
Differential Diagnoses and Management Approaches for Gastric Polyposis

Abstract
1. Introduction
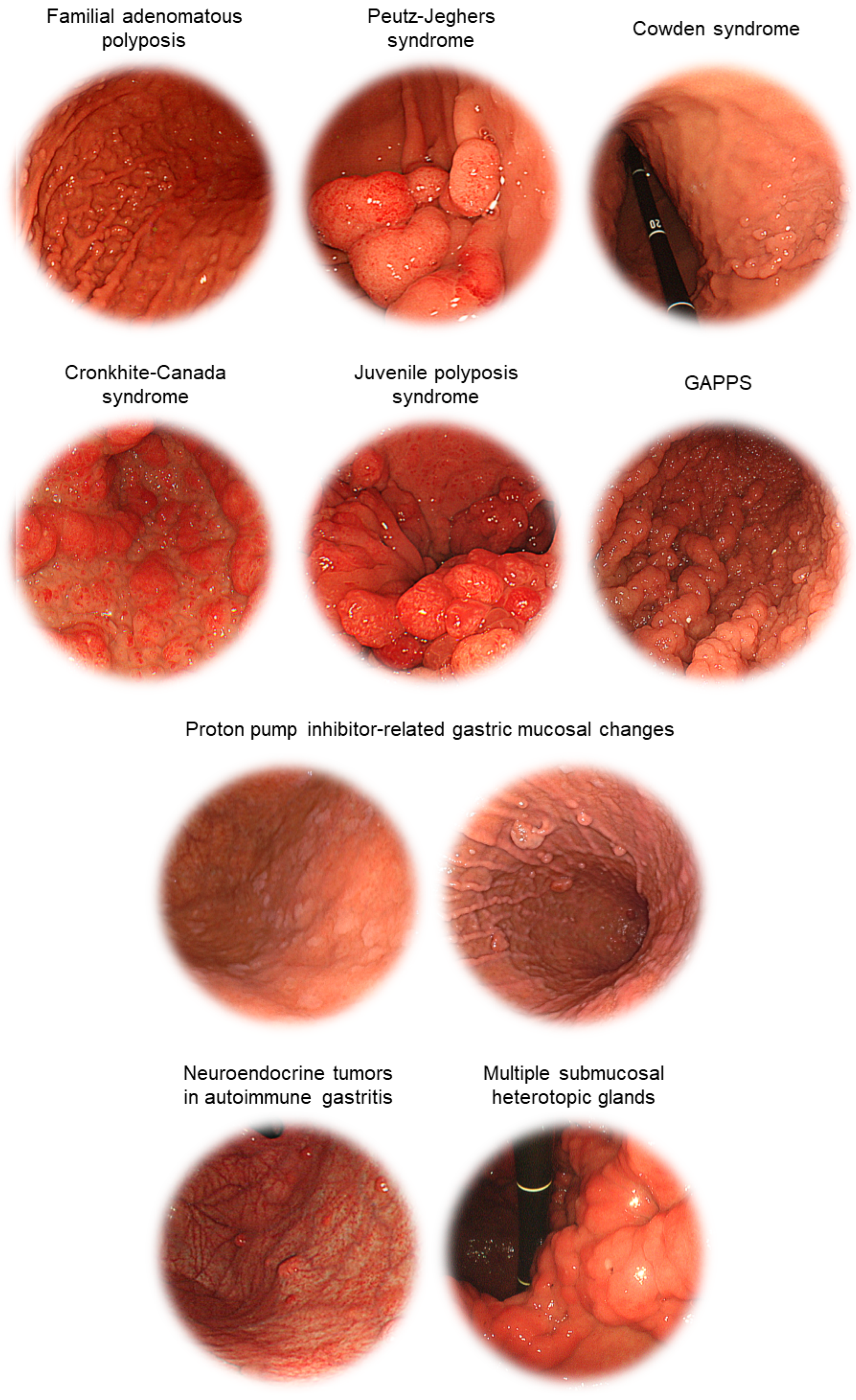
2. Gastrointestinal Polyposis Syndromes with Gastric Polyps
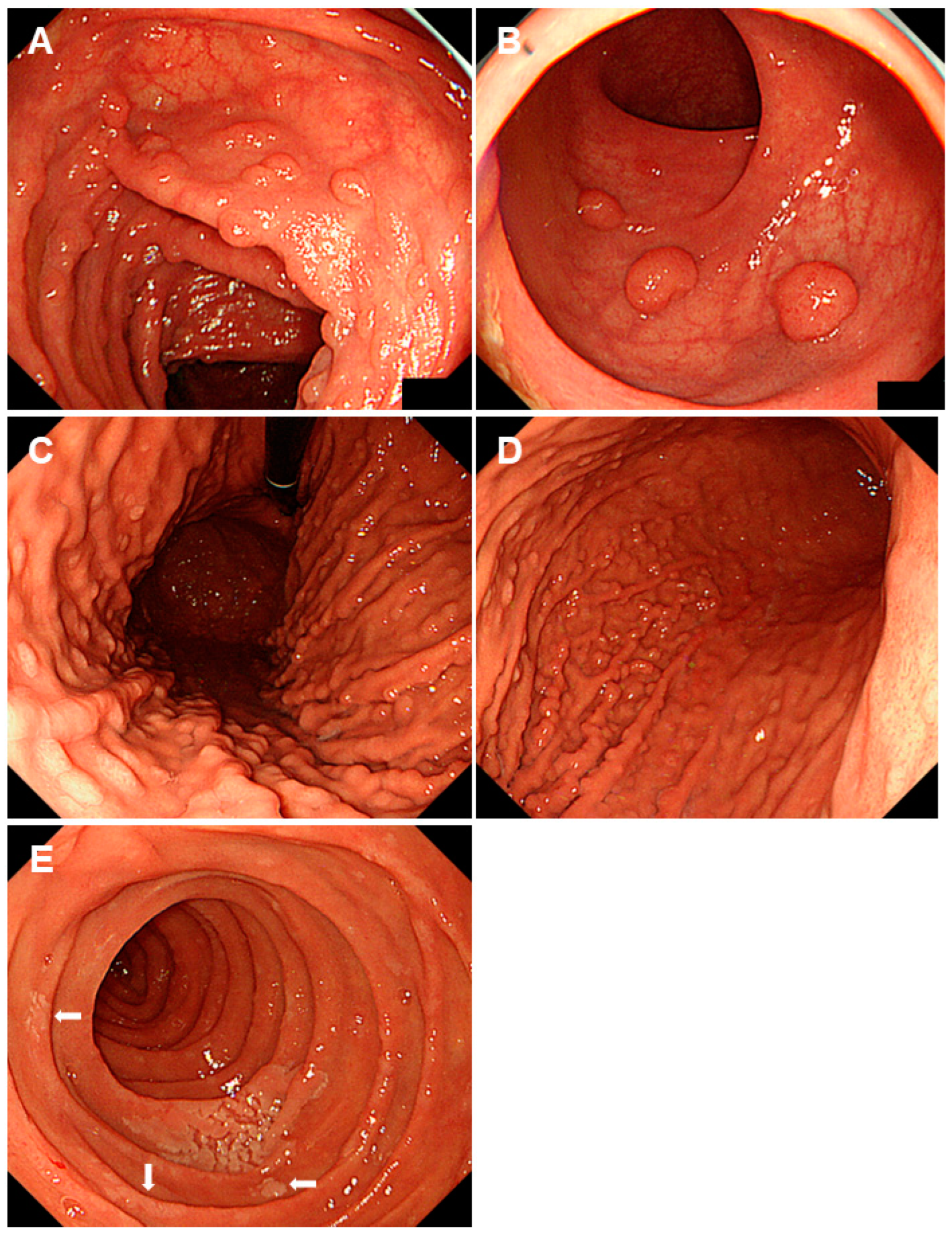
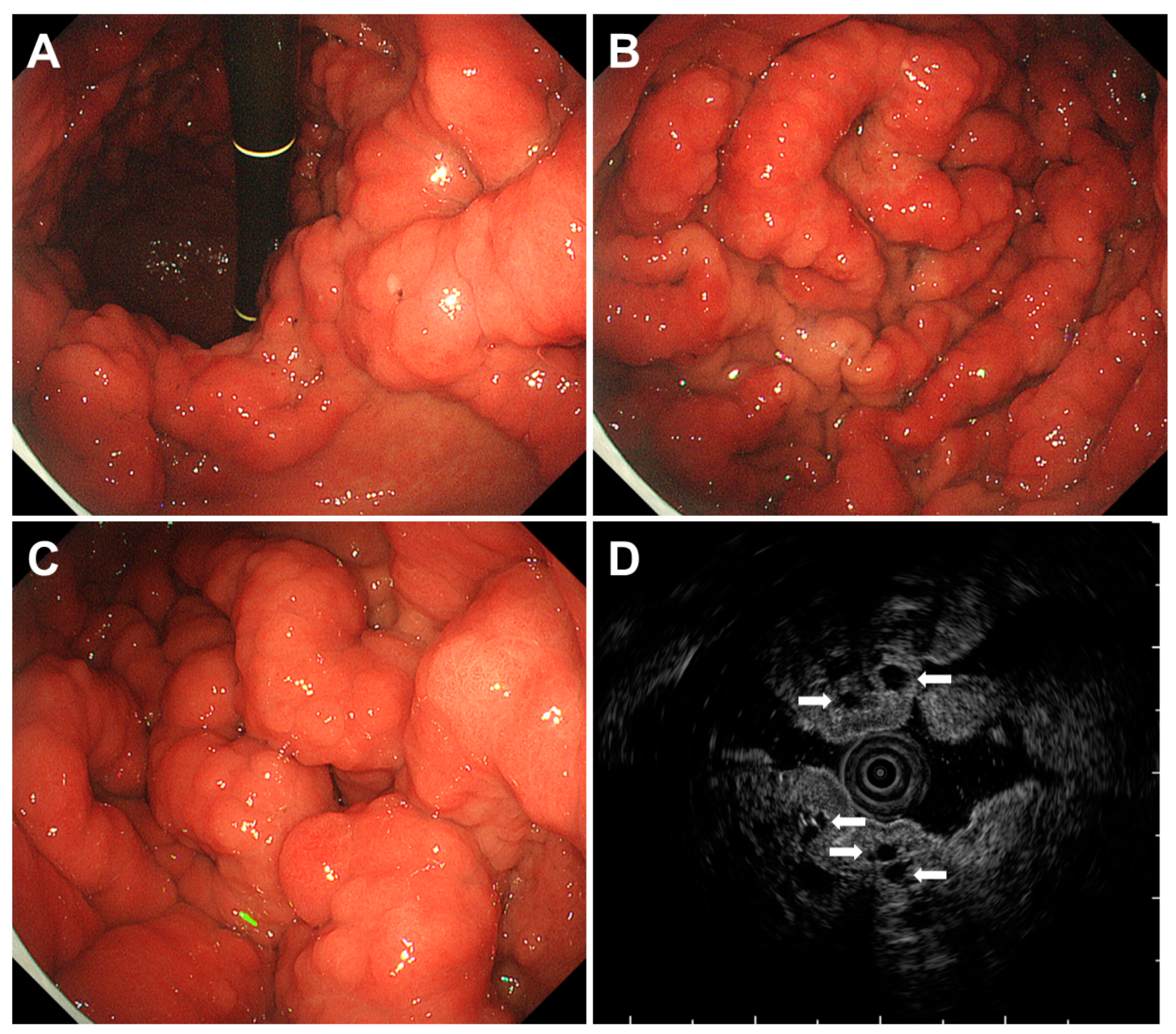
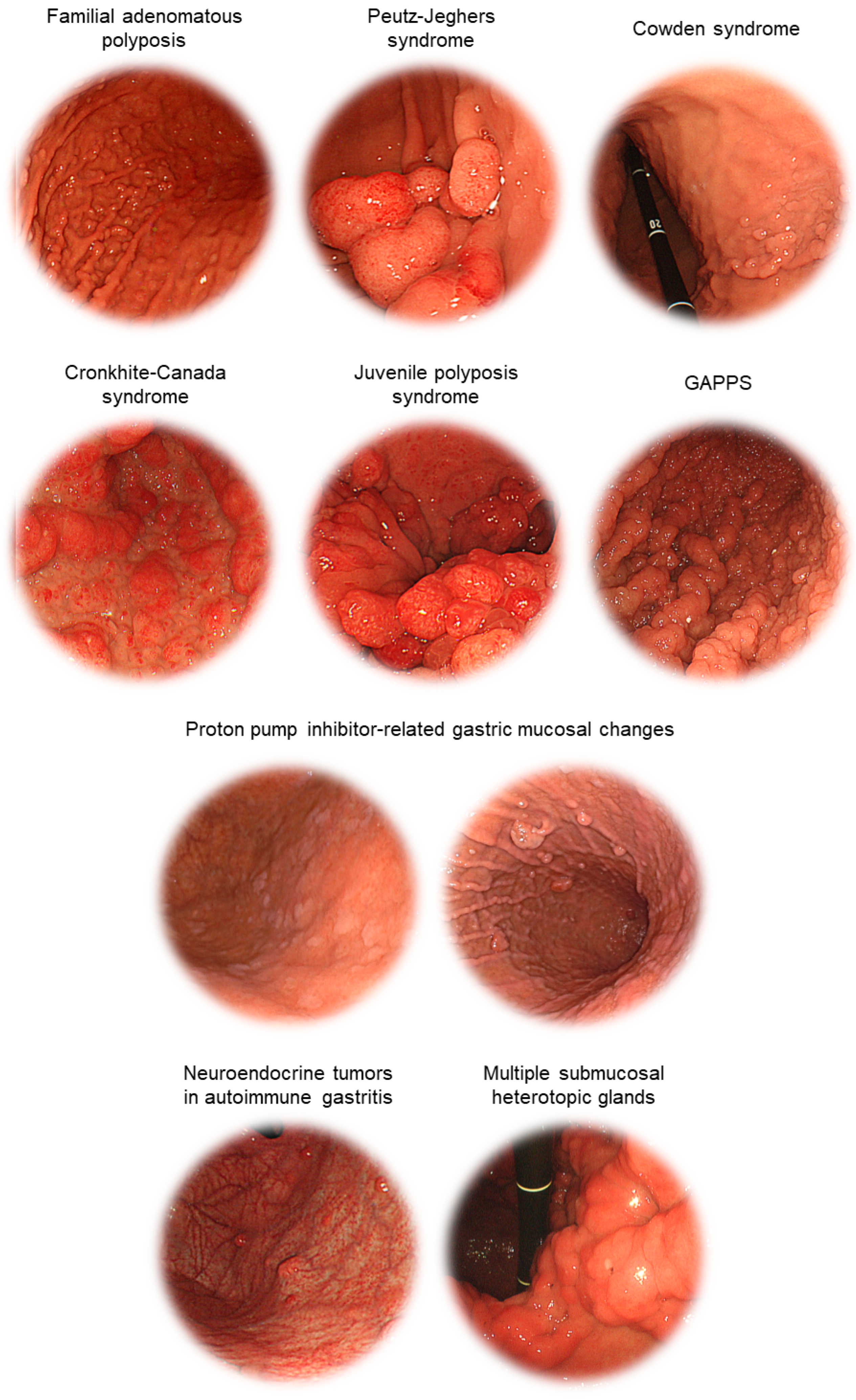
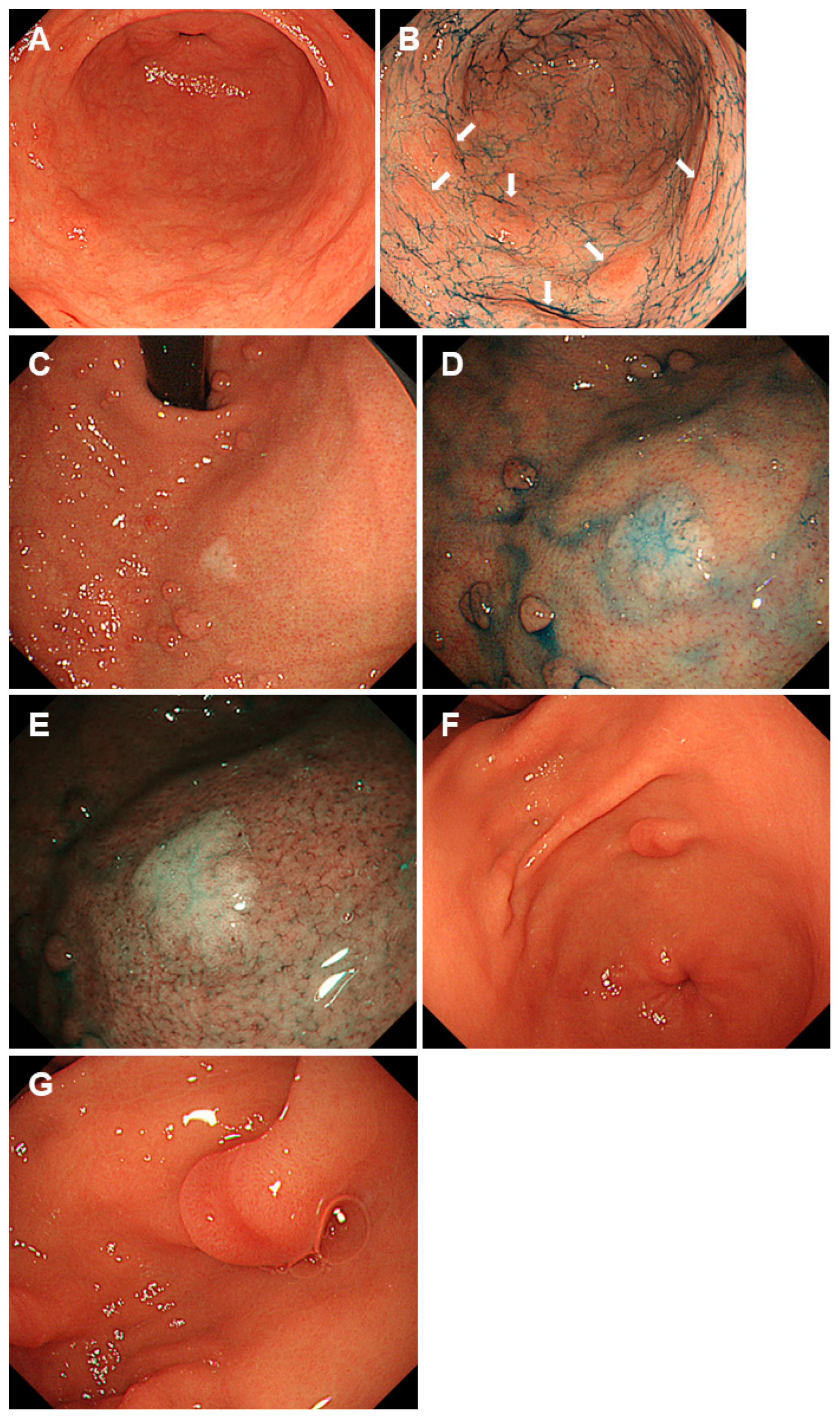
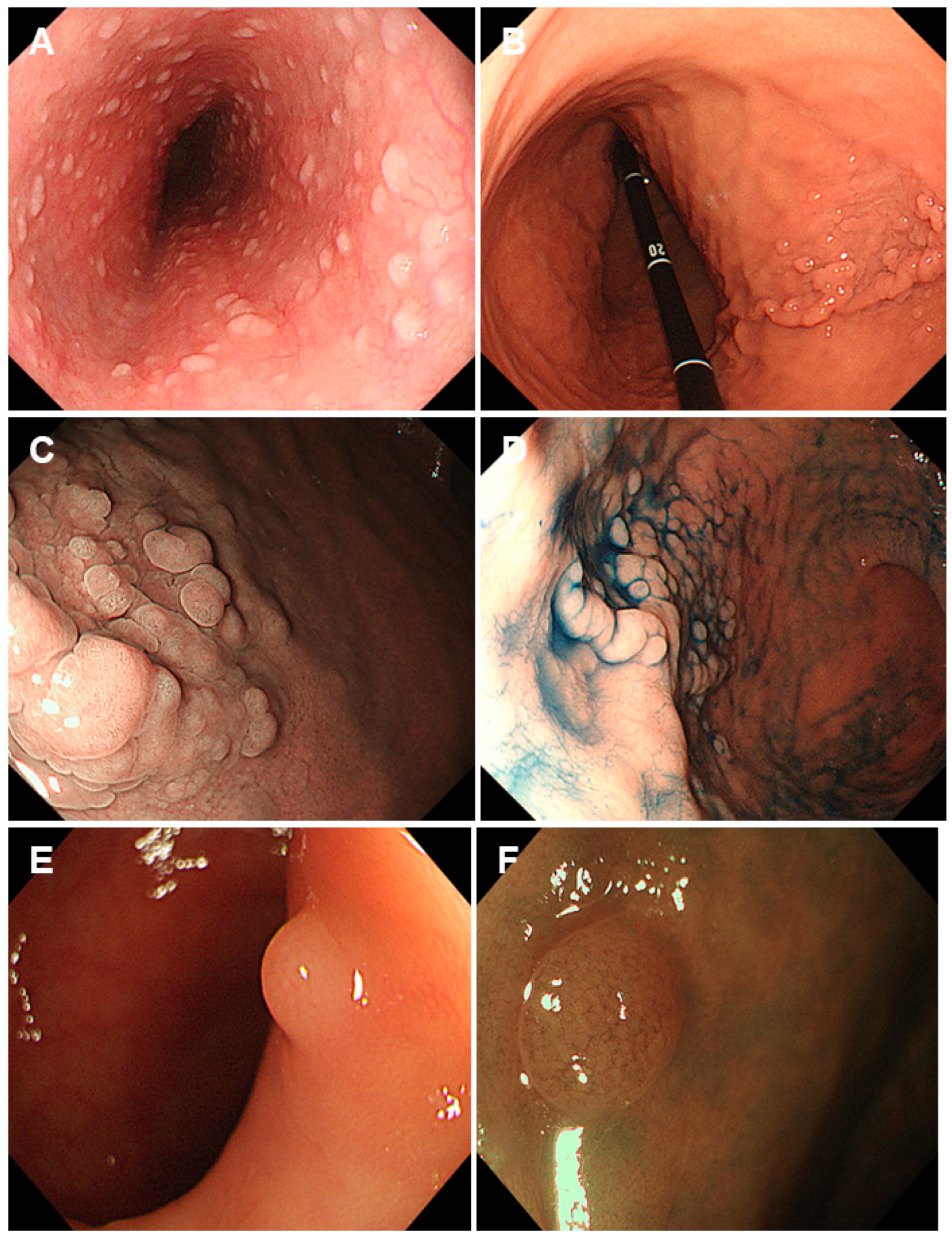
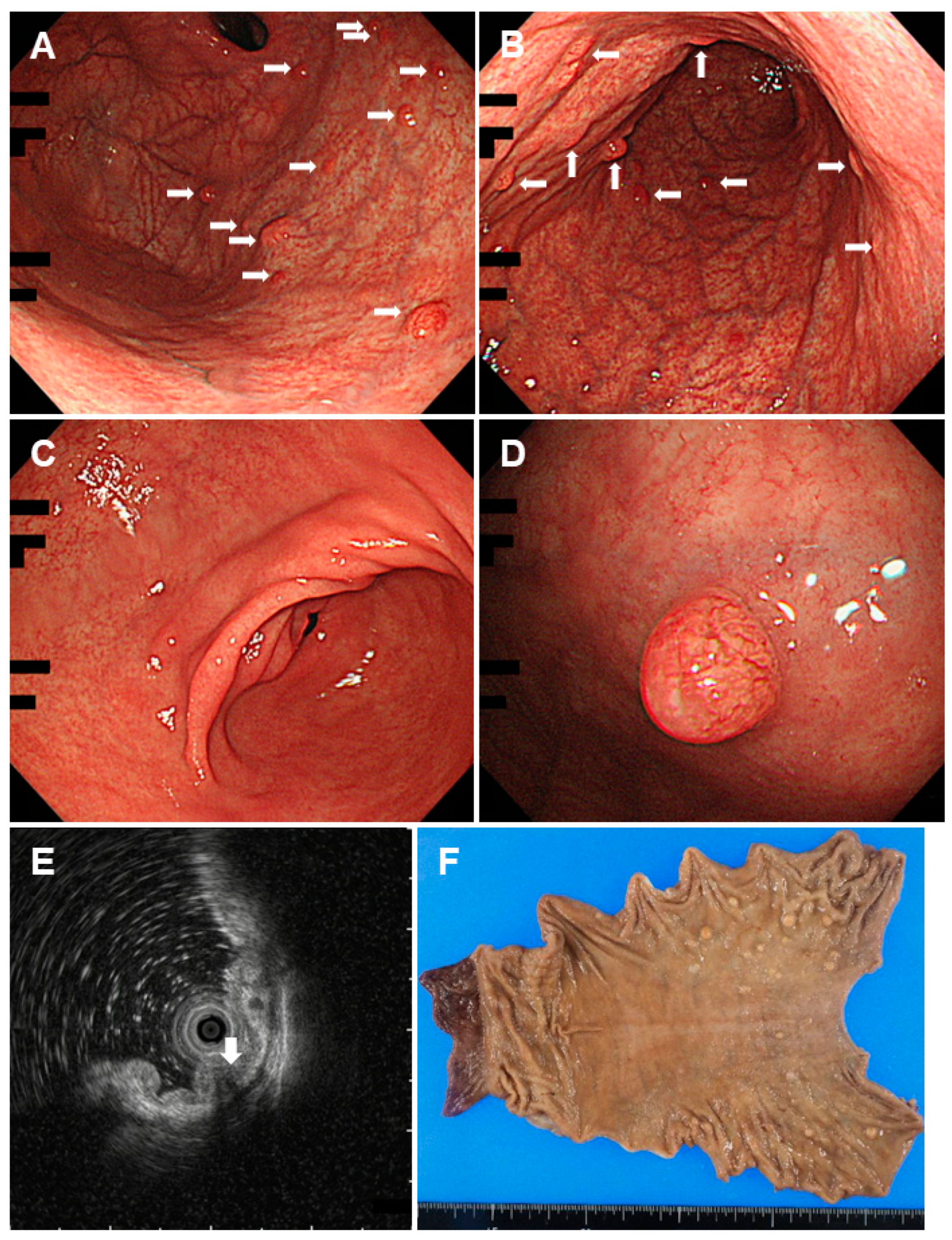
2.1. Familial Adenomatous Polyposis
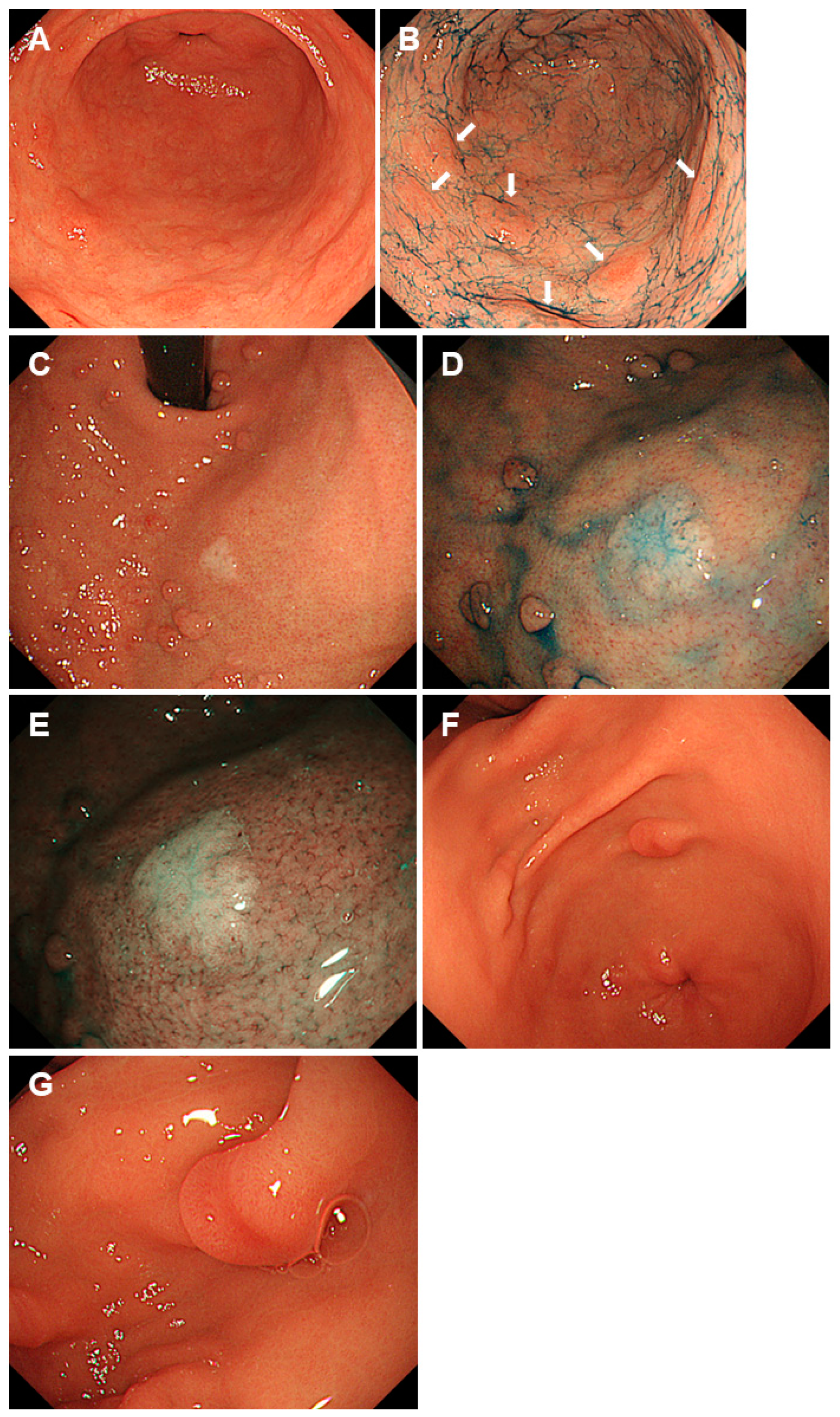
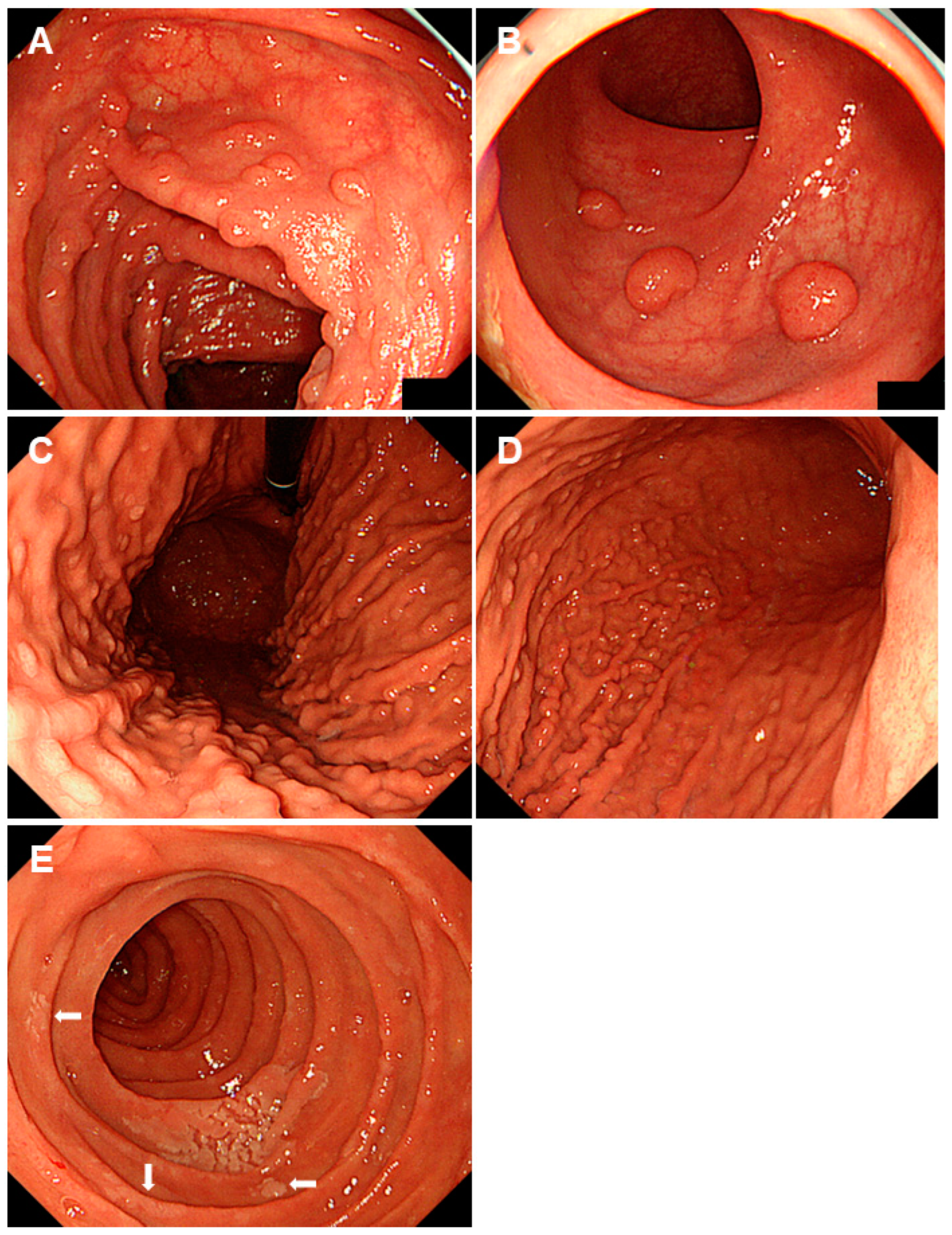
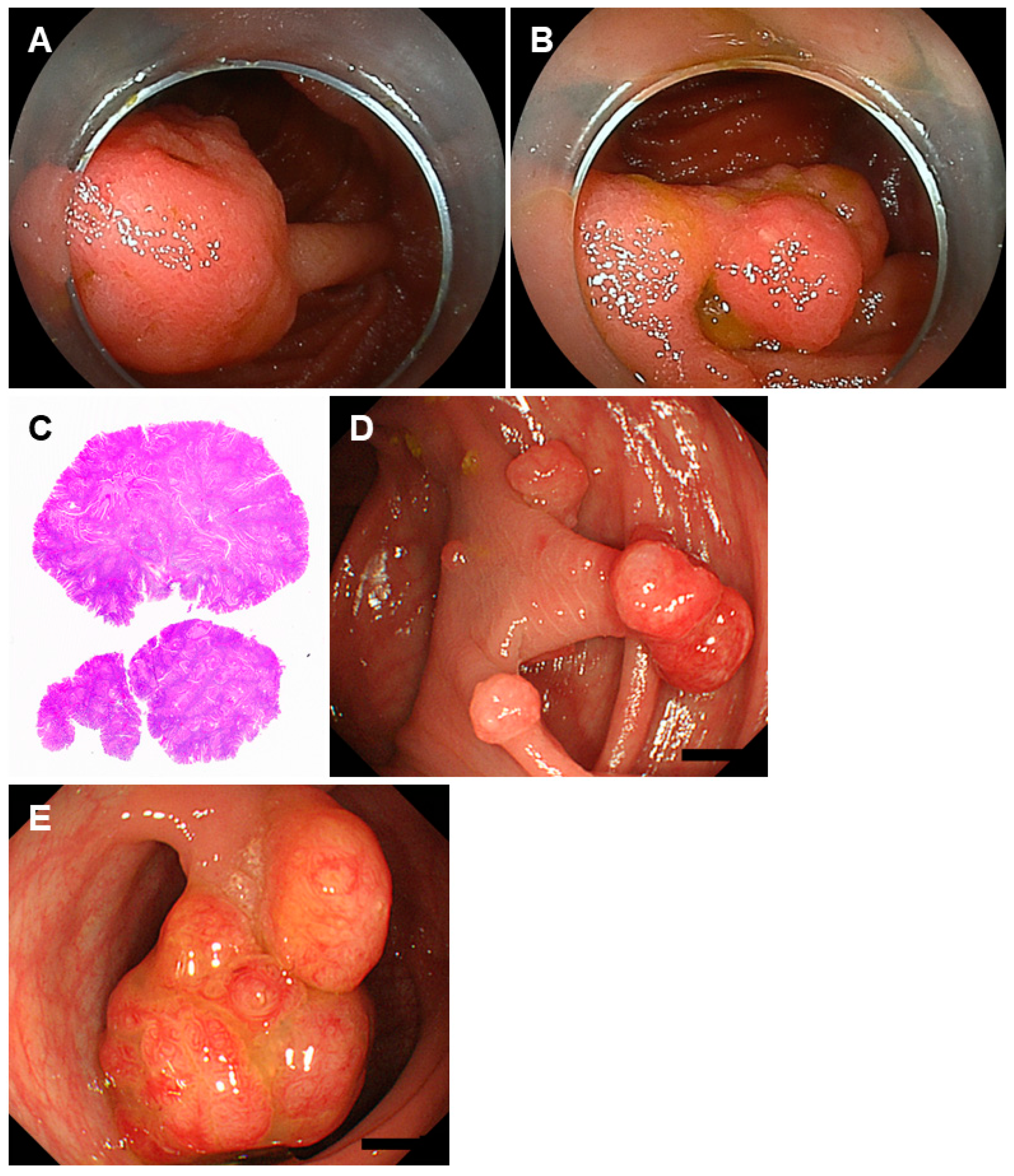
2.2. Peutz-Jeghers Syndrome
2.3. Cowden Syndrome
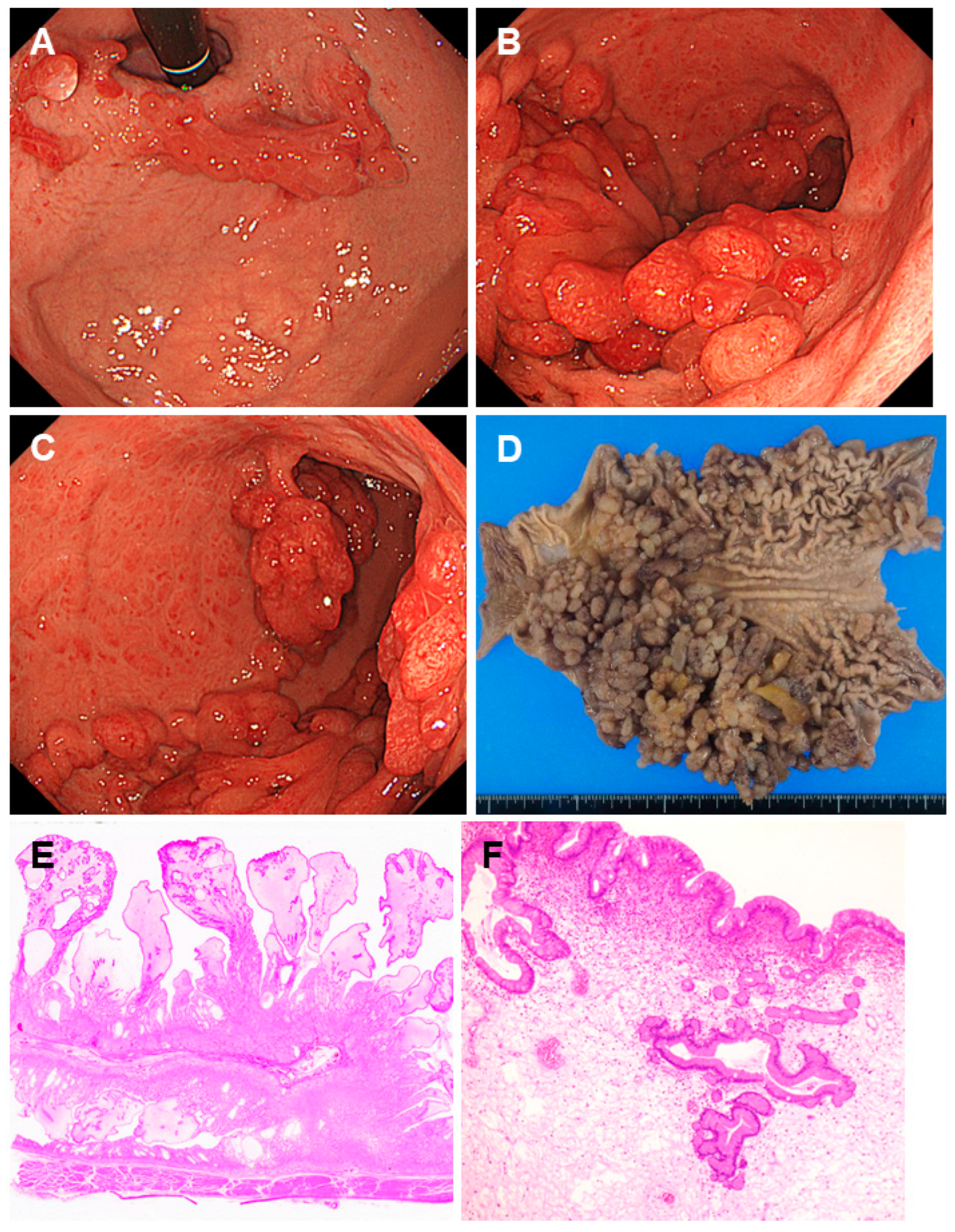
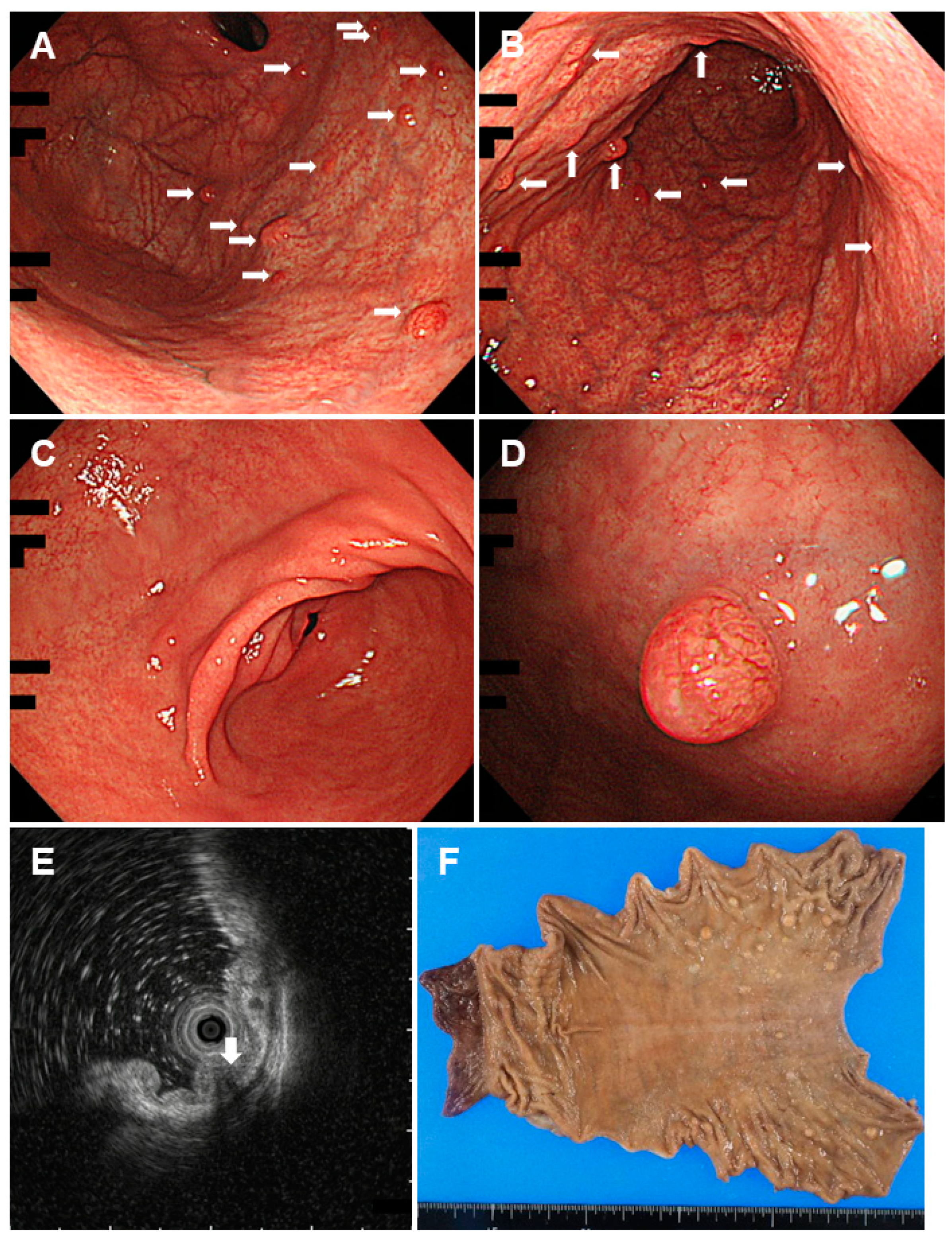
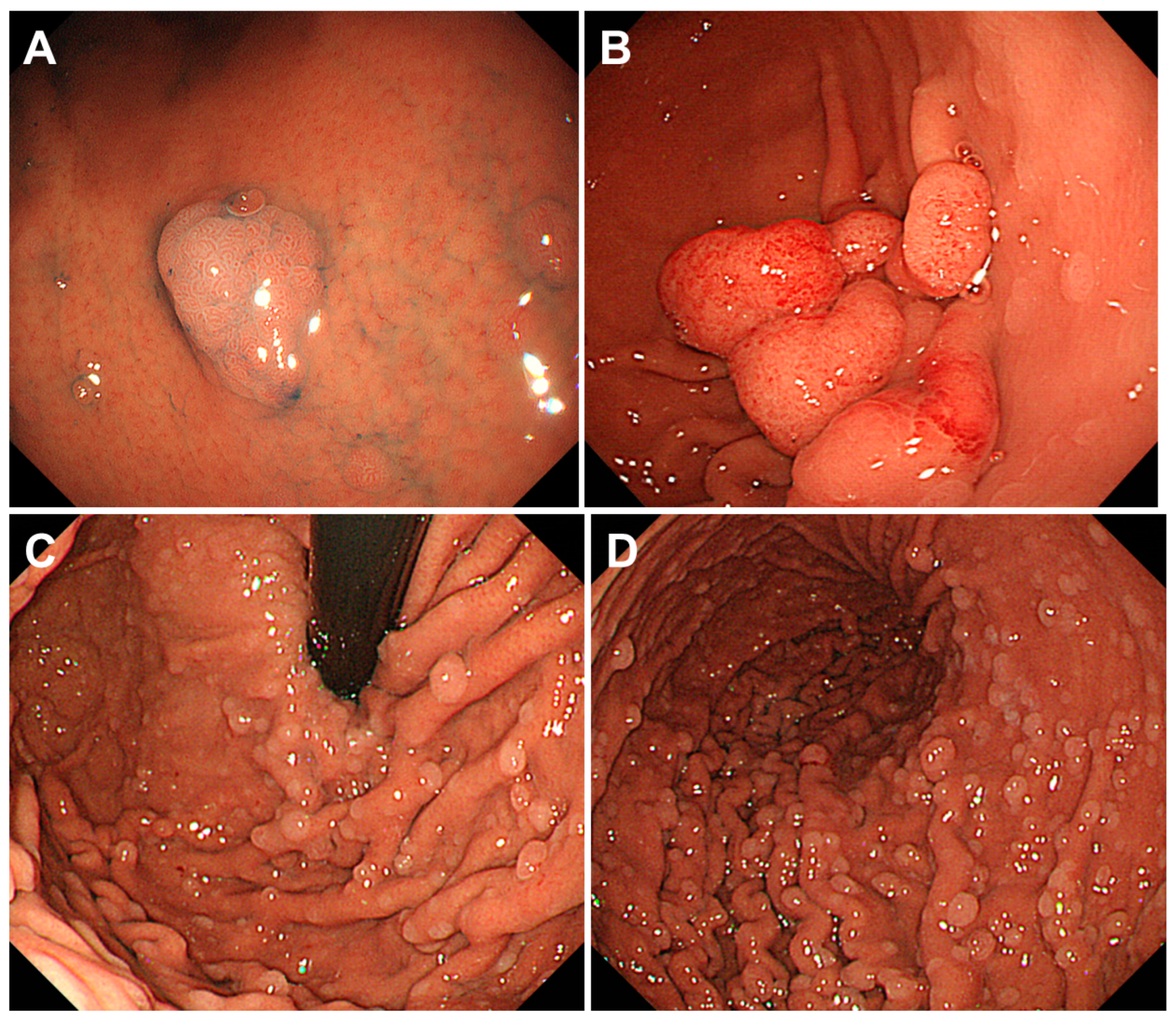
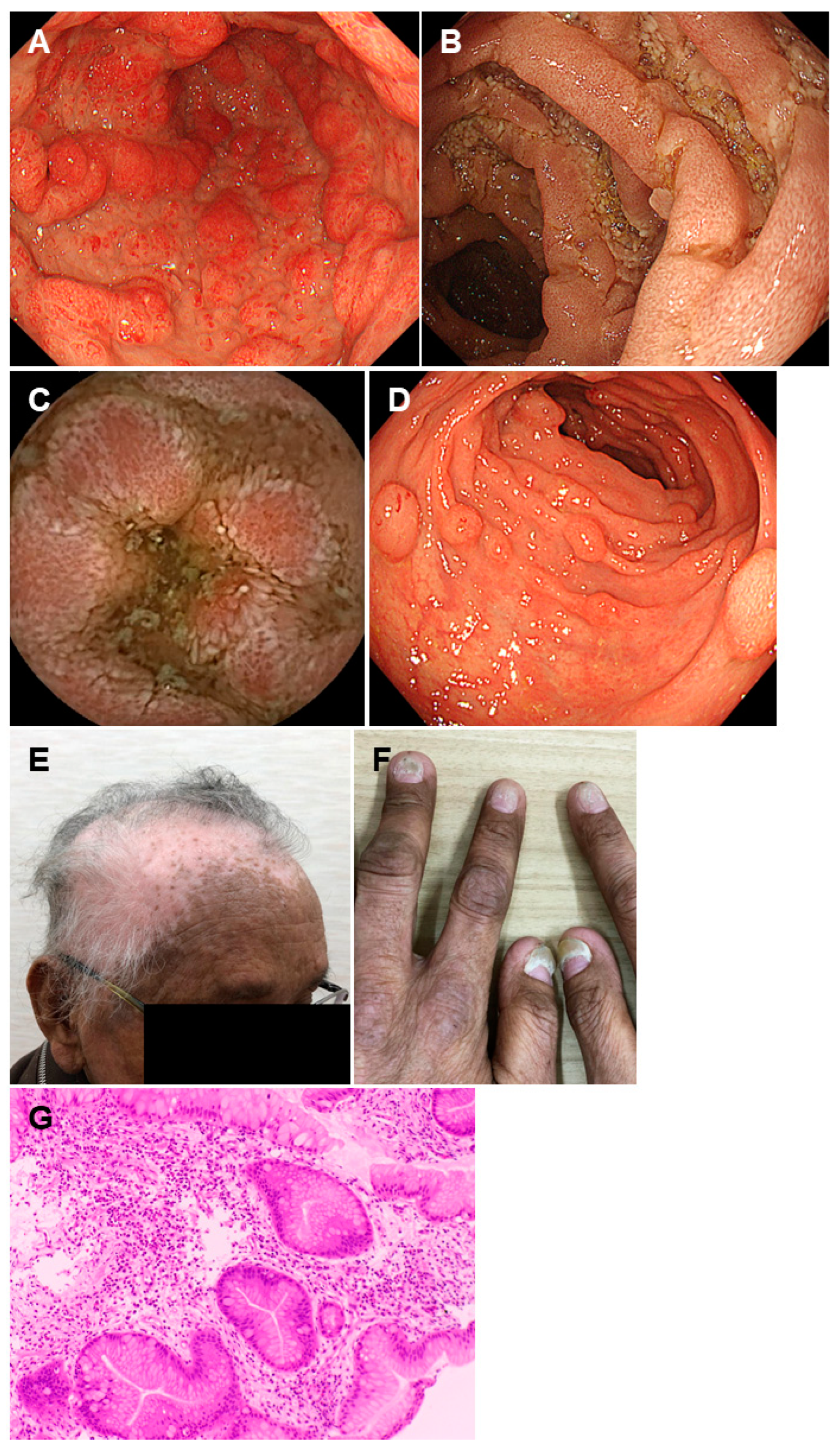
2.4. Cronkhite-Canada Syndrome
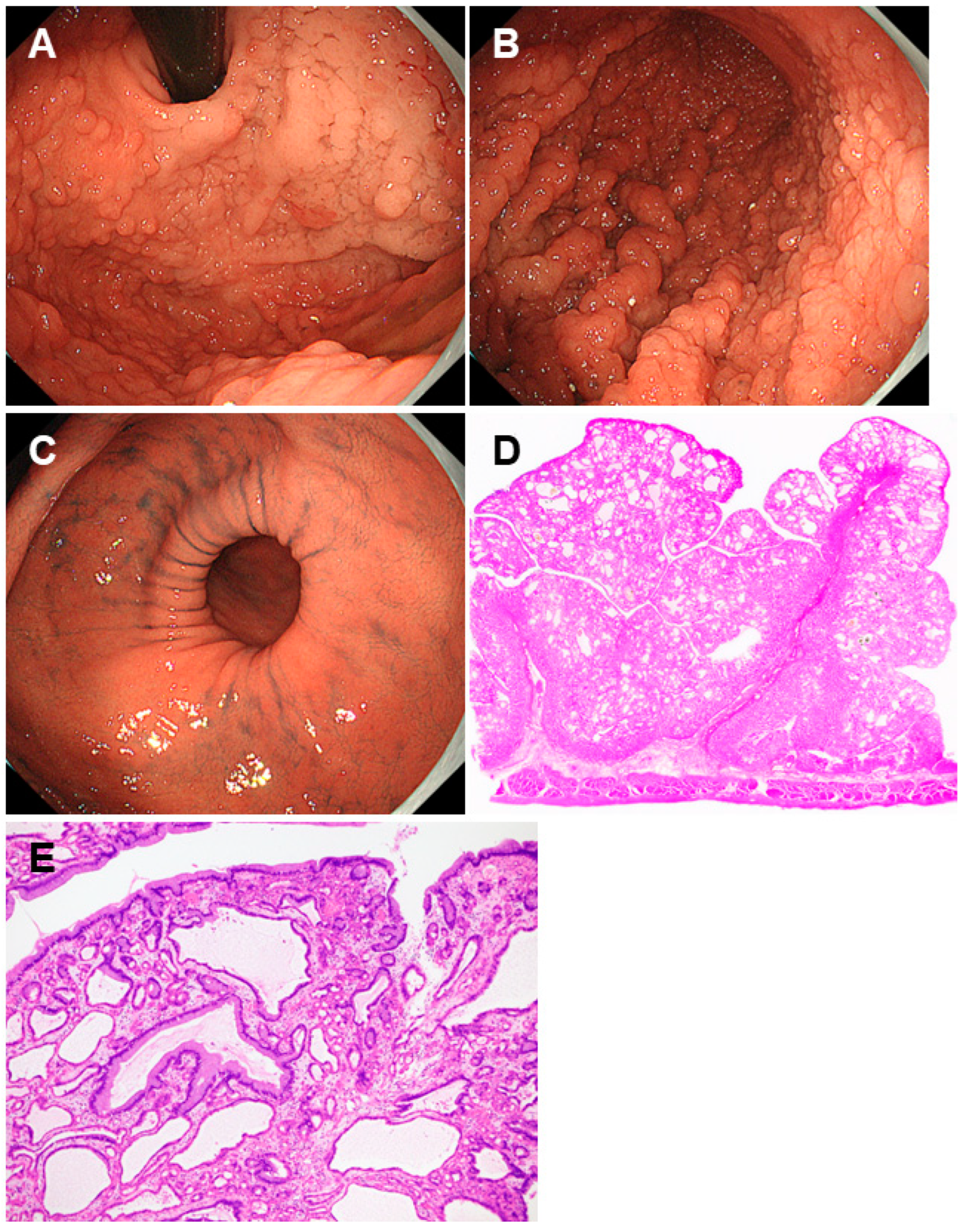
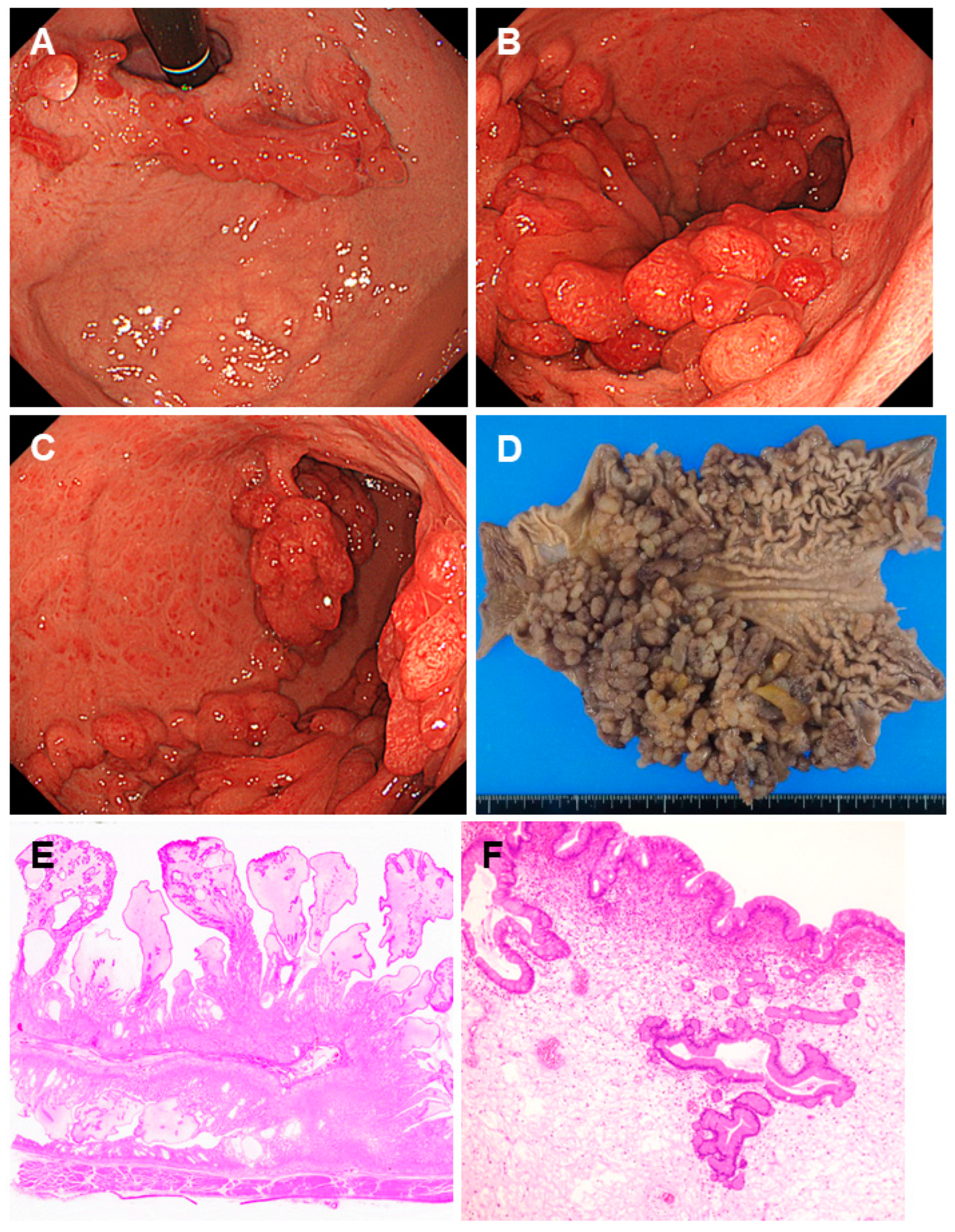
2.5. Juvenile Polyposis Syndrome
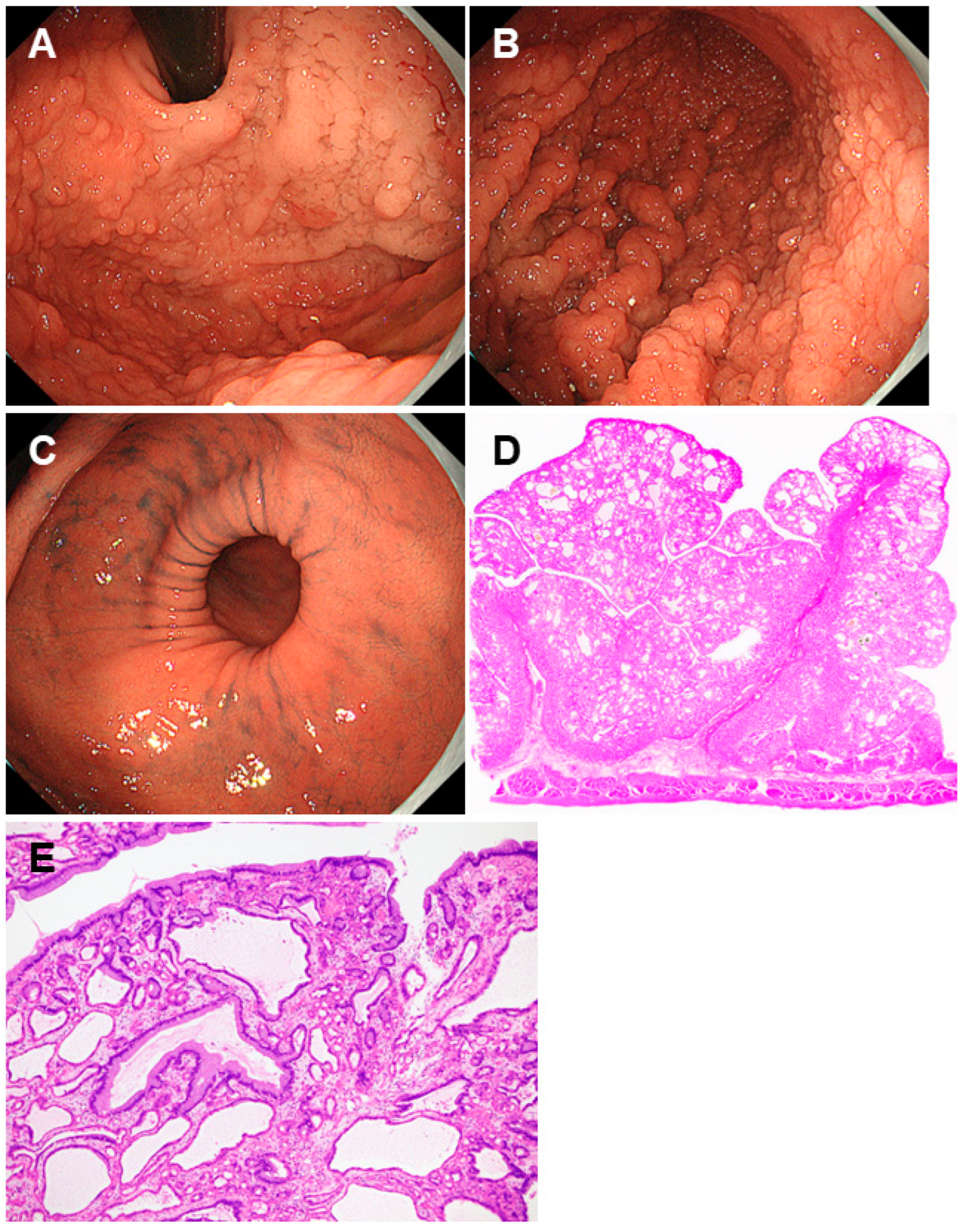
2.6. Gastric Adenocarcinoma and Proximal Polyposis of the Stomach
3. Gastric Polyposis Observed Outside of Gastrointestinal Polyposis Syndrome
3.1. Neuroendocrine Tumors in Autoimmune Gastritis
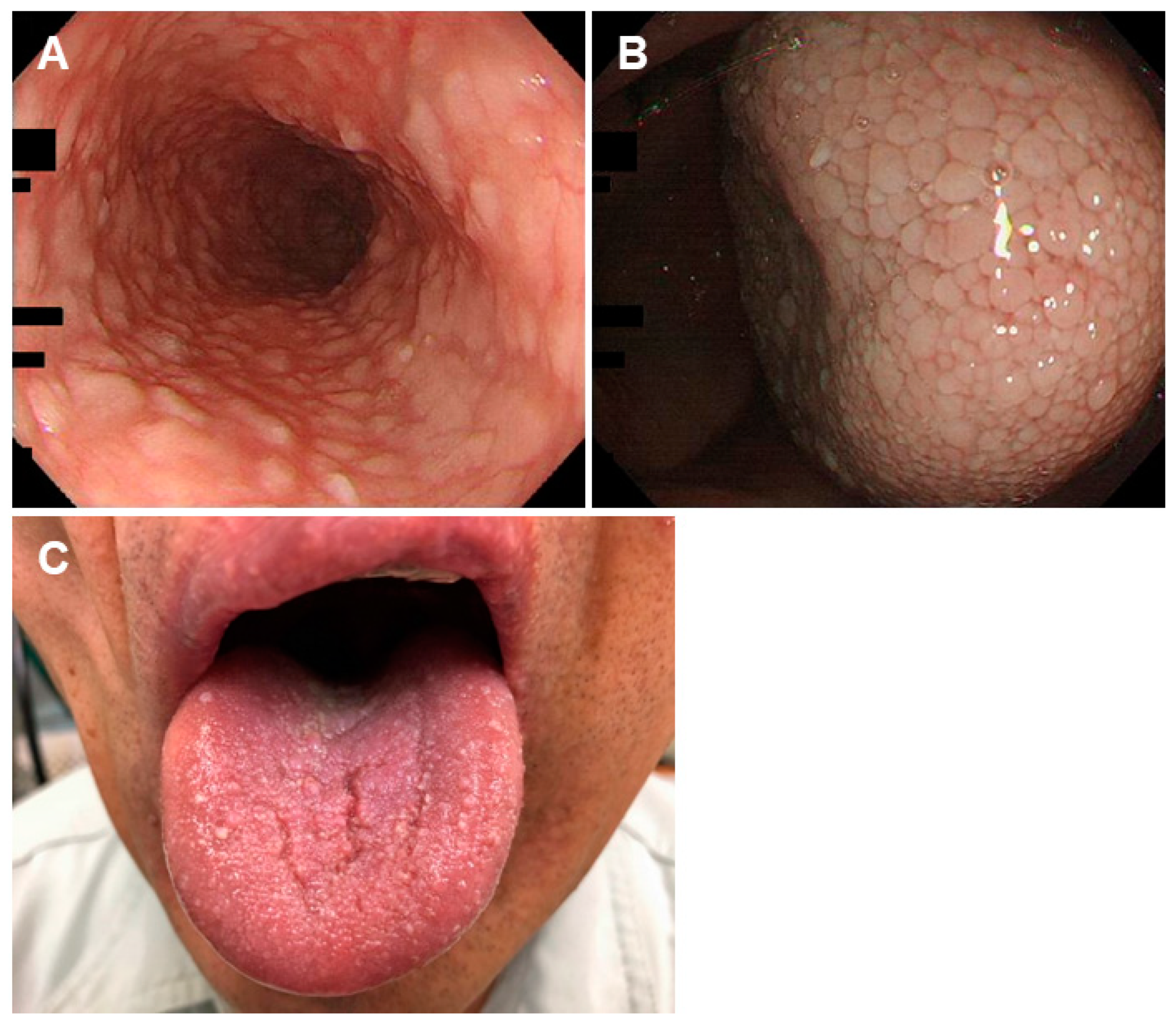
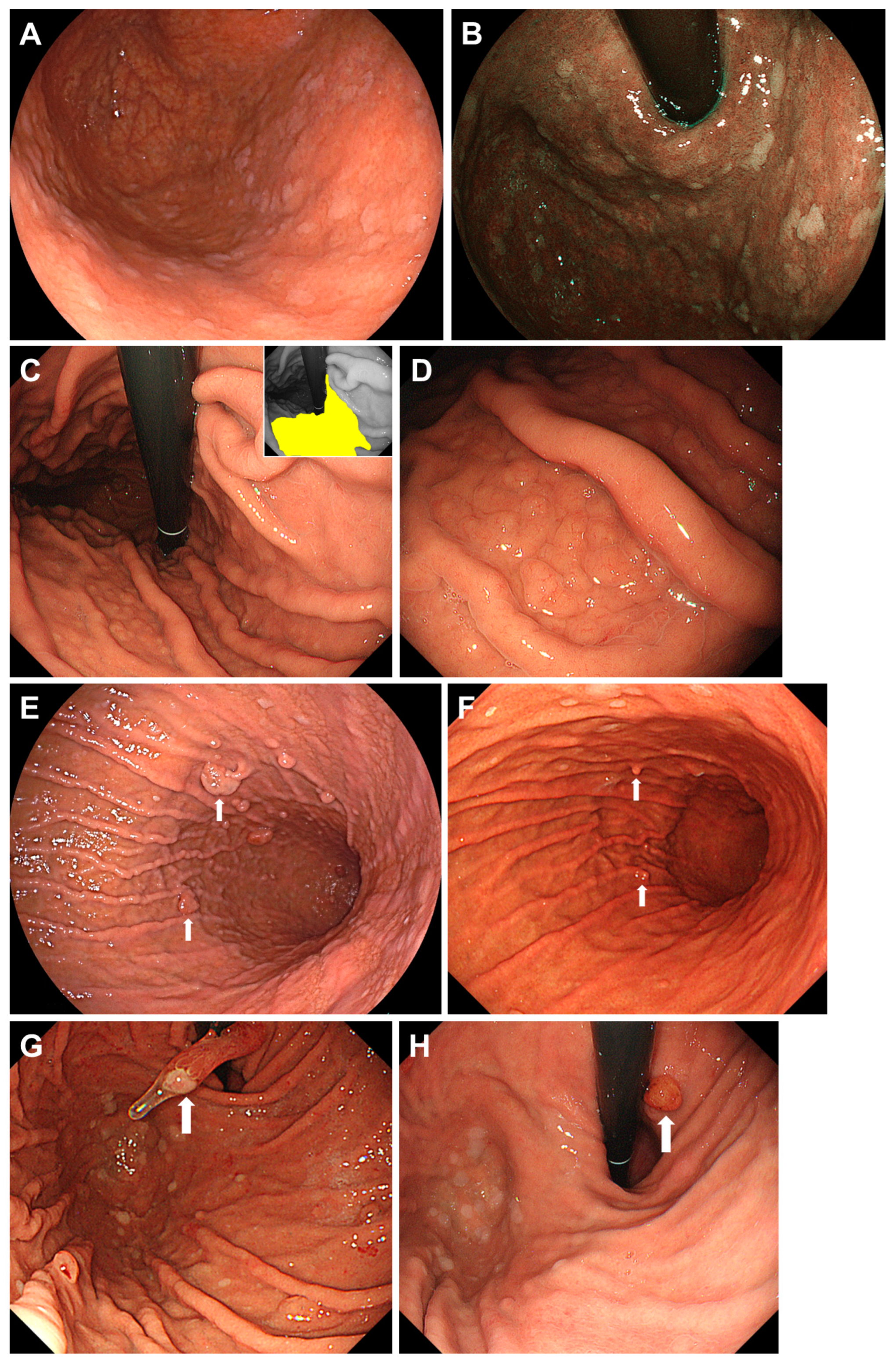
3.2. Proton Pump Inhibitor-Related Gastric Mucosal Changes
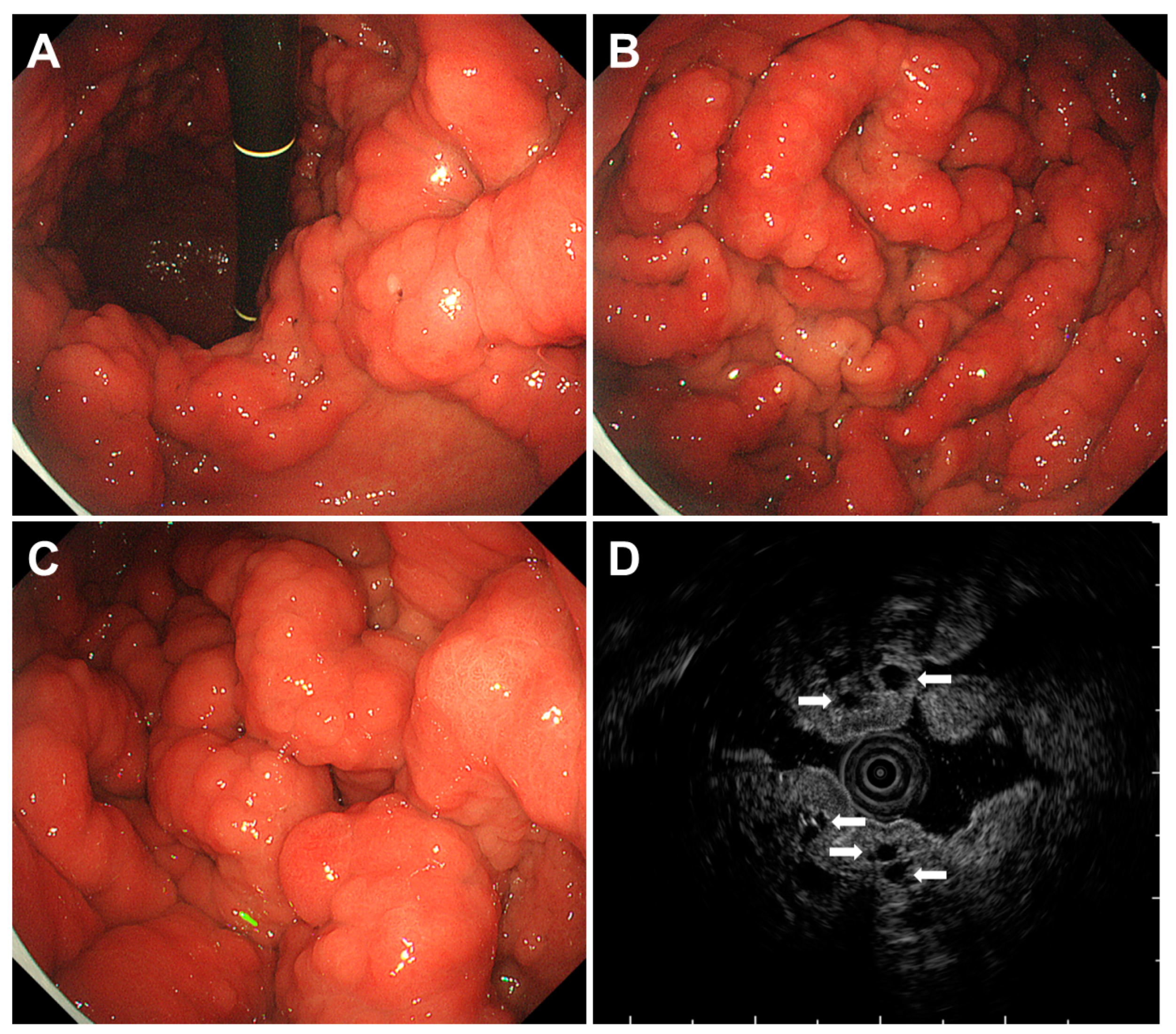
3.3. Multiple Submucosal Heterotopic Glands
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gorji, L.; Albrecht, P. Hamartomatous polyps: Diagnosis, surveillance, and management. World J. Gastroenterol. 2023, 29, 1304–1314. [Google Scholar] [CrossRef]
- Syngal, S.; Brand, R.E.; Church, J.M.; Giardiello, F.M.; Hampel, H.L.; Burt, R.W.; American College of Gastroenterology. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am. J. Gastroenterol. 2015, 110, 223–262. [Google Scholar] [CrossRef]
- Samadder, N.J.; Baffy, N.; Giridhar, K.V.; Couch, F.J.; Riegert-Johnson, D. Hereditary Cancer Syndromes—A Primer on Diagnosis and Management, Part 2: Gastrointestinal Cancer Syndromes. Mayo Clin. Proc. 2019, 94, 1099–1116. [Google Scholar] [CrossRef] [PubMed]
- Imyanitov, E.N.; Kuligina, E.S.; Sokolenko, A.P.; Suspitsin, E.N.; Yanus, G.A.; Iyevleva, A.G.; Ivantsov, A.O.; Aleksakhina, S.N. Hereditary cancer syndromes. World J. Clin. Oncol. 2023, 14, 40–68. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Idos, G.E.; Durno, C.; Giardiello, F.M.; Anderson, J.C.; Burke, C.A.; Dominitz, J.A.; Gross, S.; Gupta, S.; Jacobson, B.C.; et al. Diagnosis and Management of Cancer Risk in the Gastrointestinal Hamartomatous Polyposis Syndromes: Recommendations from the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 2022, 162, 2063–2085. [Google Scholar] [CrossRef] [PubMed]
- Tomita, N.; Ishida, H.; Tanakaya, K.; Yamaguchi, T.; Kumamoto, K.; Tanaka, T.; Hinoi, T.; Miyakura, Y.; Hasegawa, H.; Takayama, T.; et al. Japanese Society for Cancer of the Colon and Rectum (JSCCR) guidelines 2020 for the Clinical Practice of Hereditary Colorectal Cancer. Int. J. Clin. Oncol. 2021, 26, 1353–1419. [Google Scholar] [CrossRef] [PubMed]
- Aelvoet, A.S.; Buttitta, F.; Ricciardiello, L.; Dekker, E. Management of familial adenomatous polyposis and MUTYH-associated polyposis; new insights. Best Pract. Res. Clin. Gastroenterol. 2022, 58–59, 101793. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, L.K.; Burke, C.A.; Bennett, A.E.; Lopez, R.; Hasson, H.; Church, J.M. Fundic gland polyp dysplasia is common in familial adenomatous polyposis. Clin. Gastroenterol. Hepatol. 2008, 6, 180–185. [Google Scholar] [CrossRef] [PubMed]
- van Leerdam, M.E.; Roos, V.H.; van Hooft, J.E.; Dekker, E.; Jover, R.; Kaminski, M.F.; Latchford, A.; Neumann, H.; Pellisé, M.; Saurin, J.C.; et al. Endoscopic management of polyposis syndromes: European Society of Gastrointestinal Endoscopy (ESGE) Guideline. Endoscopy 2019, 51, 877–895. [Google Scholar] [CrossRef]
- Attard, T.M.; Giardiello, F.M.; Argani, P.; Cuffari, C. Fundic gland polyposis with high-grade dysplasia in a child with attenuated familial adenomatous polyposis and familial gastric cancer. J. Pediatr. Gastroenterol. Nutr. 2001, 32, 215–218. [Google Scholar]
- Garrean, S.; Hering, J.; Saied, A.; Jani, J.; Espat, N.J. Gastric adenocarcinoma arising from fundic gland polyps in a patient with familial adenomatous polyposis syndrome. Am. Surg. 2008, 74, 79–83. [Google Scholar] [CrossRef]
- Hofgärtner, W.T.; Thorp, M.; Ramus, M.W.; Delorefice, G.; Chey, W.Y.; Ryan, C.K.; Takahashi, G.W.; Lobitz, J.R. Gastric adenocarcinoma associated with fundic gland polyps in a patient with attenuated familial adenomatous polyposis. Am. J. Gastroenterol. 1999, 94, 2275–2281. [Google Scholar] [CrossRef]
- Shimamoto, Y.; Ishiguro, S.; Takeuchi, Y.; Nakatsuka, S.I.; Yunokizaki, H.; Ezoe, Y.; Nakajima, T.; Matsuno, K.; Nakahira, H.; Tanaka, K.; et al. Gastric neoplasms in patients with familial adenomatous polyposis: Endoscopic and clinicopathologic features. Gastrointest. Endosc. 2021, 94, 1030–1042.e2. [Google Scholar] [CrossRef]
- Kobashi, M.; Iwamuro, M.; Kuraoka, S.; Inoo, S.; Okanoue, S.; Satomi, T.; Hamada, K.; Abe, M.; Kono, Y.; Kanzaki, H.; et al. Endoscopic findings of gastric neoplasms in familial adenomatous polyposis are associated with the phenotypic variations and grades of dysplasia. Medicine 2022, 101, e30997. [Google Scholar] [CrossRef]
- Yang, J.; Gurudu, S.R.; Koptiuch, C.; Agrawal, D.; Buxbaum, J.L.; Abbas Fehmi, S.M.; Fishman, D.S.; Khashab, M.A.; Jamil, L.H.; Jue, T.L.; et al. American Society for Gastrointestinal Endoscopy guideline on the role of endoscopy in familial adenomatous polyposis syndromes. Gastrointest. Endosc. 2020, 91, 963–982.e2. [Google Scholar] [CrossRef] [PubMed]
- Monahan, K.J.; Bradshaw, N.; Dolwani, S.; Desouza, B.; Dunlop, M.G.; East, J.E.; Ilyas, M.; Kaur, A.; Lalloo, F.; Latchford, A.; et al. Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG). Gut 2020, 69, 411–444. [Google Scholar] [CrossRef] [PubMed]
- Klimkowski, S.; Ibrahim, M.; Ibarra Rovira, J.J.; Elshikh, M.; Javadi, S.; Klekers, A.R.; Abusaif, A.A.; Moawad, A.W.; Ali, K.; Elsayes, K.M. Peutz-Jeghers Syndrome and the Role of Imaging: Pathophysiology, Diagnosis, and Associated Cancers. Cancers 2021, 13, 5121. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.L.; Zhang, Z.; Zhang, Y.H.; Yu, P.F.; Dong, Z.W.; Yang, H.R.; Yuan, Y. Detection and analysis of common pathogenic germline mutations in Peutz-Jeghers syndrome. World J. Gastroenterol. 2021, 27, 6631–6646. [Google Scholar] [CrossRef] [PubMed]
- Jelsig, A.M.; van Overeem Hansen, T.; Gede, L.B.; Qvist, N.; Christensen, L.L.; Lautrup, C.K.; Frederiksen, J.H.; Sunde, L.; Ousager, L.B.; Ljungmann, K.; et al. Survival, surveillance, and genetics in patients with Peutz-Jeghers syndrome: A nationwide study. Clin. Genet. 2023, 104, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.X.; Jiang, L.X.; Chen, Y.R.; Zhang, Y.H.; Zhang, Z.; Yu, P.F.; Dong, Z.W.; Yang, H.R.; Gu, G.L. Clinical features, diagnosis, and treatment of Peutz-Jeghers syndrome: Experience with 566 Chinese cases. World J. Gastroenterol. 2023, 29, 1627–1637. [Google Scholar] [CrossRef]
- Bartholomew, L.G.; Dahlin, D.C.; Waugh, J.M. Intestinal polyposis associated with mucocutaneous melanin pigmentation Peutz-Jeghers syndrome; review of literature and report of six cases with special reference to pathologic findings. Gastroenterology 1957, 32, 434–451. [Google Scholar] [CrossRef]
- Iwamuro, M.; Toyokawa, T.; Matsueda, K.; Hori, S.; Yoshioka, M.; Moritou, Y.; Tanaka, T.; Mizuno, M.; Okada, H. Two Types of Polyp Shape Observed in the Stomach of Patients with Peutz-Jeghers Syndrome. Acta Medica Okayama 2021, 75, 471–477. [Google Scholar]
- Yamamoto, H.; Sakamoto, H.; Kumagai, H.; Abe, T.; Ishiguro, S.; Uchida, K.; Kawasaki, Y.; Saida, Y.; Sano, Y.; Takeuchi, Y.; et al. Clinical Guidelines for Diagnosis and Management of Peutz-Jeghers Syndrome in Children and Adults. Digestion 2023, 104, 335–347. [Google Scholar] [CrossRef]
- Wagner, A.; Aretz, S.; Auranen, A.; Bruno, M.J.; Cavestro, G.M.; Crosbie, E.J.; Goverde, A.; Jelsig, A.M.; Latchford, A.; Leerdam, M.E.V.; et al. The Management of Peutz-Jeghers Syndrome: European Hereditary Tumour Group (EHTG) Guideline. J. Clin. Med. 2021, 10, 473. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kim, E.R.; Park, J.J.; Kim, E.S.; Goong, H.J.; Kim, K.O.; Nam, J.H.; Park, Y.; Lee, S.P.; Jang, H.J.; et al. Cancer risk in patients with Peutz-Jeghers syndrome in Korea: A retrospective multi-center study. Korean J. Intern. Med. 2023, 38, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Yehia, L.; Heald, B.; Eng, C. Clinical Spectrum and Science Behind the Hamartomatous Polyposis Syndromes. Gastroenterology 2023, 164, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Z.; Wang, Y.; Wu, J.; Yu, Z.; Chen, C.; Chen, J.; Wu, B.; Chen, Y. High risk and early onset of cancer in Chinese patients with Peutz-Jeghers syndrome. Front. Oncol. 2022, 12, 900516. [Google Scholar] [CrossRef] [PubMed]
- Pilarski, R.; Burt, R.; Kohlman, W.; Pho, L.; Shannon, K.M.; Swisher, E. Cowden syndrome and the PTEN hamartoma tumor syndrome: Systematic review and revised diagnostic criteria. J. Natl. Cancer Inst. 2013, 105, 1607–1616. [Google Scholar] [CrossRef]
- Daly, M.B.; Pilarski, R.; Yurgelun, M.B.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Garber, J.E.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 1.2020: Featured updates to the NCCN guidelines. J. Natl. Compr. Cancer Netw. 2020, 18, 380–391. [Google Scholar] [CrossRef]
- McGarrity, T.J.; Wagner Baker, M.J.; Ruggiero, F.M.; Thiboutot, D.M.; Hampel, H.; Zhou, X.P.; Eng, C. GI polyposis and glycogenic acanthosis of the esophagus associated with PTEN mutation positive Cowden syndrome in the absence of cutaneous manifestations. Am. J. Gastroenterol. 2003, 98, 1429–1434. [Google Scholar] [CrossRef]
- Ha, M.; Chung, J.W.; Hahm, K.B.; Kim, Y.J.; Lee, W.; An, J.; Kim, D.K.; Kim, M.G. A case of Cowden syndrome diagnosed from multiple gastric polyposis. World J. Gastroenterol. 2012, 18, 861–864. [Google Scholar] [CrossRef]
- Marra, G.; Armelao, F.; Vecchio, F.M.; Percesepe, A.; Anti, M. Cowden’s disease with extensive gastrointestinal polyposis. J. Clin. Gastroenterol 1994, 18, 42–47. [Google Scholar] [CrossRef]
- Stanich, P.P.; Francis, D.L.; Sweetser, S. The spectrum of findings in Cowden syndrome. Clin. Gastroenterol. Hepatol. 2011, 9, e2–e3. [Google Scholar] [CrossRef] [PubMed]
- Plamper, M.; Gohlke, B.; Woelfle, J. PTEN hamartoma tumor syndrome in childhood and adolescence-a comprehensive review and presentation of the German pediatric guideline. Mol. Cell. Pediatr. 2022, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, C.; Komoto, S.; Tomita, K.; Hokari, R.; Tanaka, M.; Hirata, I.; Hibi, T.; Kaunitz, J.D.; Miura, S. Endoscopic and clinical evaluation of treatment and prognosis of Cronkhite-Canada syndrome: A Japanese nationwide survey. J. Gastroenterol. 2016, 51, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Y.; Sang, L.X.; Chang, B. Cronkhite-Canada syndrome: From clinical features to treatment. Gastroenterol. Rep. 2020, 8, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Shao, Y.; Zhao, D.H.; Xue, F.; Ma, X.B.; Li, Q.; Liu, C.X. Endoscopic and Pathological Characteristics of Cronkhite-Canada Syndrome: A Retrospective Analysis of 76 Cases. Turk. J. Gastroenterol. 2022, 33, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Huang, F.; Wang, Y.; Zhou, J.; Zhao, Q.; Liu, L. Clinical and Endoscopic Characteristics of Chinese Cronkhite-Canada Syndrome Patients: A Retrospective Study of 103 Cases. Dig. Dis. 2021, 39, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Kopáčová, M.; Urban, O.; Cyrany, J.; Laco, J.; Bureš, J.; Rejchrt, S.; Bártová, J.; Tachecí, I. Cronkhite-Canada syndrome: Review of the literature. Gastroenterol. Res. Pract. 2013, 2013, 856873. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, U.; Chaudhary, A.; Karim, M.A.; Anwar, H.; Merrell, N. Cronkhite-Canada Syndrome: A Rare Cause of Chronic Diarrhea. Gastroenterol. Res. 2017, 10, 196–198. [Google Scholar] [CrossRef]
- Zhao, Y.; Lv, F.; Yang, X.; Wang, Y.; Zhang, S.; Li, P. Cronkhite-Canada Syndrome Associated with Superficial Esophageal Carcinoma: A Case Report and Literature Review. Front. Med. 2022, 9, 855336. [Google Scholar] [CrossRef]
- Yun, S.H.; Cho, J.W.; Kim, J.W.; Kim, J.K.; Park, M.S.; Lee, N.E.; Lee, J.U.; Lee, Y.J. Cronkhite-Canada syndrome associated with serrated adenoma and malignant polyp: A case report and a literature review of 13 cronkhite-Canada syndrome cases in Korea. Clin. Endosc. 2013, 46, 301–305. [Google Scholar] [CrossRef]
- Kaneko, Y.; Kato, H.; Tachimori, Y.; Watanabe, H.; Ushio, K.; Yamaguchi, H.; Itabashi, M.; Noguchi, M. Triple carcinomas in Cronkhite-Canada syndrome. Jpn. J. Clin. Oncol. 1991, 21, 194–202. [Google Scholar]
- Isobe, T.; Kobayashi, T.; Hashimoto, K.; Kizaki, J.; Miyagi, M.; Aoyagi, K.; Koufuji, K.; Shirouzu, K. Cronkhite-Canada syndrome complicated with multiple gastric cancers and multiple colon adenomas. Am. J. Case Rep. 2013, 14, 120–128. [Google Scholar]
- Brosens, L.A.; Langeveld, D.; van Hattem, W.A.; Giardiello, F.M.; Offerhaus, G.J. Juvenile polyposis syndrome. World J. Gastroenterol. 2011, 17, 4839–4844. [Google Scholar] [CrossRef] [PubMed]
- MacFarland, S.P.; Ebrahimzadeh, J.E.; Zelley, K.; Begum, L.; Bass, L.M.; Brand, R.E.; Dudley, B.; Fishman, D.S.; Ganzak, A.; Karloski, E.; et al. Phenotypic Differences in Juvenile Polyposis Syndrome with or without a Disease-causing SMAD4/BMPR1A Variant. Cancer Prev. Res. 2021, 14, 15–222. [Google Scholar] [CrossRef] [PubMed]
- Worthley, D.L.; Phillips, K.D.; Wayte, N.; Schrader, K.A.; Healey, S.; Kaurah, P.; Shulkes, A.; Grimpen, F.; Clouston, A.; Moore, D.; et al. Gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS): A new autosomal dominant syndrome. Gut 2012, 61, 774–779. [Google Scholar] [CrossRef]
- Iwamuro, M.; Kawano, S.; Otsuka, M. An Unusual Case of Gastric Polyposis. Gastroenterology 2023, 165, 1110–1113. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki, M.; Matsumoto, C.; Mimori, K.; Baba, H. The comprehensive review of gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS) from diagnosis and treatment. Ann. Gastroenterol. Surg. 2023, 7, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Gullo, I.; van der Post, R.S.; Carneiro, F. Recent advances in the pathology of heritable gastric cancer syndromes. Histopathology 2021, 78, 125–147. [Google Scholar] [CrossRef] [PubMed]
- Foretová, L.; Navrátilová, M.; Svoboda, M.; Grell, P.; Nemec, L.; Sirotek, L.; Obermannová, R.; Novotný, I.; Sachlova, M.; Fabian, P.; et al. GAPPS–Gastric Adenocarcinoma and Proximal Polyposis of the Stomach Syndrome in 8 Families Tested at Masaryk Memorial Cancer Institute–Prevention and Prophylactic Gastrectomies. Klin Onkol 2019, 32 (Suppl. 2), 109–117. [Google Scholar] [CrossRef]
- Salami, A.C.; Stone, J.M.; Greenberg, R.H.; Leighton, J.C.; Miick, R.; Zavala, S.R.; Zeitzer, K.L.; Bakhis, C.T. Early Prophylactic Gastrectomy for the Management of Gastric Adenomatous Proximal Polyposis Syndrome (GAPPS). ACS Case Rev. Surg. 2022, 3, 62–68. [Google Scholar] [PubMed]
- Yen, T.; Stanich, P.P.; Axell, L.; Patel, S.G. APC-Associated Polyposis Conditions; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2023. [Google Scholar]
- Dottori, L.; Pivetta, G.; Annibale, B.; Lahner, E. Update on Serum Biomarkers in Autoimmune Atrophic Gastritis. Clin. Chem. 2023, 69, 1114–1131. [Google Scholar] [CrossRef] [PubMed]
- Iwamuro, M.; Tanaka, T.; Otsuka, M. Update in Molecular Aspects and Diagnosis of Autoimmune Gastritis. Curr. Issues Mol. Biol. 2023, 45, 5263–5275. [Google Scholar] [CrossRef] [PubMed]
- Neumann, W.L.; Coss, E.; Rugge, M.; Genta, R.M. Autoimmune atrophic gastritis–pathogenesis, pathology and management. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Massironi, S.; Gallo, C.; Elvevi, A.; Stegagnini, M.; Coltro, L.A.; Invernizzi, P. Incidence and prevalence of gastric neuroendocrine tumors in patients with chronic atrophic autoimmune gastritis. World J. Gastrointest. Oncol. 2023, 15, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Namikawa, K.; Kamada, T.; Fujisaki, J.; Sato, Y.; Murao, T.; Chiba, T.; Kaizaki, Y.; Ishido, K.; Ihara, Y.; Kurahara, K.; et al. Clinical characteristics and long-term prognosis of type 1 gastric neuroendocrine tumors in a large Japanese national cohort. Dig. Endosc. 2023, 35, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Kiso, M.; Ito, M.; Boda, T.; Kotachi, T.; Masuda, K.; Hata, K.; Sasaki, A.; Kawamura, T.; Yoshihara, M.; Tanaka, S.; et al. Endoscopic findings of the gastric mucosa during long-term use of proton pump inhibitor—A multicenter study. Scand. J. Gastroenterol. 2017, 52, 828–832. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kato, M.; Tsuda, M.; Matsuda, K.; Muranaka, T.; Abiko, S.; Ono, M.; Mizushima, T.; Omori, S.; Yamamoto, K.; et al. Gastric mucosal cracked and cobblestone-like changes resulting from proton pump inhibitor use. Dig. Endosc. 2017, 29, 307–313. [Google Scholar] [CrossRef]
- Majima, K.; Muraki, Y.; Shimamoto, T. Multiple White and Flat Elevated Lesions Observed in the Stomach: A Prospective Study of Clinical Characteristics and Risk Factors. Intern. Med. 2018, 57, 2613–2619. [Google Scholar] [CrossRef]
- Iwamuro, M.; Shiraha, H.; Okada, H. Gastric polyps’ regression after potassium-competitive acid blocker cessation. J. Gen. Fam. Med. 2022, 23, 358–359. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Hashimoto, T.; Naka, T.; Yatabe, Y.; Oda, I.; Saito, Y.; Yoshikawa, T.; Sekine, S. Activating KRAS and GNAS mutations in heterotopic submucosal glands of the stomach. J. Gastroenterol. 2022, 57, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Namikawa, T.; Ishida, N.; Yokota, K.; Munekage, E.; Iwabu, J.; Munekage, M.; Uemura, S.; Maeda, H.; Kitagawa, H.; Kobayashi, M.; et al. Early gastric cancer with multiple submucosal heterotopic gastric gland: A case report. Mol. Clin. Oncol. 2019, 10, 583–586. [Google Scholar] [CrossRef]
- Inaba, T.; Mizuno, M.; Kawai, K.; Okada, H.; Tsuji, T. Diffuse submucosal cysts of the stomach: Report of two cases in association with development of multiple gastric cancers during endoscopic follow up. J. Gastroenterol. Hepatol. 1999, 14, 1161–1165. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Name | Causative Factor | Characteristics of Gastric Polyps |
|---|---|---|
| Familial adenomatous polyposis | Mutations in the APC gene | Multiple fundic gland polyps are typical. Intestinal-type adenomas and early gastric cancers exhibit reddish depressed lesions, while gastric-type adenomas display whitish elevated lesions. |
| Peutz-Jeghers syndrome | Mutations in the STK11 gene | Solitary or sporadic polyps larger than 5 mm exhibiting a reddish color with a sessile or semi-pedunculated morphology (hyperplastic polyps and Peutz-Jeghers polyps), and multiple sessile polyps of 5 mm or smaller, mirroring the color of the peripheral mucosa (fundic gland polyps and hyperplastic polyps). |
| Cowden syndrome | Mutations in the PTEN gene | Polyps generally exhibit a color similar to the peripheral mucosa but may also be reddish (hyperplastic, hamartomatous, inflammatory, lipomatous, fibromatous, and adenomatous polyps). |
| Cronkhite-Canada syndrome | Not yet elucidated | Gastric polyps ranging from several millimeters to 20 mm in size are observed diffusely as sessile or slightly pedunculated elevations with a dense distribution. Edema with inflammation is present in the intervening mucosa between the polyps. |
| Juvenile polyposis syndrome | Mutations in the SMAD4 or BMPR1A gene | Papillary or tongue-like polyps, multiple and reddish in color of varying sizes with swollen or edematous features. |
| GAPPS | Mutations in the APC exon 1B promotor region | Polyps localized in the gastric body and fornix, predominantly constituted by fundic gland polyps manifesting as regions of dysplasia and adenocarcinoma in some patients. |
| Neuroendocrine tumors in autoimmune gastritis | Autoimmune gastritis | Yellow or red tumors are identified in the gastric body with marked or prevailing atrophy localized within the gastric body while sparing the gastric antrum. |
| Proton pump inhibitor-related gastric mucosal changes | Prolonged intake of proton pump inhibitors | Multiple white and flat elevated lesions, cobblestone-like mucosa, the emergence and enlargement of the fundic gland, and hyperplastic polyps. |
| Multiple submucosal heterotopic glands | Chronic inflammation, such as infection of Helicobacter pylori | Polypoid lesions with cystic areas beneath the gastric mucosa visualized during an endoscopic ultrasound examination. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwamuro, M.; Kawano, S.; Otsuka, M. Differential Diagnoses and Management Approaches for Gastric Polyposis. Gastroenterol. Insights 2024, 15, 122-144. https://doi.org/10.3390/gastroent15010009
Iwamuro M, Kawano S, Otsuka M. Differential Diagnoses and Management Approaches for Gastric Polyposis. Gastroenterology Insights. 2024; 15(1):122-144. https://doi.org/10.3390/gastroent15010009
Chicago/Turabian StyleIwamuro, Masaya, Seiji Kawano, and Motoyuki Otsuka. 2024. "Differential Diagnoses and Management Approaches for Gastric Polyposis" Gastroenterology Insights 15, no. 1: 122-144. https://doi.org/10.3390/gastroent15010009
APA StyleIwamuro, M., Kawano, S., & Otsuka, M. (2024). Differential Diagnoses and Management Approaches for Gastric Polyposis. Gastroenterology Insights, 15(1), 122-144. https://doi.org/10.3390/gastroent15010009