
Metagenomic Insight into the Microbiome and Virome Associated with Aedes aegypti Mosquitoes in Manado (North Sulawesi, Indonesia)

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
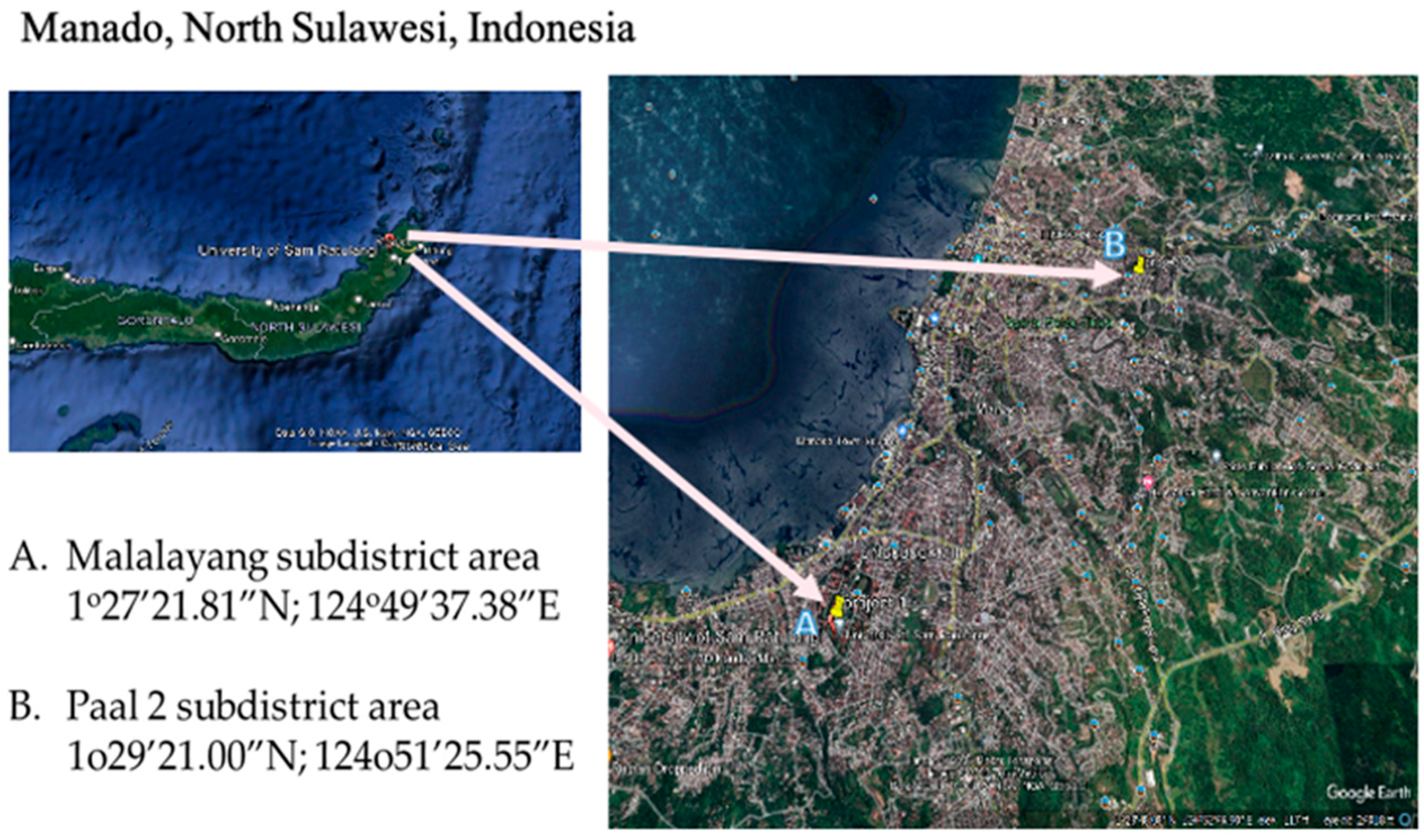
2.1. Sampling Locations
2.2. Sample Preparation
2.2.1. gDNA and RNA Extraction
2.2.2. Whole-Genome Shotgun Metagenomics (Wf-Metagenomics)
2.2.3. Bioinformatics Analysis
2.2.4. Microbial Community Analysis
2.2.5. Alpha Diversity Analysis
3. Results and Discussion
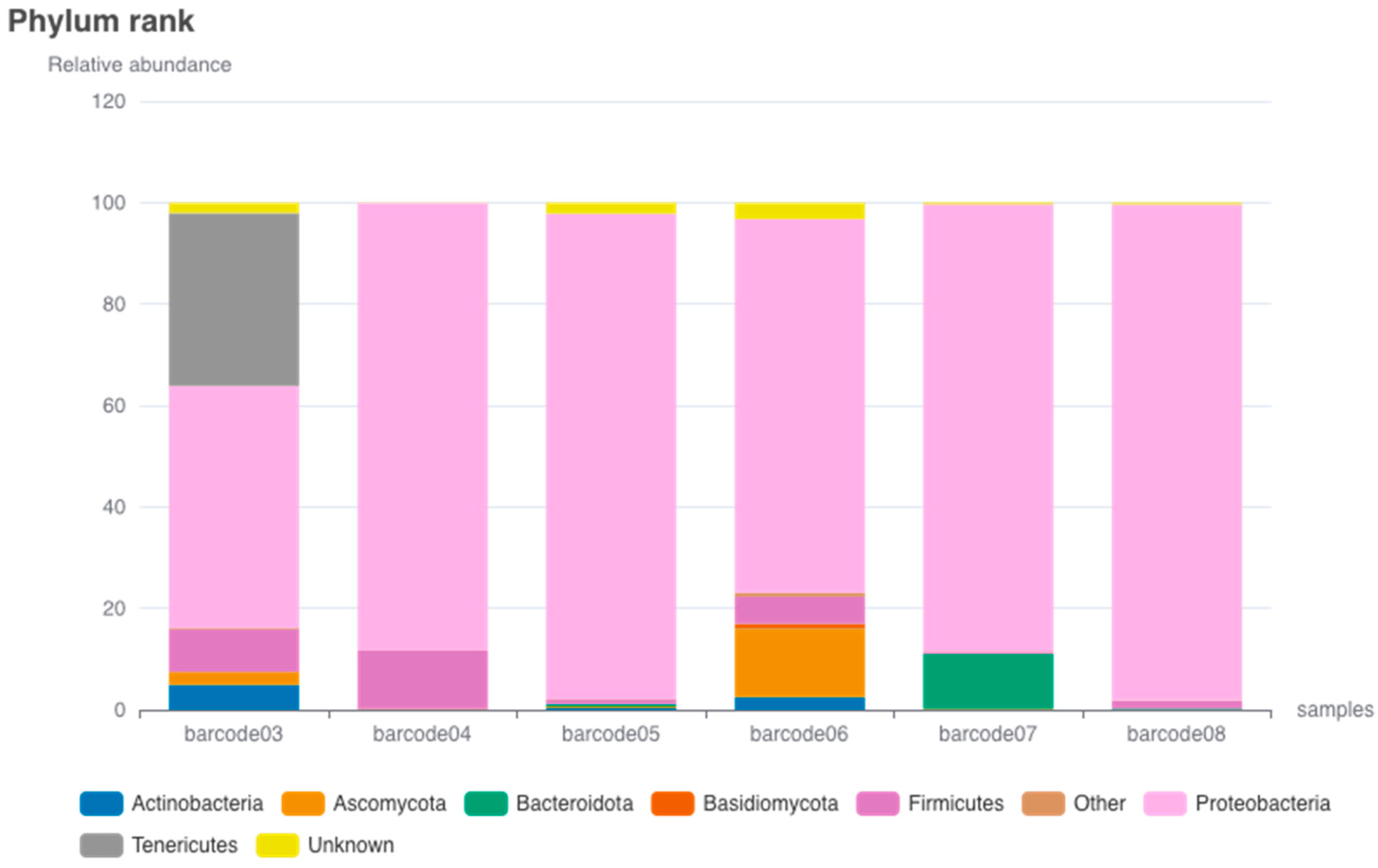
3.1. Microbial Relative Abundance
3.2. Microbial Alpha Diversity
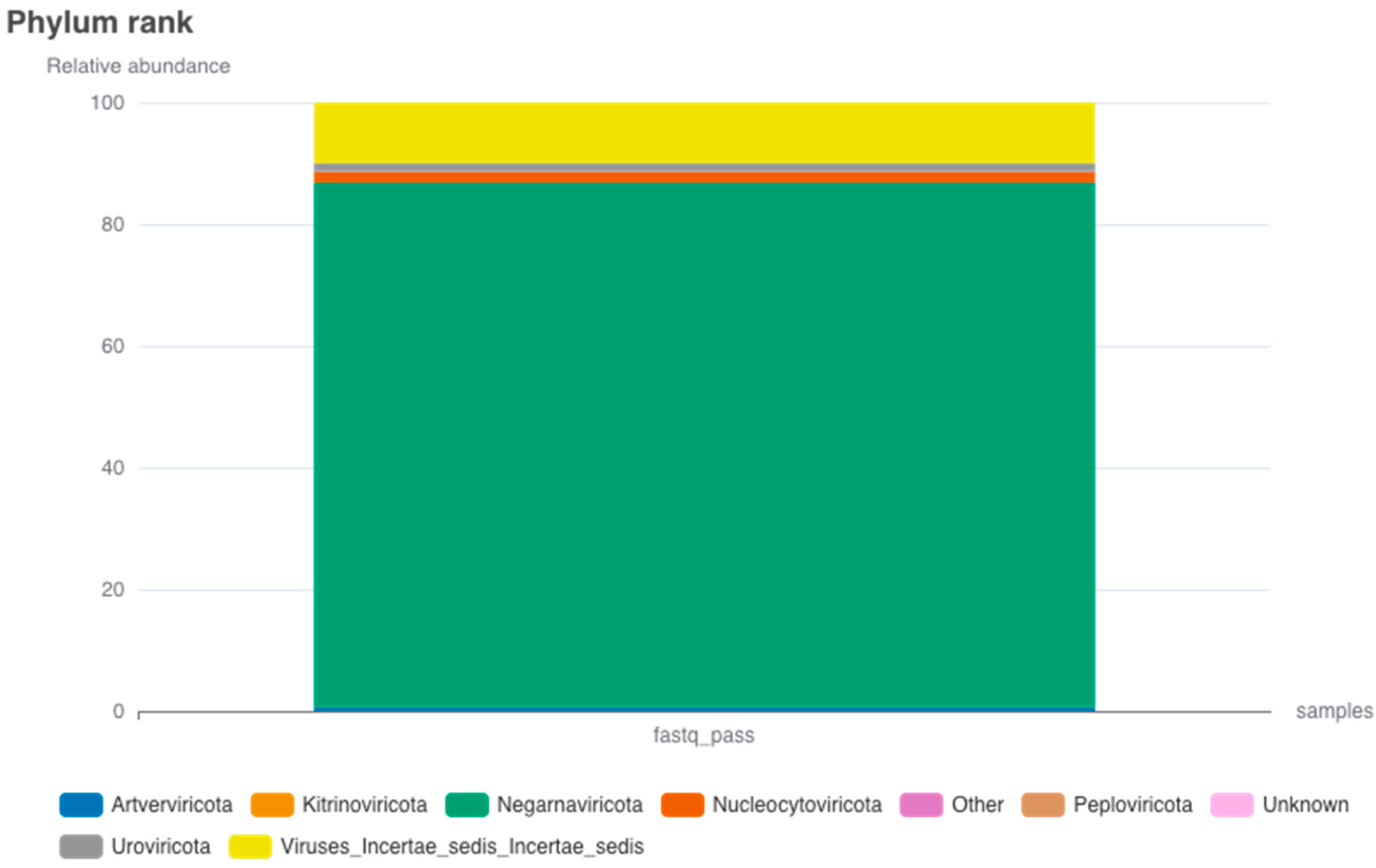
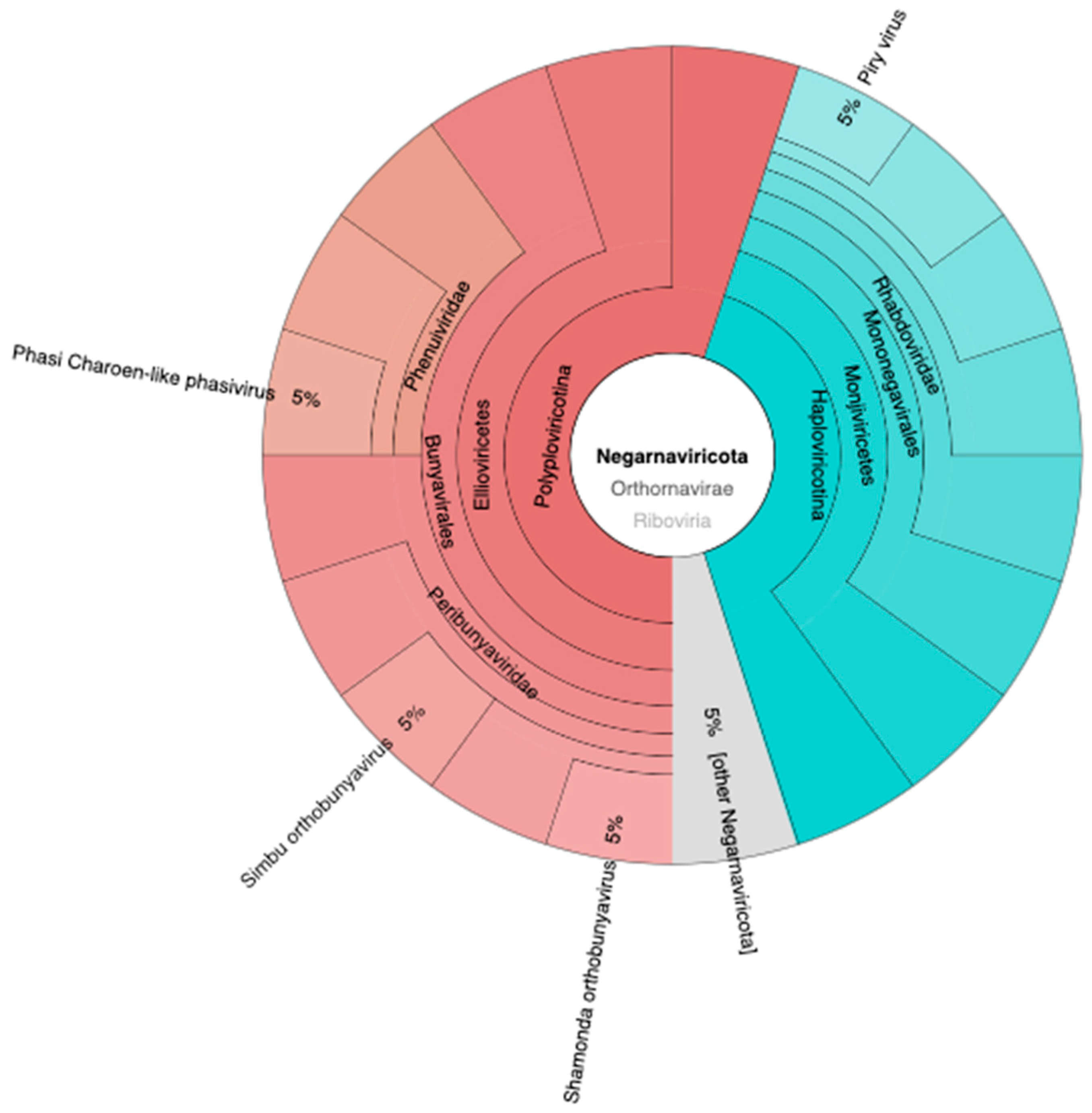
3.3. Viral Relative Abundance
3.4. Viral Alpha Diversity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kurnia, N.; Kaitana, Y.; Salaki, C.L.; Mandey, L.C.; Tuda, J.S.B.; Tallei, T.E. Study of Dengue Virus Transovarial Transmission in Aedes spp. in Ternate City Using Streptavidin-Biotin-Peroxidase Complex Immunohistochemistry. Infect. Dis. Rep. 2022, 14, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Mubbashir, H.; Munir, S.; Kashif, R.; Nawaz, H.B.; Abdul, B.; Baharullah, K. Characterization of Dengue Virus in Aedes aegypti and Aedes albopictus spp. of Mosquitoes: A Study in Khyber Pakhtunkhwa, Pakistan. Mol. Biol. Res. Commun. 2018, 7, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Trojánek, M.; Grebenyuk, V.; Manďáková, Z.; Sojková, N.; Zelená, H.; Roháčová, H.; Stejskal, F. Epidemiology of Dengue, Chikungunya and Zika Virus Infections in Travellers: A 16-Year Retrospective Descriptive Study at a Tertiary Care Centre in Prague, Czech Republic. PLoS ONE 2023, 18, e0281612. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.R. Mosquito-Borne Human Viral Diseases: Why Aedes Aegypti? Am. J. Trop. Med. Hyg. 2018, 98, 1563–1565. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Yu, X.; Cheng, G. Impact of the Microbiome on Mosquito-Borne Diseases. Protein Cell 2023, pwad021. [Google Scholar] [CrossRef]
- Leach, C.B.; Hoeting, J.A.; Pepin, K.M.; Eiras, A.E.; Hooten, M.B.; Webb, C.T. Linking Mosquito Surveillance to Dengue Fever through Bayesian Mechanistic Modeling. PLoS Negl. Trop. Dis. 2020, 14, e0008868. [Google Scholar] [CrossRef]
- Caragata, E.P.; Tikhe, C.V.; Dimopoulos, G. Curious Entanglements: Interactions between Mosquitoes, Their Microbiota, and Arboviruses. Curr. Opin. Virol. 2019, 37, 26–36. [Google Scholar] [CrossRef]
- Rückert, C.; Ebel, G.D. How Do Virus-Mosquito Interactions Lead to Viral Emergence? Trends Parasitol. 2018, 34, 310–321. [Google Scholar] [CrossRef]
- Siriphanitchakorn, T.; Kini, R.M.; Ooi, E.E.; Choy, M.M. Revisiting Dengue Virus-Mosquito Interactions: Molecular Insights into Viral Fitness. J. Gen. Virol. 2021, 102, 001693. [Google Scholar] [CrossRef]
- Nanfack-Minkeu, F.; Mitri, C.; Bischoff, E.; Belda, E.; Casademont, I.; Vernick, K.D. Interaction of RNA Viruses of the Natural Virome with the African Malaria Vector, Anopheles Coluzzii. Sci. Rep. 2019, 9, 6319. [Google Scholar] [CrossRef]
- Cottis, S.; Blisnick, A.A.; Failloux, A.-B.; Vernick, K.D. Determinants of Chikungunya and O’Nyong-Nyong Virus Specificity for Infection of Aedes and Anopheles Mosquito Vectors. Viruses 2023, 15, 589. [Google Scholar] [CrossRef]
- Zhou, T.-F.; Lai, Z.-T.; Liu, S.; Zhou, J.-Y.; Liu, Y.; Wu, Y.; Xu, Y.; Wu, K.; Gu, J.-B.; Cheng, G.; et al. Susceptibility and Interactions between Aedes Mosquitoes and Zika Viruses. Insect Sci. 2021, 28, 1439–1451. [Google Scholar] [CrossRef]
- Kerkhof, L.J. Is Oxford Nanopore Sequencing Ready for Analyzing Complex Microbiomes? FEMS Microbiol. Ecol. 2021, 97, fiab001. [Google Scholar] [CrossRef]
- Tyler, A.D.; Mataseje, L.; Urfano, C.J.; Schmidt, L.; Antonation, K.S.; Mulvey, M.R.; Corbett, C.R. Evaluation of Oxford Nanopore’s MinION Sequencing Device for Microbial Whole Genome Sequencing Applications. Sci. Rep. 2018, 8, 10931. [Google Scholar] [CrossRef]
- Lu, J.; Rincon, N.; Wood, D.E.; Breitwieser, F.P.; Pockrandt, C.; Langmead, B.; Salzberg, S.L.; Steinegger, M. Metagenome Analysis Using the Kraken Software Suite. Nat. Protoc. 2022, 17, 2815–2839. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive Metagenomic Visualization in a Web Browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Ren, H. TaxonKit: A Practical and Efficient NCBI Taxonomy Toolkit. J. Genet. Genom. 2021, 48, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cobas, A.E.; Gomez-Valero, L.; Buchrieser, C. Metagenomic Approaches in Microbial Ecology: An Update on Whole-Genome and Marker Gene Sequencing Analyses. Microb. Genom. 2020, 6, e000409. [Google Scholar] [CrossRef]
- Yadav, K.K.; Bora, A.; Datta, S.; Chandel, K.; Gogoi, H.K.; Prasad, G.B.K.S.; Veer, V. Molecular Characterization of Midgut Microbiota of Aedes Albopictus and Aedes Aegypti from Arunachal Pradesh, India. Parasit. Vectors 2015, 8, 641. [Google Scholar] [CrossRef]
- Arévalo-Cortés, A.; Mejia-Jaramillo, A.M.; Granada, Y.; Coatsworth, H.; Lowenberger, C.; Triana-Chavez, O. The Midgut Microbiota of Colombian Aedes Aegypti Populations with Different Levels of Resistance to the Insecticide Lambda-Cyhalothrin. Insects 2020, 11, 584. [Google Scholar] [CrossRef]
- Guégan, M.; Martin, E.; Valiente Moro, C. Comparative Analysis of the Bacterial and Fungal Communities in the Gut and the Crop of Aedes Albopictus Mosquitoes: A Preliminary Study. Pathogens 2020, 9, 628. [Google Scholar] [CrossRef]
- Tawidian, P.; Coon, K.L.; Jumpponen, A.; Cohnstaedt, L.W.; Michel, K. Host-Environment Interplay Shapes Fungal Diversity in Mosquitoes. mSphere 2021, 6, e0064621. [Google Scholar] [CrossRef]
- Araújo, J.P.M.; Hughes, D.P. Chapter One—Diversity of Entomopathogenic Fungi: Which Groups Conquered the Insect Body? In Genetics and Molecular Biology of Entomopathogenic Fungi; Lovett, B., St. Leger, R.J., Eds.; Advances in Genetics; Academic Press: Cambridge, MA, USA, 2016; Volume 94, pp. 1–39. [Google Scholar]
- Fatimawali; Kepel, B.J.; Gani, M.A.; Tallei, T.E. Comparison of Bacterial Community Structure and Diversity in Traditional Gold Mining Waste Disposal Site and Rice Field by Using a Metabarcoding Approach. Int. J. Microbiol. 2020, 2020, 1858732. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Brigham, C.A.; Hoeksema, J.D.; Lyons, K.G.; Mills, M.H.; van Mantgem, P.J. Linking Biodiversity to Ecosystem Function: Implications for Conservation Ecology. Oecologia 2000, 122, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Tallei, T.E.; Fatimawali; Pelealu, J.J. The Data on Metagenomic Profile of Bacterial Diversity Changes in the Different Concentration of Fermented Romaine Lettuce Brine. Data Brief 2019, 25, 104190. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Hawkins, C.P. Weighting Effective Number of Species Measures by Abundance Weakens Detection of Diversity Responses. J. Appl. Ecol. 2019, 56, 1200–1209. [Google Scholar] [CrossRef]
- Tallei, V.R.; Saroyo; Tallei, T.E. Diversity of Recorded Wild Mammals in Mount Tumpa Forest Park, North Sulawesi, Indonesia. J. Plant Sci. 2016, 12, 39–45. [Google Scholar] [CrossRef]
- Niode, N.J.; Adji, A.; Rimbing, J.; Tulung, M.; Tallei, T.E. Composition and Diversity of Bacteria from Giant Asian Honeybee Apis Dorsata Gut. Biodiversitas 2021, 22, 906–912. [Google Scholar] [CrossRef]
- Pielou, E.C. The Measurement of Diversity in Different Types of Biological Collections. J. Theor. Biol. 1966, 13, 131–144. [Google Scholar] [CrossRef]
- Liu, Q.; Cui, F.; Liu, X.; Fu, Y.; Fang, W.; Kang, X.; Lu, H.; Li, S.; Liu, B.; Guo, W.; et al. Association of Virome Dynamics with Mosquito Species and Environmental Factors. Microbiome 2023, 11, 101. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Adkins, S.; Agwanda, B.R.; Al Kubrusli, R.; Alkhovsky, S.V.; Amarasinghe, G.K.; Avšič-Županc, T.; Ayllón, M.A.; Bahl, J.; Balkema-Buschmann, A.; et al. 2021 Taxonomic Update of Phylum Negarnaviricota (Riboviria: Orthornavirae), Including the Large Orders Bunyavirales and Mononegavirales. Arch. Virol. 2021, 166, 3513–3566. [Google Scholar] [CrossRef]
- Kanojia, A.; Sharma, M.; Shiraz, R.; Tripathi, S. Flavivirus-Host Interaction Landscape Visualized through Genome-Wide CRISPR Screens. Viruses 2022, 14, 2164. [Google Scholar] [CrossRef]
- Teo, D.; Ng, L.C.; Lam, S. Is Dengue a Threat to the Blood Supply? Transfus. Med. 2009, 19, 66–77. [Google Scholar] [CrossRef]
- Duong, V.; Lambrechts, L.; Paul, R.E.; Ly, S.; Lay, R.S.; Long, K.C.; Huy, R.; Tarantola, A.; Scott, T.W.; Sakuntabhai, A.; et al. Asymptomatic Humans Transmit Dengue Virus to Mosquitoes. Proc. Natl. Acad. Sci. USA 2015, 112, 14688–14693. [Google Scholar] [CrossRef]
- Brackney, D.E.; LaReau, J.C.; Smith, R.C. Frequency Matters: How Successive Feeding Episodes by Blood-Feeding Insect Vectors Influences Disease Transmission. PLoS Pathog. 2021, 17, e1009590. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Pools | Total Counts | Richness | Shannon Diversity Index (H) | Effective N Species | Simpson’s Index (D) | Inverse of D | Pielou Evenness |
|---|---|---|---|---|---|---|---|
| Barcode03 | 2775 | 237 | 3.14 | 23.01 | 0.86 | 1.17 | 0.57 |
| Barcode04 | 37,019 | 629 | 3.94 | 51.46 | 0.93 | 1.08 | 0.61 |
| Barcode05 | 3310 | 241 | 3.17 | 23.84 | 0.85 | 1.17 | 0.58 |
| Barcode06 | 2485 | 223 | 3.53 | 34.00 | 0.93 | 1.08 | 0.65 |
| Barcode07 | 11,265 | 365 | 3.31 | 27.40 | 0.82 | 1.22 | 0.56 |
| Barcode08 | 13,438 | 540 | 3.91 | 49.80 | 0.92 | 1.09 | 0.64 |
| Sample Pools | Total Counts | Richness | Shannon Diversity Index (H) | Effective N Species | Simpson’s Index (D) | Inverse of D | Pielou Evenness |
|---|---|---|---|---|---|---|---|
| P1 | 12,788 | 66 | 0.86 | 2.37 | 0.46 | 2.18 | 0.21 |
| P2 | 14,248 | 44 | 0.85 | 2.34 | 0.51 | 1.95 | 0.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernadus, J.B.B.; Pelealu, J.; Kandou, G.D.; Pinaria, A.G.; Mamahit, J.M.E.; Tallei, T.E. Metagenomic Insight into the Microbiome and Virome Associated with Aedes aegypti Mosquitoes in Manado (North Sulawesi, Indonesia). Infect. Dis. Rep. 2023, 15, 549-563. https://doi.org/10.3390/idr15050054
Bernadus JBB, Pelealu J, Kandou GD, Pinaria AG, Mamahit JME, Tallei TE. Metagenomic Insight into the Microbiome and Virome Associated with Aedes aegypti Mosquitoes in Manado (North Sulawesi, Indonesia). Infectious Disease Reports. 2023; 15(5):549-563. https://doi.org/10.3390/idr15050054
Chicago/Turabian StyleBernadus, Janno Berty Bradly, Jantje Pelealu, Grace Debbie Kandou, Arthur Gehart Pinaria, Juliet Merry Eva Mamahit, and Trina Ekawati Tallei. 2023. "Metagenomic Insight into the Microbiome and Virome Associated with Aedes aegypti Mosquitoes in Manado (North Sulawesi, Indonesia)" Infectious Disease Reports 15, no. 5: 549-563. https://doi.org/10.3390/idr15050054
APA StyleBernadus, J. B. B., Pelealu, J., Kandou, G. D., Pinaria, A. G., Mamahit, J. M. E., & Tallei, T. E. (2023). Metagenomic Insight into the Microbiome and Virome Associated with Aedes aegypti Mosquitoes in Manado (North Sulawesi, Indonesia). Infectious Disease Reports, 15(5), 549-563. https://doi.org/10.3390/idr15050054