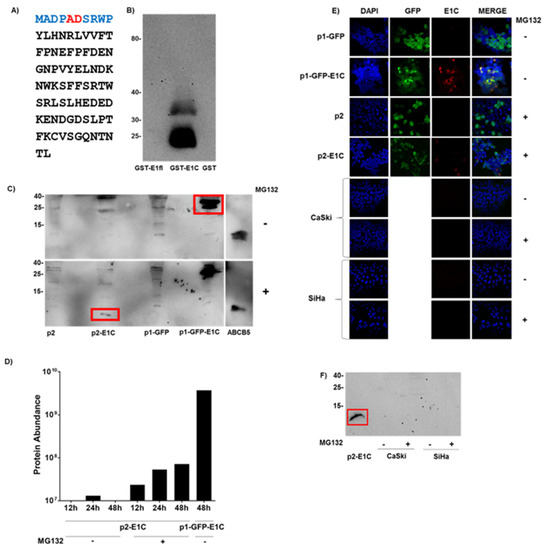
Figure 1.
Detection of E1C protein. (A) Sequence of the putative E1C protein in black showing the 10 aa E1C-specific peptide used for mouse immunization in blue and the splice junction-derived peptide fusion ‘AD’ in red. (B) Reactivity of E1C mAb (hybridoma sub-clone 61/1/2) in Western blot with E. coli lysates overexpressing GST, GST-E1C and GST-E1 full-length (fl) fusion proteins. (C) E1C mAb-stained Western blot of lysates (100 μg total protein, prepared 48 h post transfection) from HEK-293T cells transfected with expression plasmids p2, p2-E1C, p1-GFP or p1-GFP-E1C with (bottom) or without (top) proteasome inhibitor MG132 treatment. Size marker (in kDa) is shown on the left and 50 ng of truncated ATP-Binding Cassette sub-family B member 5 (ABCB5)-6XHis protein as positive control for detection of very small proteins on the right. Expected positions of E1C and GFP-E1C proteins are marked by red boxes. See supplementary information (Figure S3) for 12 h and 24 h post transfection results. (D) Mass spectrometry analysis of trypsin-digested lysates from HEK-293T cells transfected with p2-E1C or p1-GFP-E1C, harvested 12, 24 and 48 h post transfection with or without MG132 treatment. Y-axis (log10 scale) shows the combined abundance (protein abundance) of four HPV16 E1C peptides. (E) Immunofluorescence reactivity of E1C mAb (hybridoma sub-clone 61/1/2) in HEK-293T cells transfected with p1-GFP or p1-GFP-E1C and E1C immunofluorescence of HEK-293T cells transfected with p2 or p2-E1C as well as HPV16-positive human cervical cancer cell lines CaSki and SiHa with or without MG132 treatment. Nuclei were stained by DAPI (blue), GFP fluorescence (green), E1C mAb fluorescence (red) as well as a merge of all the fluorescence are shown. (F) E1C mAb-stained Western blot of lysates (100 μg total protein) from HPV16-positive human cervical cancer cell lines CaSki and SiHa with or without proteasome inhibitor MG132 treatment and, as positive control, from MG132-treated HEK-293T cells transfected with p2-E1C. Expected position of E1C is marked by a red box. See Figure S4 for all the Western blots with the molecular marker.
2.2. Cell Culture
Cell lines W12-20850 (W12-epi) and W12-20831 (W12-int) [16] were obtained from Paul F. Lambert (University of Wisconsin-Madison, Madison, WI, USA), and were cultured with mitomycin C-treated NIH-3T3 mouse fibroblasts as previously described [17]. Cell lines CaSki [18], SiHa [19], and HEK293T [20], were cultured in Dulbecco’s modified Eagle medium (DMEM) containing low glucose supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Darmstadt, Germany) and 1% penicillin/streptomycin. For all cell lines, authenticity [21] and absence of microbial or viral contaminations [22] were verified (Multiplexion, Heidelberg, Germany).
To inhibit proteasomal protein degradation, cell cultures were treated with 10 µM MG132 (Cayman Chemical Company, Ann Arbor, MI, USA) in DMSO (0.5% final concentration, Merck, Darmstadt, Germany) or DMSO alone for 6 h prior to cell lysis. The highest MG132 concentration (10 µM) tolerated by these cell lines had been predetermined in a dilution series of 2.5, 5, 10 and 20 µM by monitoring morphological differences and cell death under the microscope every 2 h over 10 h. Proteasome inhibition by MG132 in HEK-293T cells was monitored by polyubiquitin Western blot with a rabbit polyubiquitin mAb (D9D5, 1:1000 dilution, Cell Signaling Technology, Beverly, MA, USA).
2.3. Transient Transfections
For transient overexpression of E1C alone or in fusion with GFP, expression plasmids p2-E1C and p1-GFP-E1C were constructed. For p2-E1C, the HPV16 E1C coding sequence (N865-N879 fused to N2583-N2814 of HPV16 sequence (GenBank accession number AY686583.1)) was synthesized and inserted into eukaryotic expression vector p2 replacing the Gaussia luciferase coding sequence [23]. For p1-GFP-E1C, the E1C sequence of p2-E1C was inserted into the BamHI-HindIII sites of vector pBCHGS containing GFP and HIS tag in frame (p1-GFP) [24]. HEK-293T cells at 70% confluency (about 1.5 × 106 cells/10 cm-dish) were transiently transfected with 10 µg of these plasmids using 0.9 mg polyethylenimine (0.3 µg/µL, Polysciences, Warrington, PA, USA), 4% water and 94% DMEM. The transfection mix was vortexed for 10 s and incubated at room temperature for 10 min followed by the addition of pre-warmed DMEM and 5% fetal bovine serum before transfecting the cells. The transfection mix was replaced 12–14 h post transfection by growth medium and cells were lysed 48 h post transfection.
2.4. Luciferase Reporter Assay
To investigate the effect of E1C on the HPV16 URR, a Dual-Luciferase® Reporter Assay (DLRA) kit (Promega, Madison, WI, USA) was used. HEK-293T cells were transiently co-transfected with 100–200 ng of various expression constructs, 400 ng p-HPV16-URR-FLuc, a reporter plasmid with an HPV16 URR fragment (N7007–N102) upstream of the firefly luciferase gene and, to normalize for transfection efficiency, 2.5 ng p-TATAbox-RLuc, a reporter plasmid with a TATA box fragment upstream of a Renilla luciferase gene [25]. The co-transfected expression construct p-HPV16-E1C contained the HPV16 E1C coding sequence, p-HPV16-E1C-RNAmut contained a random RNA sequence with the same GC content and 45% homology as that of E1C RNA (Figure S1), p-HPV16-E1C-Promut contained the E1C sequence with the first 3 codons replaced by the 3 stop codon sequence TGA TAG TAA, p-HPV16-E2 contained the HPV16 E2 coding sequence (N2726-N3853), all inserted downstream of the CMV promoter into the empty pcDNA 3.1 (−) vector backbone (p-EV) (Thermo Fisher Scientific, Darmstadt, Germany). Forty-eight hours post transfection, cells were lysed following the manufacturer’s instructions. Firefly luciferase activity of 20 µL lysate in chimney-well white Lumitrac 96-well microplates (Greiner Bio-one, Frickenhausen, Germany) was measured in a luminometer (Biotek, Winooski, VT, USA) using Gen5TM software 30 s after addition of 50 µL firefly luciferase substrate. Subsequently, Renilla luciferase activity was measured 30 s after addition of 50 µL of Renilla luciferase substrate.
2.5. Lentiviral Transduction
Effects of overexpressed E1C on levels of HPV16 URR early promoter-driven RNAs were analyzed in HPV16-positive cell lines stably transduced with lentiviral vectors. To generate lentiviral expression vector LV-E1C, the HPV16 E1C coding sequence flanked by attL recombination sites was shuttled with Gateway® recombination technology from a pMX plasmid (Thermo Fisher Scientific, Waltham, MA, USA) into empty lentiviral expression vector LV (rwpLX305, R. W., unpublished), a puromycin resistance-conferring derivative of pLX304 [26]. For the generation of lentiviral particles, LV and LV-E1C were co-transfected with 2nd generation viral packaging plasmids VSV.G (Addgene #14888, Watertown, MA, USA) and psPAX2 (Addgene #12260) as described previously [27]. Cells were transduced with lentiviral particles in 10 μg/mL polybrene (Merck) and 24 h later, the selection was applied with puromycin (Santa Cruz Biotechnology, Dallas, TX, USA) at predetermined minimal toxic concentrations of 700 ng/mL (W12-epi and SiHa), 800 ng/mL (W12-int) and 500 ng/mL (CaSki). When non-transduced cells had completely detached, transduced cells were harvested.
2.6. Western Blot
Cells were suspended in lysis buffer (10 mM PIPES (pH 6.8), 300 mM NaCl, 1 mM EDTA (pH 8.0), 300 mM sucrose, 1 mM MgCl2, 0.5% Triton X-100) and incubated on ice for 20 min. Supernatant proteins after centrifugation (20,800× g, 10 min) were denatured with SDS at 95°C for 5 min, separated by electrophoresis in 20% polyacrylamide gels and blotted onto nitrocellulose membranes at 250 mA for 35 min. After blocking in 10% milk for 1 h, membranes were incubated with primary Ab (E1C mAb, 0.02 mg/mL, or 6X-His tag mAb [24], tissue culture supernatant) overnight at 4 °C and with secondary Ab (goat anti-mouse pox, 1:10,000 dilution, Dianova, Hamburg, Germany) for 2 h at room temperature followed by treatment with Amersham™ ECL Select™ Western Blotting Detection Reagent (GE Healthcare, Chicago, IL, USA) and reading in an INTAS chemocam imager (Göttingen, Germany). Prestained Protein Ladder, Broad Range (10–230 kDa) (New England Biolabs, Frankfurt am Main, Germany) or Spectra™ Multicolor Low Range Protein Ladder (Thermo Fisher Scientific, Waltham, MA, USA) were used as protein size markers. Protein ABCB5-6XHis (10 kDa) consists of a truncated sequence of ABCB5 protein from amino acids 599–678 [28]. It was purified over nickel columns after overexpression in E. coli (plasmid pQH, Qiagen, Hilden, Germany) and was used as the positive control for Western blot detection of small proteins.
2.7. HPV16 E1C Immunofluorescence
Cells grown on glass cover slips were fixed in 3.7% formaldehyde for 10 min, washed with PBS, 30 mM Glycine, for 5 min to quench formaldehyde, and permeabilized with PBS, 0.1% Triton X-100 for 10 min. After blocking in PBS, 2% horse serum, 0.1% Triton X-100 for 45 min, E1C mAb staining (0.03 mg/mL) followed by DAPI (1 µg/µL, Roche #10236276001, Sigma Aldrich, Darmstadt, Germany) and secondary Ab staining (Alexa Fluor™ 594, 1:500 dilution, Thermo Fisher Scientific, Eugene, OR, USA) were performed for 2 h each at room temperature in the dark with washing steps in between using PBS, 0.1% Triton X-100. Afterwards, cells were again washed with PBS for 5 min, rinsed in deionized water, dried and mounted on glass slides, and fluorescence signals were visualized in a Zeiss LSM 710 ConfoCor 3 (Zeiss, Oberkochen, Germany) using Zen (Black edition) software.
2.8. Mass Spectrometry
Cell lysate proteins were loaded on SDS-gel and run a short distance of 0.5 cm. After Coomassie staining, the total sample was cut out unfractionated and digested with trypsin as described by Shevchenko et al. [29] using a DigestProMSi robotic system (INTAVIS Bioanalytical Instruments, Cologne, Germany), loaded on a cartridge trap column, packed with Acclaim PepMap300 C18, 5 µm, 300Å wide pore (Thermo Fisher Scientific, Waltham, MA, USA) and separated in a 180 min gradient from 3% to 40% ACN on a nanoEase MZ Peptide analytical column (300Å, 1.7 µm, 75 µm × 200 mm, Waters) and UltiMate 3000 UHPLC system. Eluting peptides were analyzed by an online-coupled Q-Exactive-HF-X mass spectrometer (Thermo Fisher Scientific) running in a data-dependent acquisition mode where one full scan was followed by up to 12 MSMS scans of eluting peptides. Data were analyzed by MaxQuant [30] using an organism-specific database extracted from Uniprot.org under default settings. Identified FDR (false discovery rate) cutoffs were 0.01 at the peptide level and 0.01 at the protein level. The match between runs option was enabled to transfer peptide identifications across raw files based on accurate retention time and m/z. Quantification was performed using a label-free quantification approach based on the MaxLFQ algorithm [31]. A minimum of 2 quantified peptides per protein was required for protein quantification. Data were further processed by in-house compiled R-scripts to plot data and the Perseus software package (version 1.6.2.5, Max Planck Institute of Biochemistry, Martinsried, Germany) using default settings for filtering, imputation of missing values and statistical analysis [32].
2.9. RNA Extraction and RT-qPCR
Total RNA was extracted with the RNeasy Mini Kit (QIAGEN, Hilden, Germany) including DNaseI treatment according to the manufacturer’s instructions. RNA was eluted in 30 µL RNase-free water and the concentration was measured in a NanoDrop™ 1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Specific transcript levels were determined by RT-qPCR in a Roche cobas® z 480 analyzer system (Roche, Mannheim, Germany) using the LightCycler® 480 RNA Master Hydrolysis Probes kit (Roche). E6*I, E1^E4 and ubC transcripts were measured in a triplex RT-qPCR and E1C and E7 (N562-N858, forward primer 5′-TTTGCAACCAGAGACAACTGAT-3′, reverse primer 5′-TGTTGCAAGTGTGACTCTACGCT-3′, Sigma Aldrich, Darmstadt, Germany; Probe 5′-AGAGCCCATTACAATATTGTA-3′, TIB MOLBIOL, Berlin, Germany) transcripts in a singleplex RT-qPCR with conditions as previously described [9]. For quantification standards, dilution series of E6*I, E1^E4, E1C and ubC in vitro transcripts were used as described [9] and for E7, a dilution series of full-length HPV16 DNA in 50 ng/μL human placenta DNA. E1C RNA copies/cell were calculated assuming a total RNA content of 20 pg per cell.
2.10. Statistical Analysis
Data are expressed as mean ± SD. Means were tested for statistical significance using a paired t test. A significance level of p < 0.05 was applied when comparing experimental and control transfections in luciferase reporter assays and lentiviral transductions. Statistical analyses were performed using GraphPad Prism 6 (San Diego, CA, USA).
3. Results
3.1. Detection of E1C Protein
We first analyzed whether E1C RNA in eukaryotic cells can be translated into a detectable protein. Since no immunological reagents for the detection of putative E1C protein have been described so far, we developed an E1C-specific murine mAb. We generated lymphocyte hybridomas from mice immunized with a peptide encompassing the first ten amino acids of the E1C protein sequence with the fusion peptide encoded by the E1C splice junction in the central position (Figure 1A). A stable hybridoma (Clone 61/1/2) was isolated which, in the Western blot specifically, reacted with E. coli GST fusion proteins of E1C (35 kDa) but not HPV16 full length E1 (95 kDa) (Figure 1B). Additional 25–28 kDa bands were observed in the GST-E1C lane, probably products of N- or C-terminal protein degradation or a premature translation stop after the E1C-specific peptide sequence.
Upon overexpression in eukaryotic HEK-293T cells, E1C protein fused to GFP was readily detected in the Western blot (Figure 1C). E1C was also overexpressed (1.3 × 104 E1C RNA copies/cell, Table S1) without a fusion partner but protein was detectable only in cells treated with proteasomal protein degradation inhibitor MG132 and harvested 48 h and not 12 and 24 h post transfection, which hints towards the instability of the E1C protein (Figure 1C).
In HEK-293T cells 48 h post transfection with GFP-E1C expression, plasmid E1C-specific immunofluorescence was detected in the cytoplasm (Figure 1E) co-localizing with the fluorescence of the GFP fusion partner. When E1C was expressed without a fusion partner, E1C-specific immunofluorescence was detected only with MG132 treatment. The E1C signals appeared to be located in the cytoplasm, indicating some cytosolic function. Only 31 out of 175 GFP-positive cells were E1C-positive (18%). E1C positivity appeared not to be correlated with the extent of GFP fluorescence (Figure 1E).
To detect E1C protein by a non-immunological technique, mass spectrometry was applied to these HEK-293T cell lysates. Four tryptic peptides common to E1C and E1 were present in high abundance in GFP-E1C fusion protein-overexpressing cells (Figure 1D). The same peptides were, however, present at >50-fold lower levels in E1C-overexpressing cells treated with MG132. In contrast to Western blot, the E1C peptides were detected 12 h post transfection, with their abundance increasing three-fold 48 h post transfection. In cells without MG132 treatment, a signal close to the sensitivity threshold was detectable at 24 h, while no signals were detected 12 and 48 h post transfection. Hence, overexpressed E1C protein was also detectable in mass spectrometry but, again, under proteasomal inhibition conditions.
3.2. URR Activation Is Not Mediated by E1C Protein
Using a chloramphenicol acetyltransferase reporter assay, Alloul and Sherman [10] have shown that an HPV16 E1C plasmid activates HPV16 URR activity in the absence or presence of HPV16 URR-repressing HPV16 E2 plasmid. To investigate the effect of wild type (p-HPV16-E1C) and mutated E1C (p-HPV16-E1C-RNAmut and p-HPV16-E1C-Promut) on URR activity in the absence or presence of E2 (p-HPV16-E2), we used a firefly luciferase reporter under the transcription control of HPV16 URR (p-HPV16-URR-FLuc) in co-transfection experiments in HEK-293T cells. In order to normalize for transfection efficiency, we additionally co-transfected Renilla luciferase under transcription control of a TATA box (p-TATAbox-RLuc). However, Renilla luciferase expression was systematically repressed by co-transfected E2 plasmid (data not shown) and thus, could not be used for normalization. We therefore performed an additional set of parallel experiments without p-TATAbox-RLuc co-transfection. Since firefly luciferase activities were not systematically different in the presence or absence of p-TATAbox-RLuc (Figure S2), results from both experimental groups were analyzed together (Figure 2).
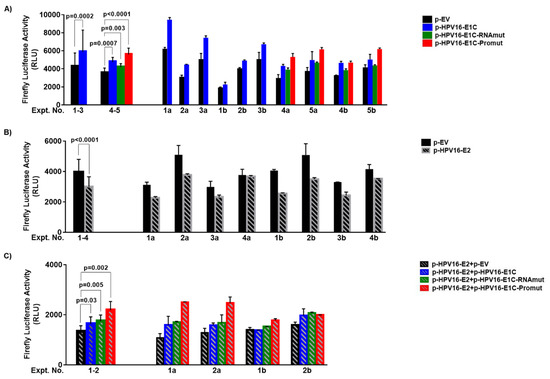
Figure 2.
Effect of wild type and mutant E1C expression plasmids on HPV16 upstream regulatory region (URR) without and with E2 plasmid co-transfection. HEK-293T cells were co-transfected with 400 ng of p-HPV16-URR-FLuc reporter plasmid and (A) 100 ng of either p-EV, p-HPV16-E1C (number of experiments, n = 6), p-HPV16-E1C-RNAmut (n = 4) or p-HPV16-E1C-Promut (n = 4), (B) 100 ng of p-HPV16-E2 (n = 8), (C) the same plasmids as in (A) always together with 100 ng of p-HPV16-E2 (n = 4). Firefly luciferase signals (RLU, Relative Light Units) are shown on the y-axis. Mean and standard deviation of all experiments combined and of duplicates from the individual experiments (numbered below x-axis) are shown on the left and right, respectively. p values are from paired t test. Letters a and b in experimental numbers indicate experiments without and with additional co-transfection of 2.5 ng of p-TATAbox-RLuc, respectively.
In all six co-transfection experiments, firefly luciferase activity was always higher with p-HPV16-E1C (2102–9622 Relative Light Units (RLU)) in comparison to the empty vector (p-EV) (1908–6335 RLU). The mean of all p-HPV16-E1C values (5886 RLU) was 38% higher than that of all p-EV values (4256 RLU) (p = 0.0002) (Figure 2A, Expt. No. 1–3) indicating that p-HPV16-E1C, also in this experimental design, activates the URR.
To study whether this URR activation is mediated by E1C protein or E1C RNA, we also co-transfected p-HPV16-E1C-Promut having the first three E1C codons replaced by TGA, TAG and TAA stop codons to abolish translation of E1C protein and p-HPV16-E1C-RNAmut transcribing a random sequence with the same size, GC content and 45% homology to E1C RNA. In all four co-transfection experiments, p-HPV16-E1C-Promut (range 4578–6314 RLU) and p-HPV16-E1C-RNAmut (3765–4733 RLU) also showed higher firefly luciferase activity in comparison to p-EV (2725–4370). The mean of all p-HPV16-E1C-Promut values (5598 RLU) was 57% and that of all p-HPV16-E1C-RNAmut values (4210 RLU) was 18% higher than that of all p-EV values (3557 RLU) (p < 0.0001 and p = 0.003, respectively) (Figure 2A, Expt. No. 4–5). In comparison to p-HPV16-E1C, the firefly luciferase activities with co-transfected p-HPV16-E1C-Promut were 18% higher (p = 0.0083) and that of co-transfected p-HPV16-E1C-RNAmut 11% lower (p = 0.0197). In summary, both p-HPV16-E1C-Promut and p-HPV16-E1C-RNAmut activated URR in comparison to p-EV. Consequently, URR activation is not mediated by E1C protein and is not specific to the whole E1C RNA sequence, indicating that special RNA motifs might be responsible for URR activation.
URR-repression by E2 alone was seen in seven out of eight co-transfection experiments with p-HPV16-E2 (2201–3829 RLU) in comparison to the empty vector (p-EV) (2725–5610 RLU). The mean of all p-HPV16-E2 values (3004 RLU) was 24% lower than that of all p-EV values (3949 RLU) (p < 0.0001) (Figure 2B). Thus, p-HPV16-E2 represses the URR.
In the presence of E2, co-transfected p-HPV16-E1C (1344–2160 RLU), p-HPV16-E1C-Promut (1722–2639 RLU) or p-HPV16-E1C-RNAmut (1457–2093 RLU) in all four experiments increased firefly luciferase activities in comparison to p-EV. The corresponding mean values (1634, 2184, 1744 RLU) were 22% (p = 0.03), 63% (p = 0.002) and 30% (p = 0.005) higher than that of p-EV (Figure 2C). Thus, p-HPV16-E1C as well as both p-HPV16-E1C-Promut and p-HPV16-E1C-RNAmut appeared to activate URR despite E2-induced URR-repression. Furthermore, URR activation by p-HPV16-E1C does not appear to require the whole E1C RNA sequence.
3.3. Overexpression of E1C in HPV16-Positive Cell Lines Decreases E1^E4 RNA Expression
To extend the observation described above that HPV16 E1C activates the HPV16 URR, we analyzed effects of E1C on levels of HPV16-URR early promoter-driven transcripts, in an experimental setting closer to the natural situation. As examples representing different developmental stages of HPV16-driven cell transformation, we selected four cell lines: W12, derived from a cervical intraepithelial neoplasia grade 1 which, in early passage, harbors HPV16 genomes as episomes (W12-epi), and in late passage, integrated into the host genome (W12-int), and invasive cervical carcinoma-derived cell lines CaSki and SiHa. To obtain steady-state experimental E1C expression, we selected sublines stably transduced by lentiviral vectors. In pairs of lentivirally transduced sublines with or without experimental E1C overexpression (3–7 independent transductions), we quantified HPV16 E6*I and E7 RNA, which are upregulated in HPV16-transformed cells and encode the E7 oncoprotein, and E1^E4, which is down-regulated in HPV16-transformed cells and encodes the E4 protein. SiHa cells cannot express E1^E4 because the splice acceptor is deleted in the single integrated HPV16 genome [33,34].
In all cell lines analyzed, overexpression of E1C did not affect the expression levels of E6*I and E7 RNA (Figure 3). In contrast, E1^E4 expression was significantly reduced by a mean of 30% in W12-epi and 20% in W12-int and CaSki.
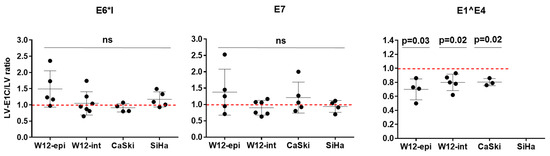
Figure 3.
Effect of overexpression of HPV16 E1C on HPV16 transcripts. HPV16-positive cell lines W12-epi, W12-int, CaSki and SiHa were stably transduced with lentiviral vector expressing E1C (LV-E1C) or empty vector (LV) and HPV16 transcripts E6*I, E7, and E1^E4 were quantified. Ratios of transcript quantities in LV-E1C- to LV-transduced cells (LV-E1C/LV ratio) are shown on the y-axis. For reference, ratio 1.0 is indicated by red broken lines. Bars show mean with standard deviation from 3 to 7 independent experiments; ns—not significant, p—two-tailed p value from paired t test. Extreme outliers (n = 15 of 108 total data points (14%)) defined as LV-E1C/LV ratios < 0.5 x median or >2.0 x median (Table S3) were excluded from this analysis.
3.4. Discussion and Conclusions
Spliced HPV16 E1C RNA was found to be almost exclusively associated with high-grade CxCa precursor lesions and therefore, possess the potential as a biomarker to distinguish from low-grade lesions [8,9]. Despite the low endogenous levels of E1C RNA, Höfler et al. have shown the prevalence of E1C transcript peaks in CIN3 lesions (54% of 158 cervical cell samples) [9]. Additionally, Schmitt et al. have identified the strong upregulation of E1C transcript in HSIL and CxCa, which clearly manifests its biological relevance [8]. Hence, we aimed to biologically characterize E1C RNA by investigating E1C protein, employing detailed analysis of the effects of E1C on the HPV16 URR with mutant constructs and by quantifying HPV16 RNA involved in cell cycle progression and transformation after experimental E1C overexpression.
This is the first study which has extensively looked for the translated product of E1C RNA. Here, we developed, for the first time, an immunological reagent, i.e., E1C mAb detecting the splice junction-derived fusion peptide of E1C protein. The specificity of E1C mAb was validated by detecting E. coli GST fusion protein of HPV16 E1C and not full length E1. In eukaryotic HEK-293T cells, experimentally overexpressed E1C protein without a fusion partner was not detectable in Western blot and mass spectrometry, even with 1.3 × 104 E1C RNA copies/cell. However, E1C protein was visible in these cells after proteasomal protein degradation inhibition with MG132. A GFP fusion of E1C protein was readily detectable with greater abundance in both techniques without any MG132 treatment. This points towards the instability of E1C protein and its lower abundance is in part due to the proteasomal degradation in comparison to highly abundant, not degraded GFP-E1C protein. Several factors such as thermal stability, sequence composition, folding, post translational modifications, protein–protein interactions, cellular environment, etc., determine the stability of a protein [35]. The significance of post translational modifications shows, for example, the reduced half-life of HPV16 E2 when phosphorylated at serine 243 compared to wild type E2 [36]. In contrast, the serine 33 phosphorylated form of HPV E7 possesses longer half-life than the de-phosphorylated form. When it comes to cellular environment, HPV E7 expression increases in the presence of hydrocortisone [37]. Proteasomal degradation also plays a role in balancing the concentrations of regulatory proteins [38]. The reason for instability of E1C protein and consequent degradation needs to be further investigated.
Alloul and Sherman, who have studied in vitro the E1C translational capacity of an HPV16 polycistronic RNA with 880/2581 splice junction, could not detect E1C protein. They performed in vitro translation in wheat germ extract, producing 35S-labelled translation products which were run on SDS-PAGE gels for protein detection [11,39]. The following reasons were proposed for non-detection of E1C protein: (a) E1C ORF overlaps with E2 ORF in this polycistronic RNA and therefore, E1C translation termination followed by E2 translation re-initiation is difficult; (b) low expression levels; or (c) protein instability [10,11]. However, they did not conduct any proteasome degradation inhibition.
GFP-E1C fusion protein and E1C protein without any fusion partner after MG132 treatment were found to be localized in the cytoplasm. However, only 18% of the cells overexpressing E1C stained positive for E1C. We assume this could be due to the proteasomal degradation inhibition heterogeneity in cells after MG132 treatment, leading to more proteasomal degradation inhibition in some cells and less in others. However, this demands further investigation, and no literature was found addressing the issue of proteasomal degradation inhibition heterogeneity by MG132.
Endogenous E1C protein in CaSki and SiHa cells was not detectable even after MG132 treatment. These cells contain low endogenous E1C RNA copies (CaSki—0.014 copies/cell, SiHa—0.001 copies/cell). Moreover, our finding that E1C protein could be instable might even make it less likely to detect endogenous E1C protein in these cells.
Since activation of the HPV16 URR increases oncoprotein expression, our second aim was to perform the functional analysis of E1C by studying its effect on the HPV16 URR. Firstly, in a luciferase reporter assay, we independently reproduced the principal findings of Alloul and Sherman [10], which were the activation and repression of URR by E1C and E2, respectively, and the reduced repression by E2 and E1C together. Next, we analyzed whether these effects of E1C were mediated by E1C RNA or E1C protein by utilizing one RNA and one protein mutant. Both the mutants activated URR, with the RNA mutant showing lower URR activation when compared to E1C. The protein mutant showed higher URR activation when compared to E1C. Additionally, both E1C mutants reduced the URR repressive effect of E2. Thus, URR activation is not mediated by E1C protein which is in agreement with the finding that E1C protein is instable. The cytoplasmic localization of overexpressed E1C protein in MG132-treated HEK-293T cells is also in accordance with these data. However, URR activation by E1C RNA was also found not to be specific to the whole RNA sequence. To understand which RNA motifs are necessary for URR activation, a detailed E1C sequence mutant analysis with different RNA mutants needs to be performed. Over the last few decades, scientists have demonstrated the paramount significance of functional RNAs such as small and long noncoding RNAs (lncRNA) that do also exist as bifunctional RNAs with coding and noncoding potential [40]. LncRNA encoded by Kaposi’s sarcoma-associated herpesvirus genome, for example, can maintain cellular transformation by affecting cellular gene expression [41] and bifunctional RNAs have been shown to disrupt tumor suppressor gene p53 [42]. If E1C RNA is a bifunctional RNA and mediates URR activation by binding, this needs further investigation.
The absence of a normalization control was a technical difficulty which we faced in our luciferase reporter assays. Co-transfected HPV16 E2 plasmid had a repressive effect on the minimal TATA box promoter of the renilla luciferase normalization plasmid. Alloul and Sherman used β-galactosidase plasmid under the control of CMV promoter for normalization in their co-transfection experiments. However, no normalization issues similar to ours were reported in their studies [10]. The overall URR activation effects by E1C and E1C mutants were not very strong and therefore, multiple repetitions of these complex co-transfection experiments were performed to confirm the consistency in the findings due to the inter-experimental variations observed.
In all the cell lines analyzed, overexpression of E1C did not affect the expression levels of E6*I and E7 RNA. This means the transformation pathways that are affected by altered E6*I and E7 expression levels are not further affected by overexpressed E1C levels. The prevalence of E6*I was found to be 8% in NIL/M, rising to more than 95% in CIN3 and CxCa [9]. Zheng’s lab has shown that E6*I transcripts are highly abundant in CaSki and SiHa cells [43] and the majority of the E7 proteins are translated from E6*I mRNAs in these cells. Duerksen-Hughes and colleagues have shown that E6*I overexpression in SiHa cells increases the expression of p53 and sensitized the cells to apoptosis [44]. E7 protein is one of the two viral oncoproteins and it binds and degrades the cellular tumor suppressor protein pRb, induces genome instability and has immortalization and transformation functions. Although E1C is described as a specific marker for ≥CIN3 cases [9], the occurrence of the transcript in severe lesions seems to have no transformation promoting role and works independent of E6*I and E7 transcript levels. Alternatively, as E6*I protein possesses an anti-oncogenic function, an increase in E6*I expression could inhibit cell proliferation and vice versa. This might be because E1C expression maintains the cell cycle progression in CxCa cell lines by maintaining the homeostatic levels of E6*I and E7. However, this hypothesis demands further investigation.
In contrast, E1^E4 expression was significantly reduced by a mean of 30% in W12-epi and 20% in W12-int and CaSki. This is in line with endogenous E1^E4 copies/cell in these cell lines, the lowest being in W12-epi in comparison to W12-int and CaSki (Table S4). Davy and colleagues have previously demonstrated the ability of HPV16 E1^E4 protein to inhibit cell proliferation [45]. Therefore, a lower E1^E4 RNA expression induced by E1C could prevent the cells from G2 arrest and maintain cell cycle progression in these cells. It would be interesting to analyze the expression of the CDK inhibitors of p21 and p27 in E1C overexpressed cells to understand whether E1C induces changes in cell cycle kinetics. So far, the data point towards an indirect role of E1C in transformation by not changing the RNA expression levels of the major transformation transcripts such as E6*I and E7 but by reducing E1^E4 levels, thereby helping the cells remain in the proliferative stage. To shed more light on the other key players that orchestrate either upstream or downstream of E1C to execute its functions, as next steps, it would be interesting to look at the entire HPV16 as well as cellular transcriptomes, for example, by the RNA-seq technique to analyze the changes in the RNA expression profiles of both virus and host upon E1C overexpression.
In summary, E1C RNA is active and might contribute to transformation independent of the canonical HPV oncogenesis pathway, i.e., by not affecting E6*I or E7 expression. However, weak effects in the expression of HPV16 RNA observed after E1C overexpression and the presence of less endogenous E1C copies in cervical cancer cell lines point towards a minimal role of E1C transcript in natural HPV transformation.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/microbiolres12030038/s1, Figure S1: cDNA sequence alignments of HPV16 E1C, HPV16 E1C-RNAmut and HPV16 E1C-Promut; Figure S2: Effect of wild type and mutant E1C expression plasmids on HPV16 upstream regulatory region (URR) without and with E2 plasmid co-transfection; Figure S3: Detection of E1C protein; Figure S4:All the Western blots in the manuscript with the molecular marker; Table S1: Copies/cell of endogenous and experimentally overexpressed E1C RNA; Table S2: Effect of wild type (p-HPV16-E1C) and mutant E1C expression plasmids (p-HPV16-E1C-RNAmut or p-HPV16-E1C-Promut) on HPV16 upstream regulatory region (URR) without and with E2 expression plasmid (p-HPV16-E2) co-transfection; Table S3: Effect of overexpression of HPV16 E1C on HPV16 transcripts - Results of individual experiments; Table S4: Copies/cell of endogenous E1^E4 RNA.
Author Contributions
Conceptualization: C.S.V., M.P., D.H.; Investigation: C.S.V., R.W., C.T., I.H., B.H., M.P., D.H.; Methodology: R.W.; Supervision: M.P., D.H.; Writing—original draft preparation: C.S.V., M.P., D.H.; Writing—review and editing: C.S.V., M.P., D.H., R.W., C.T., I.H., B.H. All authors have read and agreed to the published version of the manuscript.
Funding
C.S.V. was funded by the DKFZ International PhD Program. The authors received no specific funding for this work.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
We are grateful to Paul F. Lambert for W12-20850 and W12-20831 cell lines, to Martin Müller and Frank Rösl for HPV 16 E2 pcDNA 3.1 (-) and HPV 16 URR plasmids, to Birgit Aengeneyndt for excellent technical assistance, and to Martina Niebler and Maria Polycarpou-Schwarz for their scientific input in MG132 experiments and HPV16 E1C immunofluorescence.
Conflicts of Interest
M.P. is an inventor on a patent application to the European Patent Office (Europe patent application EP08168608.1-1222). M.P. and D.H. have received research support through cooperation contracts of DKFZ with Robert Bosch GmbH in the field of HPV diagnostics. The other authors have declared that no competing interests exist.
References
- Guan, P.; Howell-Jones, R.; Li, N.; Bruni, L.; de Sanjosé, S.; Franceschi, S.; Clifford, G.M. Human papillomavirus types in 115,789 HPV-positive women: A meta-analysis from cervical infection to cancer. Int. J. Cancer 2012, 131, 2349–2359. [Google Scholar] [CrossRef] [PubMed]
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjosé, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef]
- zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef]
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; De Sanjosé, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Primers 2016, 2, 16086. [Google Scholar] [CrossRef]
- Schmitt, M.; Pawlita, M. The HPV transcriptome in HPV16 positive cell lines. Mol. Cell Probes 2011, 25, 108–113. [Google Scholar] [CrossRef]
- Burley, M.; Roberts, S.; Parish, J.L. Epigenetic regulation of human papillomavirus transcription in the productive virus life cycle. Semin. Immunopathol. 2020, 42, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30 (Suppl. 5), F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Dalstein, V.; Waterboer, T.; Clavel, C.; Gissmann, L.; Pawlita, M. Diagnosing cervical cancer and high-grade precursors by HPV16 transcription patterns. Cancer Res. 2010, 70, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Höfler, D.; Böhmer, G.; Von Wasielewski, R.; Neumann, H.; Halec, G.; Holzinger, D.; Dondog, B.; Gissmann, L.; Pawlita, M.; Schmitt, M. HPV16 RNA patterns defined by novel high-throughput RT-qPCR as triage marker in HPV-based cervical cancer precursor screening. Gynecol. Oncol. 2015, 138, 676–682. [Google Scholar] [CrossRef]
- Alloul, N.; Sherman, L. Transcription-modulatory activities of differentially spliced cDNAs encoding the E2 protein of human papillomavirus type 16. J. Gen. Virol. 1999, 80 Pt 9, 2461–2470. [Google Scholar] [CrossRef]
- Alloul, N.; Sherman, L. The E2 protein of human papillomavirus type 16 is translated from a variety of differentially spliced polycistronic mRNAs. J. Gen. Virol. 1999, 80 Pt 1, 29–37. [Google Scholar] [CrossRef][Green Version]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Sehr, P.; Zumbach, K.; Pawlita, M. A generic capture ELISA for recombinant proteins fused to glutathione S-transferase: Validation for HPV serology. J. Immunol. Methods 2001, 253, 153–162. [Google Scholar] [CrossRef]
- Combes, J.D.; Pawlita, M.; Waterboer, T.; Hammouda, D.; Rajkumar, T.; Vanhems, P.; Snijders, P.; Herrero, R.; Franceschi, S.; Clifford, G. Antibodies against high-risk human papillomavirus proteins as markers for invasive cervical cancer. Int. J. Cancer 2014, 135, 2453–2461. [Google Scholar] [CrossRef] [PubMed]
- EH, D.L. Antibodies: A Laboratory Manual, 1st ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1988. [Google Scholar]
- Jeon, S.; Allen-Hoffmann, B.L.; Lambert, P.F. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 1995, 69, 2989–2997. [Google Scholar] [CrossRef]
- Lee, D.; Norby, K.; Hayes, M.; Chiu, Y.F.; Sugden, B.; Lambert, P.F. Using Organotypic Epithelial Tissue Culture to Study the Human Papillomavirus Life Cycle. Curr. Protoc. Microbiol. 2016, 41, 14B-8. [Google Scholar] [CrossRef] [PubMed]
- Pattillo, R.A.; Hussa, R.O.; Story, M.T.; Ruckert, A.C.; Shalaby, M.R.; Mattingly, R.F. Tumor antigen and human chorionic gonadotropin in CaSki cells: A new epidermoid cervical cancer cell line. Science 1977, 196, 1456–1458. [Google Scholar] [CrossRef]
- Friedl, F.; Kimura, I.; Osato, T.; Ito, Y. Studies on a new human cell line (SiHa) derived from carcinoma of uterus. I. Its establishment and morphology. Proc. Soc. Exp. Biol. Med. 1970, 135, 543–545. [Google Scholar] [CrossRef]
- Pear, W.S.; Nolan, G.P.; Scott, M.L.; Baltimore, D. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA 1993, 90, 8392–8396. [Google Scholar] [CrossRef]
- Castro, F.; Dirks, W.G.; Fahnrich, S.; Hotz-Wagenblatt, A.; Pawlita, M.; Schmitt, M. High-throughput SNP-based authentication of human cell lines. Int. J. Cancer 2013, 132, 308–314. [Google Scholar] [CrossRef]
- Schmitt, M.; Pawlita, M. High-throughput detection and multiplex identification of cell contaminations. Nucleic Acids Res. 2009, 37, e119. [Google Scholar] [CrossRef] [PubMed]
- Sehr, P.; Rubio, I.; Seitz, H.; Putzker, K.; Ribeiro-Müller, L.; Pawlita, M.; Müller, M. High-throughput pseudovirion-based neutralization assay for analysis of natural and vaccine-induced antibodies against human papillomaviruses. PLoS ONE 2013, 8, e75677. [Google Scholar] [CrossRef]
- Zentgraf, H.; Frey, M.; Schwinn, S.; Tessmer, C.; Willemann, B.; Samstag, Y.; Velhagen, I. Detection of histidine-tagged fusion proteins by using a high-specific mouse monoclonal anti-histidine tag antibody. Nucleic Acids Res. 1995, 23, 3347–3348. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De-Castro Arce, J.; Gockel-Krzikalla, E.; Rosl, F. Silencing of multi-copy HPV16 by viral self-methylation and chromatin occlusion: A model for epigenetic virus-host interaction. Hum. Mol. Genet. 2012, 21, 1693–1705. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yang, X.; Boehm, J.S.; Yang, X.; Salehi-Ashtiani, K.; Hao, T.; Shen, Y.; Lubonja, R.; Thomas, S.R.; Alkan, O.; Bhimdi, T.; et al. A public genome-scale lentiviral expression library of human ORFs. Nat. Methods 2011, 8, 659–661. [Google Scholar] [CrossRef] [PubMed]
- Haller, F.; Bieg, M.; Will, R.; Körner, C.; Weichenhan, D.; Bott, A.; Ishaque, N.; Lutsik, P.; Moskalev, E.A.; Mueller, S.K.; et al. Enhancer hijacking activates oncogenic transcription factor NR4A3 in acinic cell carcinomas of the salivary glands. Nat. Commun. 2019, 10, 368. [Google Scholar] [CrossRef] [PubMed]
- Frank, N.Y.; Pendse, S.S.; Lapchak, P.H.; Margaryan, A.; Shlain, D.; Doeing, C.; Sayegh, M.H.; Frank, M.H. Regulation of progenitor cell fusion by ABCB5 P-glycoprotein, a novel human ATP-binding cassette transporter. J. Biol. Chem. 2003, 278, 47156–47165. [Google Scholar] [CrossRef]
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef]
- Tyanova, S.; Cox, J. Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research. Methods Mol. Biol. 2018, 1711, 133–148. [Google Scholar]
- Baker, C.C.; Phelps, W.C.; Lindgren, V.; Braun, M.J.; Gonda, M.A.; Howley, P.M. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J. Virol. 1987, 61, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Chotewutmontri, S.; Wolf, S.; Klos, U.; Schmitz, M.; Dürst, M.; Schwarz, E. Multiplex Identification of Human Papillomavirus 16 DNA Integration Sites in Cervical Carcinomas. PLoS ONE 2013, 8, e66693. [Google Scholar] [CrossRef] [PubMed]
- Parsell, D.A.; Sauer, R.T. The structural stability of a protein is an important determinant of its proteolytic susceptibility in Escherichia coli. J. Biol. Chem. 1989, 264, 7590–7595. [Google Scholar] [CrossRef]
- Chang, S.W.; Liu, W.C.; Liao, K.Y.; Tsao, Y.P.; Hsu, P.H.; Chen, S.L. Phosphorylation of HPV-16 E2 at serine 243 enables binding to Brd4 and mitotic chromosomes. PLoS ONE 2014, 9, e110882. [Google Scholar] [CrossRef] [PubMed]
- Selvey, L.A.; Dunn, L.A.; Tindle, R.W.; Park, D.S.; Frazer, I.H. Human papillomavirus (HPV) type 18 E7 protein is a short-lived steroid-inducible phosphoprotein in HPV-transformed cell lines. J. Gen. Virol. 1994, 75, 1647–1653. [Google Scholar] [CrossRef]
- Schrader, E.K.; Harstad, K.G.; Matouschek, A. Targeting proteins for degradation. Nat. Chem. Biol. 2009, 5, 815–822. [Google Scholar] [CrossRef]
- Sherman, L.; Jackman, A.; Itzhaki, H.; Stoppler, M.C.; Koval, D.; Schlegel, R. Inhibition of serum- and calcium-induced differentiation of human keratinocytes by HPV16 E6 oncoprotein: Role of p53 inactivation. Virology 1997, 237, 296–306. [Google Scholar] [CrossRef]
- Nam, J.W.; Choi, S.W.; You, B.H. Incredible RNA: Dual Functions of Coding and Noncoding. Mol. Cells 2016, 39, 367–374. [Google Scholar]
- Rossetto, C.C.; Tarrant-Elorza, M.; Verma, S.; Purushothaman, P.; Pari, G.S. Regulation of viral and cellular gene expression by Kaposi’s sarcoma-associated herpesvirus polyadenylated nuclear RNA. J. Virol. 2013, 87, 5540–5553. [Google Scholar] [CrossRef] [PubMed]
- Kumari, P.; Sampath, K. cncRNAs: Bi-functional RNAs with protein coding and non-coding functions. Semin. Cell Dev. Biol. 2015, 47–48, 40–51. [Google Scholar] [CrossRef]
- Tang, S.; Tao, M.; McCoy, J.P.; Jr Zheng, Z.M. The E7 oncoprotein is translated from spliced E6*I transcripts in high-risk human papillomavirus type 16- or type 18-positive cervical cancer cell lines via translation reinitiation. J. Virol. 2006, 80, 4249–4263. [Google Scholar] [CrossRef] [PubMed]
- Filippova, M.; Evans, W.; Aragon, R.; Filippov, V.; Williams, V.M.; Hong, L.; Reeves, M.E.; Duerksen-Hughes, P. The small splice variant of HPV16 E6, E6, reduces tumor formation in cervical carcinoma xenografts. Virology 2014, 450–451, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Davy, C.E.; Jackson, D.J.; Wang, Q.; Raj, K.; Masterson, P.J.; Fenner, N.F. Identification of a G(2) arrest domain in the E1 wedge E4 protein of human papillomavirus type 16. J. Virol. 2002, 76, 9806–9818. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).