
Advances in Diagnosis, Pathological Mechanisms, Clinical Impact, and Future Therapeutic Perspectives in Tay–Sachs Disease



{kind=link}
Abstract
1. Introduction
2. Epidemiology
3. Advances in Diagnosis
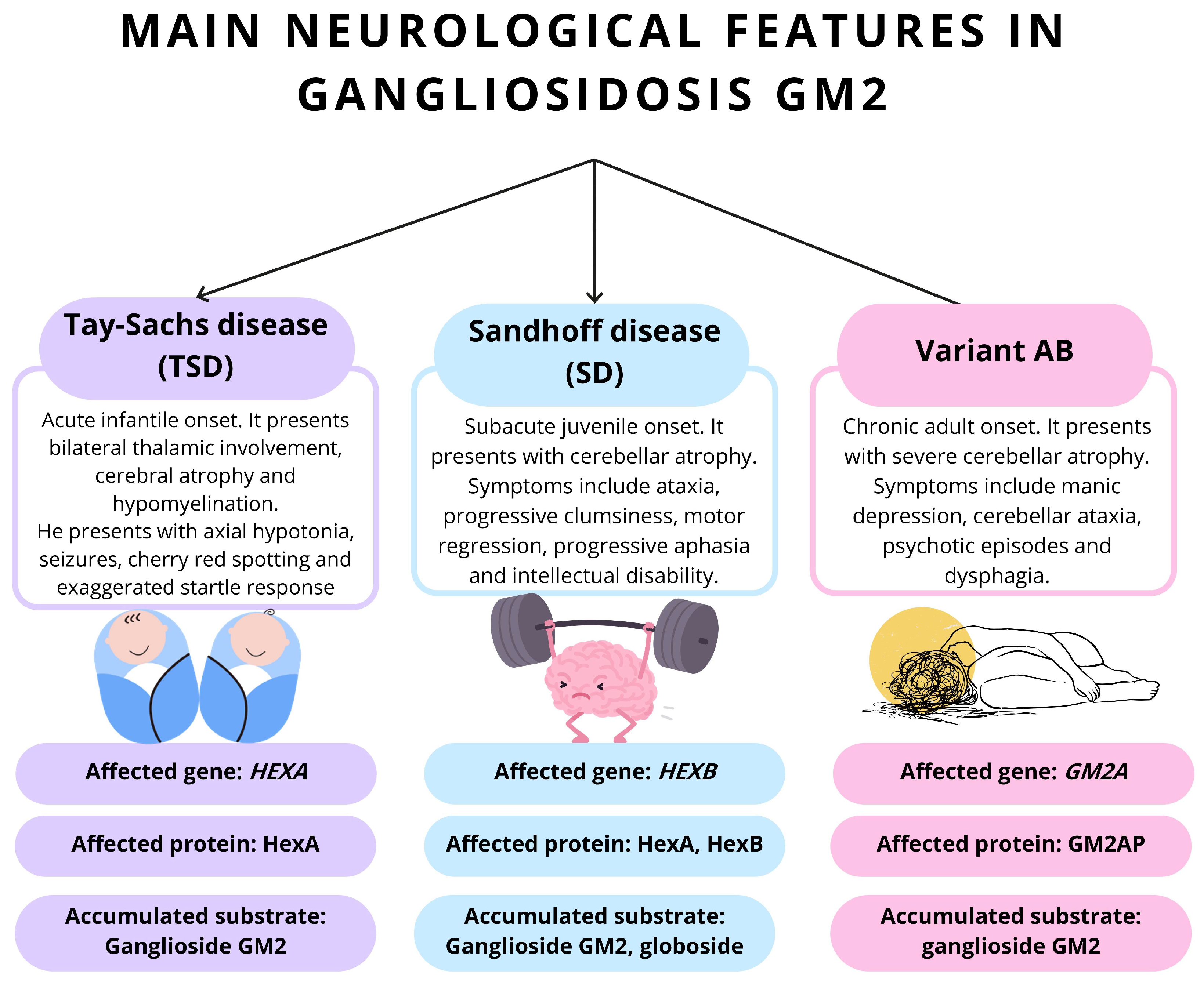
3.1. Differential Diagnosis
3.1.1. TSD Due to Activator Deficiency (AB Variant)
3.1.2. Sandhoff’s Disease and Other Lysosomal Storage Disorders
3.1.3. Differential Diagnosis of Late-Onset TSD
3.1.4. Professionals Involved After Diagnosis
4. Evaluation
4.1. Genetic Studies
4.2. Imaging Studies
5. Etiopathogenesis
5.1. Etiology of the Disease
5.2. Pathogenesis of the Disease
6. Pathophysiology
6.1. Structure and Physiological Function of Gangliosides
6.2. β-Hexosaminidases: Synthesis, Transport and Catalytic Functions
6.3. Clinical Presentations and Biochemical Correlates of GM2 Gangliosidosis
6.4. Pathophysiology of GM2 Gangliosidoses
6.5. Neurodevelopmental Process
6.6. Neuronal Death and Neuroinflammation
7. Histopathology
7.1. Neuroinflammation
7.2. Neurodegeneration
8. Clinical Manifestations
8.1. Childhood TSD
8.2. Juvenile TSD
8.3. TSD in Adults
9. Forecast
10. TSD Models
10.1. In Vitro and In Vivo Models of TSD for Therapy Development
10.2. HexA Enzyme Activity Assays
11. Treatment
11.1. Non-Pharmacological Treatment
11.2. Enzyme Replacement Therapy
11.3. Hematopoietic Stem Cell Transplantation
11.4. Pharmacological Chaperones
11.5. Substrate Reduction Therapy
11.6. Gene Therapy
11.7. Ex Vivo Gene Therapy
11.8. Pharmacological Treatment
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| BBB | Blood–brain barrier |
| CT | Computed tomography |
| EMA | European Medicines Agency |
| ERT | Enzyme replacement therapy |
| GM2 | GM2 ganglioside |
| HEXA | Hexosaminidase A gene |
| HexA | β-N-acetylhexosaminidase A |
| HEXB | Hexosaminidase B gene |
| HexB | β-N-acetylhexosaminidase B |
| HSCT | Hematopoietic stem cell transplantation |
| iPSCs | Induced pluripotent stem cells |
| KD | Ketogenic diet |
| M6PR | Mannose-6-phosphate receptor |
| MAP2 | Microtubule associated protein 2 |
| MOMA-2 | Macrophage/monocyte activity |
| MRI | Magnetic resonance imaging |
| MUG | 4-methylumbelliferyl-β-N-acetylglucosamine |
| MUGS | 4-methylumbelliferyl-β-N-acetylglucosamine-6-sulfate |
| OMIM | Online Mendelian Inheritance in Man |
| ORPHA | ORPHANET |
| PERK | Protein kinase RNA-like ER kinase |
| PG | Propagermanium |
| SD | Sandhoff disease |
| SRT | Substrate reduction therapy |
| TSD | Tay–Sachs disease |
References
- Rare Diseases at FDA. Available online: https://www.fda.gov/patients/rare-diseases-fda (accessed on 1 May 2025).
- ORDO—Orphadata. Available online: https://www.orphadata.com/ordo/ (accessed on 1 May 2025).
- Silibello, G.; Vizziello, P.; Gallucci, M.; Selicorni, A.; Milani, D.; Ajmone, P.F.; Rigamonti, C.; De Stefano, S.; Bedeschi, M.F.; Lalatta, F. Daily life changes and adaptations investigated in 154 families with a child suffering from a rare disability at a public centre for rare diseases in Northern Italy. Ital. J. Pediatr. 2016, 42, 76. [Google Scholar] [CrossRef] [PubMed]
- Uhlenbusch, N.; Lowe, B.; Harter, M.; Schramm, C.; Weiler-Normann, C.; Depping, M.K. Depression and anxiety in patients with different rare chronic diseases: A cross-sectional study. PLoS ONE 2019, 14, e0211343. [Google Scholar] [CrossRef]
- Sachs, B.; Strauss, I. The Cell Changes in Amaurotic Family Idiocy. J. Exp. Med. 1910, 12, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Friedland, J.; Schneck, L.; Saifer, A.; Pourfar, M.; Volk, B.W. Identification of Tay-Sachs disease carriers by acrylamide gel electrophoresis. Clin. Chim. Acta 1970, 28, 397–402. [Google Scholar] [CrossRef]
- Arpaia, E.; Dumbrille-Ross, A.; Maler, T.; Neote, K.; Tropak, M.; Troxel, C.; Stirling, J.L.; Pitts, J.S.; Bapat, B.; Lamhonwah, A.M.; et al. Identification of an altered splice site in Ashkenazi Tay-Sachs disease. Nature 1988, 333, 85–86. [Google Scholar] [CrossRef] [PubMed]
- Swallow, D.M.; Evans, L.; Saha, N.; Harris, H. Characterization and tissue distribution of N-acetyl hexosaminidase C: Suggestive evidence for a separate hexosaminidase locus. Ann. Hum. Genet. 1976, 40, 55–66. [Google Scholar] [CrossRef]
- Charrow, J.; Inui, K.; Wenger, D.A. Late onset GM2 gangliosidosis: An alpha-locus genetic compound with near normal hexosaminidase activity. Clin. Genet. 1985, 27, 78–84. [Google Scholar] [CrossRef]
- Ozkara, H.A.; Akerman, B.R.; Ciliv, G.; Topcu, M.; Renda, Y.; Gravel, R.A. Donor splice site mutation in intron 5 of the HEXA gene in a Turkish infant with Tay-Sachs disease. Hum. Mutat. 1995, 5, 186–187. [Google Scholar] [CrossRef]
- Orphanet: Diseases. Available online: https://www.orpha.net/en/disease (accessed on 1 May 2025).
- Suzuki, K.; Rapin, I.; Suzuki, Y.; Ishii, N. Juvenile GM2-gangliosidosis. Clinical variant of Tay-Sachs disease or a new disease. Neurology 1970, 20, 190–204. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD((R))): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef]
- Izquierdo-Martínez, M.; Avellaneda-Fernández, A. Enfermedades Raras: Un Enfoque Práctico; Instituto de Salud Carlos III: Madrid, Spain; Instituto de Enfermedades Raras: Madrid, Spain, 2004. [Google Scholar]
- Jarnes Utz, J.R.; Kim, S.; King, K.; Ziegler, R.; Schema, L.; Redtree, E.S.; Whitley, C.B. Infantile gangliosidoses: Mapping a timeline of clinical changes. Mol. Genet. Metab. 2017, 121, 170–179. [Google Scholar] [CrossRef]
- King, K.E.; Kim, S.; Whitley, C.B.; Jarnes-Utz, J.R. The juvenile gangliosidoses: A timeline of clinical change. Mol. Genet. Metab. Rep. 2020, 25, 100676. [Google Scholar] [CrossRef] [PubMed]
- Fahim, A.T. GeneReviews (R). Available online: https://cir.nii.ac.jp/crid/1370283694460643363 (accessed on 1 May 2025).
- Bibi, F.; Ullah, A.; Bourinaris, T.; Efthymiou, S.; Kriouile, Y.; Sultan, T.; Haider, S.; Salpietro, V.; Houlden, H.; Kaukab Raja, G. Tay-Sachs Disease: Two Novel Rare HEXA Mutations from Pakistan and Morocco. Klin. Padiatr. 2021, 233, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Rechnitz, G.A.; Kobos, R.K.; Riechel, S.J.; Gebauer, C.R. A bio-selective membrane electrode prepared with living bacterial cells. Anal. Chim. Acta 1977, 94, 357–365. [Google Scholar] [CrossRef]
- Staretz-Chacham, O.; Lang, T.C.; LaMarca, M.E.; Krasnewich, D.; Sidransky, E. Lysosomal storage disorders in the newborn. Pediatrics 2009, 123, 1191–1207. [Google Scholar] [CrossRef]
- Cruvinel Isaac, D.L.; Martins de Abreu, T.; Avila, M. Cherry-red Spot in Tay-Sachs Disease. Retina 2025, 386, e72. [Google Scholar] [CrossRef]
- Maegawa, G.H.; Stockley, T.; Tropak, M.; Banwell, B.; Blaser, S.; Kok, F.; Giugliani, R.; Mahuran, D.; Clarke, J.T. The natural history of juvenile or subacute GM2 gangliosidosis: 21 new cases and literature review of 134 previously reported. Pediatrics 2006, 118, e1550–e1562. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Palmeri, S.; Malandrini, A.; Fabrizi, G.; Mondelli, M.; Guazzi, G.C. The clinical aspects of adult hexosaminidase deficiencies. Dev. Neurosci. 1991, 13, 280–287. [Google Scholar] [CrossRef]
- Mitsumoto, H.; Sliman, R.J.; Schafer, I.A.; Sternick, C.S.; Kaufman, B.; Wilbourn, A.; Horwitz, S.J. Motor neuron disease and adult hexosaminidase A deficiency in two families: Evidence for multisystem degeneration. Ann. Neurol. 1985, 17, 378–385. [Google Scholar] [CrossRef]
- Lew, R.M.; Burnett, L.; Proos, A.L.; Delatycki, M.B. Tay-Sachs disease: Current perspectives from Australia. Appl. Clin. Genet. 2015, 8, 19–25. [Google Scholar] [CrossRef]
- Picache, J.A.; Zheng, W.; Chen, C.Z. Therapeutic Strategies For Tay-Sachs Disease. Front. Pharmacol. 2022, 13, 906647. [Google Scholar] [CrossRef] [PubMed]
- Rozenberg, R.; Pereira Lda, V. The frequency of Tay-Sachs disease causing mutations in the Brazilian Jewish population justifies a carrier screening program. Sao Paulo Med. J. 2001, 119, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.-S.; Choi, J.-H.; Yoo, H.-W. A Case of Tay-Sachs Disease in Korea Diagnosed by Enzyme Assay and DNA Analysis. Korean J. Pediatr. 2004, 47, 1360–1363. [Google Scholar]
- Choi, S.Y.; Park, J.H.; Lee, J.S.; Coe, C.J.; Han, S.H.; Lee, E.H. GM2 Gangliosidosis II. J. Korean Child. Neurol. Soc. 1999, 4, 244–249. [Google Scholar]
- Solovyeva, V.V.; Shaimardanova, A.A.; Chulpanova, D.S.; Kitaeva, K.V.; Chakrabarti, L.; Rizvanov, A.A. New Approaches to Tay-Sachs Disease Therapy. Front. Physiol. 2018, 9, 1663. [Google Scholar] [CrossRef]
- Rose, E.; Schreiber-Agus, N.; Bajaj, K.; Klugman, S.; Goldwaser, T. Challenges of Pre- and Post-Test Counseling for Orthodox Jewish Individuals in the Premarital Phase. J. Genet. Couns. 2016, 25, 18–24. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Howells, K.; Phillips, A.D.; Thomas, N.S.; Cooper, D.N. The Human Gene Mutation Database: 2008 update. Genome Med. 2009, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, R.N.; Pascual, J.M. Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, 6th ed.; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar]
- Jain, A.; Kohli, A.; Sachan, D. Infantile Sandhoff’s disease with peripheral neuropathy. Pediatr. Neurol. 2010, 42, 459–461. [Google Scholar] [CrossRef]
- Hall, P.; Minnich, S.; Teigen, C.; Raymond, K. Diagnosing Lysosomal Storage Disorders: The GM2 Gangliosidoses. Curr. Protoc. Hum. Genet. 2014, 83, 17.16.1–17.16.8. [Google Scholar] [CrossRef]
- Tomatsu, S.; Lavery, C.; Giugliani, R.; Harmatz, P.; Scarpa, M.; Wegrzyn, G.; Orii, T. Mucopolysaccharidoses Update (2 Volume Set); Nova Publishers: Hauppauge, NY, USA, 2018. [Google Scholar]
- Venugopalan, P.; Joshi, S.N. Cardiac involvement in infantile Sandhoff disease. J. Paediatr. Child. Health 2002, 38, 98–100. [Google Scholar] [CrossRef]
- Cutz, E.; Lowden, J.A.; Conen, P.E. Ultrastructural demonstration of neuronal storage in fetal Tay-Sachs disease. J. Neurol. Sci. 1974, 21, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, L.M.; Amsterdam, D.; Schneck, L. GM2 ganglioside in fetal Tay-Sachs disease brain cultures: A model system for the disease. Brain Res. 1976, 111, 109–117. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, H.; Kornreich, R.; Yu, C. Prenatal Diagnosis of Tay-Sachs Disease. Methods Mol. Biol. 2019, 1885, 233–250. [Google Scholar] [CrossRef] [PubMed]
- Sandhoff, K. My journey into the world of sphingolipids and sphingolipidoses. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 554–582. [Google Scholar] [CrossRef]
- Sandhoff, K.; Harzer, K. Gangliosides and gangliosidoses: Principles of molecular and metabolic pathogenesis. J. Neurosci. 2013, 33, 10195–10208. [Google Scholar] [CrossRef] [PubMed]
- Adam, N.B.; Amemiya, A.R.; Wallace, S.E.; Mahon, C.T.; Mirzaa, G.M.; Adam, M.P. Evaluation of Targeted Therapies Currently Available for Congenital Genetic Conditions Indexed in GeneReviews. Am. J. Med. Genet. C Semin. Med. Genet. 2025, 0, e32137. [Google Scholar] [CrossRef]
- Cachon-Gonzalez, M.B.; Zaccariotto, E.; Cox, T.M. Genetics and Therapies for GM2 Gangliosidosis. Curr. Gene Ther. 2018, 18, 68–89. [Google Scholar] [CrossRef]
- Okada, S.; O’Brien, J.S. Tay-Sachs disease: Generalized absence of a beta-D-N-acetylhexosaminidase component. Science 1969, 165, 698–700. [Google Scholar] [CrossRef]
- Leinekugel, P.; Michel, S.; Conzelmann, E.; Sandhoff, K. Quantitative correlation between the residual activity of beta-hexosaminidase A and arylsulfatase A and the severity of the resulting lysosomal storage disease. Hum. Genet. 1992, 88, 513–523. [Google Scholar] [CrossRef]
- Ramani, P.K.; Sankaran, B.P. Tay-Sachs Disease; StatPearls: Treasure Island, FL, USA, 2023; Available online: https://www.statpearls.com/physician/cme/activity/90733/?deg=MD (accessed on 15 June 2025).
- Hurowitz, G.I.; Silver, J.M.; Brin, M.F.; Williams, D.T.; Johnson, W.G. Neuropsychiatric aspects of adult-onset Tay-Sachs disease: Two case reports with several new findings. J. Neuropsychiatry Clin. Neurosci. 1993, 5, 30–36. [Google Scholar] [CrossRef]
- Akerman, B.R.; Zielenski, J.; Triggs-Raine, B.L.; Prence, E.M.; Natowicz, M.R.; Lim-Steele, J.S.; Kaback, M.M.; Mules, E.H.; Thomas, G.H.; Clarke, J.T.; et al. A mutation common in non-Jewish Tay-Sachs disease: Frequency and RNA studies. Hum. Mutat. 1992, 1, 303–309. [Google Scholar] [CrossRef]
- Kaplan, F. Tay-Sachs disease carrier screening: A model for prevention of genetic disease. Genet. Test. 1998, 2, 271–292. [Google Scholar] [CrossRef] [PubMed]
- Fukumizu, M.; Yoshikawa, H.; Takashima, S.; Sakuragawa, N.; Kurokawa, T. Tay-Sachs disease: Progression of changes on neuroimaging in four cases. Neuroradiology 1992, 34, 483–486. [Google Scholar] [CrossRef]
- Gupta, L.; Mirza, A.; Gulati, A.; Gulati, P. Magnetic resonance imaging findings in Tay-Sachs disease. Neurol. India 2018, 66, 1201–1202. [Google Scholar] [CrossRef] [PubMed]
- Mugikura, S.; Takahashi, S.; Higano, S.; Kurihara, N.; Kon, K.; Sakamoto, K. MR findings in Tay-Sachs disease. J. Comput. Assist. Tomogr. 1996, 20, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.J.; Chipman, S.I.; Johnston, J.M.; Acosta, M.T.; Toro, C.; Tifft, C.J. Late-onset GM2 gangliosidosis: Magnetic resonance imaging, diffusion tensor imaging, and correlational fiber tractography differentiate Tay-Sachs and Sandhoff diseases. J. Neurol. 2025, 272, 355. [Google Scholar] [CrossRef]
- Gravel, R.A.; Triggs-Raine, B.L.; Mahuran, D.J. Biochemistry and genetics of Tay-Sachs disease. Can. J. Neurol. Sci. 1991, 18, 419–423. [Google Scholar] [CrossRef]
- Bley, A.E.; Giannikopoulos, O.A.; Hayden, D.; Kubilus, K.; Tifft, C.J.; Eichler, F.S. Natural history of infantile G(M2) gangliosidosis. Pediatrics 2011, 128, e1233–e1241. [Google Scholar] [CrossRef]
- Ledeen, R.; Wu, G. Gangliosides of the Nervous System. Methods Mol. Biol. 2018, 1804, 19–55. [Google Scholar] [CrossRef]
- Schnaar, R.L. The Biology of Gangliosides. Adv. Carbohydr. Chem. Biochem. 2019, 76, 113–148. [Google Scholar] [CrossRef]
- Virgolini, M.J.; Feliziani, C.; Cambiasso, M.J.; Lopez, P.H.; Bollo, M. Neurite atrophy and apoptosis mediated by PERK signaling after accumulation of GM2-ganglioside. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Masingue, M.; Dufour, L.; Lenglet, T.; Saleille, L.; Goizet, C.; Ayrignac, X.; Ory-Magne, F.; Barth, M.; Lamari, F.; Mandia, D.; et al. Natural History of Adult Patients with GM2 Gangliosidosis. Ann. Neurol. 2020, 87, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.A.; Martin, D.R. Animal models of GM2 gangliosidosis: Utility and limitations. Appl. Clin. Genet. 2016, 9, 111–120. [Google Scholar] [CrossRef]
- Seyrantepe, V.; Demir, S.A.; Timur, Z.K.; Von Gerichten, J.; Marsching, C.; Erdemli, E.; Oztas, E.; Takahashi, K.; Yamaguchi, K.; Ates, N.; et al. Murine Sialidase Neu3 facilitates GM2 degradation and bypass in mouse model of Tay-Sachs disease. Exp. Neurol. 2018, 299, 26–41. [Google Scholar] [CrossRef]
- Bradbury, A.M.; Gurda, B.L.; Casal, M.L.; Ponder, K.P.; Vite, C.H.; Haskins, M.E. A review of gene therapy in canine and feline models of lysosomal storage disorders. Hum. Gene Ther. Clin. Dev. 2015, 26, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.E.; Brewer, R.A.; Elbakr, L.; Mollica, A.; Forguson, G.; Chan, W.S.; Ivakine, E.A. Precise template-free correction restores gene function in Tay-Sachs disease while reframing is ineffective. Mol. Ther. Nucleic Acids 2025, 36, 102401. [Google Scholar] [CrossRef]
- Yousefpour Shahrivar, R.; Karami, F.; Karami, E. Differential gene expression patterns in Niemann-Pick Type C and Tay-Sachs diseases: Implications for neurodegenerative mechanisms. PLoS ONE 2025, 20, e0319401. [Google Scholar] [CrossRef]
- Yu, R.K.; Tsai, Y.T.; Ariga, T.; Yanagisawa, M. Structures, biosynthesis, and functions of gangliosides–An overview. J. Oleo Sci. 2011, 60, 537–544. [Google Scholar] [CrossRef]
- Sonnino, S.; Chiricozzi, E.; Grassi, S.; Mauri, L.; Prioni, S.; Prinetti, A. Gangliosides in Membrane Organization. Prog. Mol. Biol. Transl. Sci. 2018, 156, 83–120. [Google Scholar] [CrossRef]
- Zeller, C.B.; Marchase, R.B. Gangliosides as modulators of cell function. Am. J. Physiol. 1992, 262, C1341–C1355. [Google Scholar] [CrossRef]
- Lopez, P.H.H.; Baez, B.B. Gangliosides in Axon Stability and Regeneration. Prog. Mol. Biol. Transl. Sci. 2018, 156, 383–412. [Google Scholar] [CrossRef] [PubMed]
- Rubovitch, V.; Zilberstein, Y.; Chapman, J.; Schreiber, S.; Pick, C.G. Restoring GM1 ganglioside expression ameliorates axonal outgrowth inhibition and cognitive impairments induced by blast traumatic brain injury. Sci. Rep. 2017, 7, 41269. [Google Scholar] [CrossRef] [PubMed]
- Groux-Degroote, S.; Rodriguez-Walker, M.; Dewald, J.H.; Daniotti, J.L.; Delannoy, P. Gangliosides in Cancer Cell Signaling. Prog. Mol. Biol. Transl. Sci. 2018, 156, 197–227. [Google Scholar] [CrossRef]
- Regina Todeschini, A.; Hakomori, S.I. Functional role of glycosphingolipids and gangliosides in control of cell adhesion, motility, and growth, through glycosynaptic microdomains. Biochim. Biophys. Acta 2008, 1780, 421–433. [Google Scholar] [CrossRef]
- Lemieux, M.J.; Mark, B.L.; Cherney, M.M.; Withers, S.G.; Mahuran, D.J.; James, M.N. Crystallographic structure of human beta-hexosaminidase A: Interpretation of Tay-Sachs mutations and loss of GM2 ganglioside hydrolysis. J. Mol. Biol. 2006, 359, 913–929. [Google Scholar] [CrossRef]
- Lahey, H.G.; Webber, C.J.; Golebiowski, D.; Izzo, C.M.; Horn, E.; Taghian, T.; Rodriguez, P.; Batista, A.R.; Ellis, L.E.; Hwang, M.; et al. Pronounced Therapeutic Benefit of a Single Bidirectional AAV Vector Administered Systemically in Sandhoff Mice. Mol. Ther. 2020, 28, 2150–2160. [Google Scholar] [CrossRef]
- Tropak, M.B.; Yonekawa, S.; Karumuthil-Melethil, S.; Thompson, P.; Wakarchuk, W.; Gray, S.J.; Walia, J.S.; Mark, B.L.; Mahuran, D. Construction of a hybrid beta-hexosaminidase subunit capable of forming stable homodimers that hydrolyze GM2 ganglioside in vivo. Mol. Ther. Methods Clin. Dev. 2016, 3, 15057. [Google Scholar] [CrossRef]
- Leal, A.F.; Benincore-Florez, E.; Solano-Galarza, D.; Garzon Jaramillo, R.G.; Echeverri-Pena, O.Y.; Suarez, D.A.; Almeciga-Diaz, C.J.; Espejo-Mojica, A.J. GM2 Gangliosidoses: Clinical Features, Pathophysiological Aspects, and Current Therapies. Int. J. Mol. Sci. 2020, 21, 6213. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.; Bigger, B.W. The role of innate immunity in mucopolysaccharide diseases. J. Neurochem. 2019, 148, 639–651. [Google Scholar] [CrossRef]
- Ballabio, A. The awesome lysosome. EMBO Mol. Med. 2016, 8, 73–76. [Google Scholar] [CrossRef]
- Darios, F.; Stevanin, G. Impairment of Lysosome Function and Autophagy in Rare Neurodegenerative Diseases. J. Mol. Biol. 2020, 432, 2714–2734. [Google Scholar] [CrossRef]
- Yim, W.W.; Mizushima, N. Lysosome biology in autophagy. Cell Discov. 2020, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Setia, H.; Muotri, A.R. Brain organoids as a model system for human neurodevelopment and disease. Semin. Cell Dev. Biol. 2019, 95, 93–97. [Google Scholar] [CrossRef]
- Allende, M.L.; Cook, E.K.; Larman, B.C.; Nugent, A.; Brady, J.M.; Golebiowski, D.; Sena-Esteves, M.; Tifft, C.J.; Proia, R.L. Cerebral organoids derived from Sandhoff disease-induced pluripotent stem cells exhibit impaired neurodifferentiation. J. Lipid Res. 2018, 59, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Kuil, L.E.; Lopez Marti, A.; Carreras Mascaro, A.; van den Bosch, J.C.; van den Berg, P.; van der Linde, H.C.; Schoonderwoerd, K.; Ruijter, G.J.G.; van Ham, T.J. Hexb enzyme deficiency leads to lysosomal abnormalities in radial glia and microglia in zebrafish brain development. Glia 2019, 67, 1705–1718. [Google Scholar] [CrossRef]
- Sargeant, T.J.; Drage, D.J.; Wang, S.; Apostolakis, A.A.; Cox, T.M.; Cachon-Gonzalez, M.B. Characterization of inducible models of Tay-Sachs and related disease. PLoS Genet. 2012, 8, e1002943. [Google Scholar] [CrossRef]
- Sargeant, T.J.; Wang, S.; Bradley, J.; Smith, N.J.; Raha, A.A.; McNair, R.; Ziegler, R.J.; Cheng, S.H.; Cox, T.M.; Cachon-Gonzalez, M.B. Adeno-associated virus-mediated expression of beta-hexosaminidase prevents neuronal loss in the Sandhoff mouse brain. Hum. Mol. Genet. 2011, 20, 4371–4380. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Q.; Trasler, J.M.; Igdoura, S.; Michaud, J.; Hanal, N.; Gravel, R.A. Apoptotic cell death in mouse models of GM2 gangliosidosis and observations on human Tay-Sachs and Sandhoff diseases. Hum. Mol. Genet. 1997, 6, 1879–1885. [Google Scholar] [CrossRef]
- Wada, R.; Tifft, C.J.; Proia, R.L. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc. Natl. Acad. Sci. USA 2000, 97, 10954–10959. [Google Scholar] [CrossRef]
- Ginzburg, L.; Li, S.C.; Li, Y.T.; Futerman, A.H. An exposed carboxyl group on sialic acid is essential for gangliosides to inhibit calcium uptake via the sarco/endoplasmic reticulum Ca2+-ATPase: Relevance to gangliosidoses. J. Neurochem. 2008, 104, 140–146. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef] [PubMed]
- Witting, A.; Muller, P.; Herrmann, A.; Kettenmann, H.; Nolte, C. Phagocytic clearance of apoptotic neurons by Microglia/Brain macrophages in vitro: Involvement of lectin-, integrin-, and phosphatidylserine-mediated recognition. J. Neurochem. 2000, 75, 1060–1070. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, S.; Nakanishi, H. Microglial clearance of focal apoptotic synapses. Neurosci. Lett. 2019, 707, 134317. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, A.M.; Peterson, T.A.; Gross, A.L.; Wells, S.Z.; McCurdy, V.J.; Wolfe, K.G.; Dennis, J.C.; Brunson, B.L.; Gray-Edwards, H.; Randle, A.N.; et al. AAV-mediated gene delivery attenuates neuroinflammation in feline Sandhoff disease. Neuroscience 2017, 340, 117–125. [Google Scholar] [CrossRef]
- Gray-Edwards, H.L.; Randle, A.N.; Maitland, S.A.; Benatti, H.R.; Hubbard, S.M.; Canning, P.F.; Vogel, M.B.; Brunson, B.L.; Hwang, M.; Ellis, L.E.; et al. Adeno-Associated Virus Gene Therapy in a Sheep Model of Tay-Sachs Disease. Hum. Gene Ther. 2018, 29, 312–326. [Google Scholar] [CrossRef]
- Ogawa, Y.; Furusawa, E.; Saitoh, T.; Sugimoto, H.; Omori, T.; Shimizu, S.; Kondo, H.; Yamazaki, M.; Sakuraba, H.; Oishi, K. Inhibition of astrocytic adenosine receptor A(2A) attenuates microglial activation in a mouse model of Sandhoff disease. Neurobiol. Dis. 2018, 118, 142–154. [Google Scholar] [CrossRef]
- Ogawa, Y.; Sano, T.; Irisa, M.; Kodama, T.; Saito, T.; Furusawa, E.; Kaizu, K.; Yanagi, Y.; Tsukimura, T.; Togawa, T.; et al. FcRgamma-dependent immune activation initiates astrogliosis during the asymptomatic phase of Sandhoff disease model mice. Sci. Rep. 2017, 7, 40518. [Google Scholar] [CrossRef]
- Chen, Y.; Allegood, J.; Liu, Y.; Wang, E.; Cachon-Gonzalez, B.; Cox, T.M.; Merrill, A.H., Jr.; Sullards, M.C. Imaging MALDI mass spectrometry using an oscillating capillary nebulizer matrix coating system and its application to analysis of lipids in brain from a mouse model of Tay-Sachs/Sandhoff disease. Anal. Chem. 2008, 80, 2780–2788. [Google Scholar] [CrossRef]
- Nathaniel, E.J.; Nathaniel, D.R. Astroglial response to degeneration of dorsal root fibers in adult rat spinal cord. Exp. Neurol. 1977, 54, 60–76. [Google Scholar] [CrossRef]
- Osher, E.; Anis, Y.; Singer-Shapiro, R.; Urshanski, N.; Unger, T.; Albeck, S.; Bogin, O.; Weisinger, G.; Kohen, F.; Valevski, A.; et al. Treating late-onset Tay Sachs disease: Brain delivery with a dual trojan horse protein. Mol. Ther. Methods Clin. Dev. 2024, 32, 101300. [Google Scholar] [CrossRef]
- Ziemens, D.; Oschmann, F.; Gerkau, N.J.; Rose, C.R. Heterogeneity of Activity-Induced Sodium Transients between Astrocytes of the Mouse Hippocampus and Neocortex: Mechanisms and Consequences. J. Neurosci. 2019, 39, 2620–2634. [Google Scholar] [CrossRef]
- Utz, J.R.; Crutcher, T.; Schneider, J.; Sorgen, P.; Whitley, C.B. Biomarkers of central nervous system inflammation in infantile and juvenile gangliosidoses. Mol. Genet. Metab. 2015, 114, 274–280. [Google Scholar] [CrossRef]
- Story, B.; Taghian, T.; Gallagher, J.; Koehler, J.; Taylor, A.; Randle, A.; Nielsen, K.; Gross, A.; Maguire, A.; Carl, S.; et al. Natural history of Tay-Sachs disease in sheep. Mol. Genet. Metab. 2021, 134, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Lui, F.; Ramani, P.K.; Parayil Sankaran, B. Tay-Sachs Disease; StatPearls: Treasure Island, FL, USA, 2025; Available online: https://www.statpearls.com/point-of-care/29887 (accessed on 15 June 2025).
- Kolter, T. Ganglioside biochemistry. ISRN Biochem. 2012, 2012, 506160. [Google Scholar] [CrossRef]
- Chen, H.; Chan, A.Y.; Stone, D.U.; Mandal, N.A. Beyond the cherry-red spot: Ocular manifestations of sphingolipid-mediated neurodegenerative and inflammatory disorders. Surv. Ophthalmol. 2014, 59, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Saito, Y.; Ishiyama, A.; Sugai, K.; Iso, T.; Inagaki, M.; Sasaki, M. Correlation of augmented startle reflex with brainstem electrophysiological responses in Tay-Sachs disease. Brain Dev. 2015, 37, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Strupp, M.; Kremmyda, O.; Adamczyk, C.; Bottcher, N.; Muth, C.; Yip, C.W.; Bremova, T. Central ocular motor disorders, including gaze palsy and nystagmus. J. Neurol. 2014, 261 (Suppl. 2), S542–S558. [Google Scholar] [CrossRef] [PubMed]
- Gowda, V.K.; Gupta, P.; Bharathi, N.K.; Bhat, M.; Shivappa, S.K.; Benakappa, N. Clinical and Laboratory Profile of Gangliosidosis from Southern Part of India. J. Pediatr. Genet. 2022, 11, 34–41. [Google Scholar] [CrossRef]
- Wang, H.P.; Wong, L.C.; Hsu, C.J.; Hu, S.C.; Chu, Y.J.; Lee, W.T. Eye motor manifestations in children with neurometabolic disorders. J. Formos. Med. Assoc. 2022, 121, 736–748. [Google Scholar] [CrossRef]
- Pavone, P.; Pratico, A.D.; Rizzo, R.; Corsello, G.; Ruggieri, M.; Parano, E.; Falsaperla, R. A clinical review on megalencephaly: A large brain as a possible sign of cerebral impairment. Medicine 2017, 96, e6814. [Google Scholar] [CrossRef]
- Lin, A.E.; Basson, C.T.; Goldmuntz, E.; Magoulas, P.L.; McDermott, D.A.; McDonald-McGinn, D.M.; McPherson, E.; Morris, C.A.; Noonan, J.; Nowak, C.; et al. Adults with genetic syndromes and cardiovascular abnormalities: Clinical history and management. Genet. Med. 2008, 10, 469–494. [Google Scholar] [CrossRef] [PubMed]
- Spyropoulos, B. Tay-Sachs carriers and tuberculosis resistance. Nature 1988, 331, 666. [Google Scholar] [CrossRef] [PubMed]
- Udwadia-Hegde, A.; Hajirnis, O. Temporary Efficacy of Pyrimethamine in Juvenile-Onset Tay-Sachs Disease Caused by 2 Unreported HEXA Mutations in the Indian Population. Child. Neurol. Open 2017, 4, 2329048X16687887. [Google Scholar] [CrossRef] [PubMed]
- Barritt, A.W.; Anderson, S.J.; Leigh, P.N.; Ridha, B.H. Late-onset Tay-Sachs disease. Pract. Neurol. 2017, 17, 396–399. [Google Scholar] [CrossRef]
- MacQueen, G.M.; Rosebush, P.I.; Mazurek, M.F. Neuropsychiatric aspects of the adult variant of Tay-Sachs disease. J. Neuropsychiatry Clin. Neurosci. 1998, 10, 10–19. [Google Scholar] [CrossRef]
- Zelnik, N.; Khazanov, V.; Sheinkman, A.; Karpati, A.M.; Peleg, L. Clinical manifestations of psychiatric patients who are carriers of tay-sachs disease. Possible role of psychotropic drugs. Neuropsychobiology 2000, 41, 127–131. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, R. Generation of HEXA-deficient hiPSCs from fibroblasts of a Tay-Sachs disease patient. Stem Cell Res. 2016, 17, 289–291. [Google Scholar] [CrossRef]
- Avior, Y.; Sagi, I.; Benvenisty, N. Pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Mol. Cell Biol. 2016, 17, 170–182. [Google Scholar] [CrossRef]
- Park, I.H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef]
- Taniike, M.; Yamanaka, S.; Proia, R.L.; Langaman, C.; Bone-Turrentine, T.; Suzuki, K. Neuropathology of mice with targeted disruption of Hexa gene, a model of Tay-Sachs disease. Acta Neuropathol. 1995, 89, 296–304. [Google Scholar] [CrossRef]
- Phaneuf, D.; Wakamatsu, N.; Huang, J.Q.; Borowski, A.; Peterson, A.C.; Fortunato, S.R.; Ritter, G.; Igdoura, S.A.; Morales, C.R.; Benoit, G.; et al. Dramatically different phenotypes in mouse models of human Tay-Sachs and Sandhoff diseases. Hum. Mol. Genet. 1996, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sango, K.; Yamanaka, S.; Hoffmann, A.; Okuda, Y.; Grinberg, A.; Westphal, H.; McDonald, M.P.; Crawley, J.N.; Sandhoff, K.; Suzuki, K.; et al. Mouse models of Tay-Sachs and Sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nat. Genet. 1995, 11, 170–176. [Google Scholar] [CrossRef]
- Demir, S.A.; Timur, Z.K.; Ates, N.; Martinez, L.A.; Seyrantepe, V. GM2 ganglioside accumulation causes neuroinflammation and behavioral alterations in a mouse model of early onset Tay-Sachs disease. J. Neuroinflamm. 2020, 17, 277. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.R.; Chen, C.H. Bone biomarker for the clinical assessment of osteoporosis: Recent developments and future perspectives. Biomark. Res. 2017, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.F.; Almeciga-Diaz, C.J. Efficient CRISPR/Cas9 nickase-mediated genome editing in an in vitro model of mucopolysaccharidosis IVA. Gene Ther. 2023, 30, 107–114. [Google Scholar] [CrossRef]
- Leal, A.F.; Cifuentes, J.; Torres, C.E.; Suarez, D.; Quezada, V.; Gomez, S.C.; Cruz, J.C.; Reyes, L.H.; Espejo-Mojica, A.J.; Almeciga-Diaz, C.J. Delivery and assessment of a CRISPR/nCas9-based genome editing system on in vitro models of mucopolysaccharidoses IVA assisted by magnetite-based nanoparticles. Sci. Rep. 2022, 12, 15045. [Google Scholar] [CrossRef]
- Zeng, B.J.; Torres, P.A.; Viner, T.C.; Wang, Z.H.; Raghavan, S.S.; Alroy, J.; Pastores, G.M.; Kolodny, E.H. Spontaneous appearance of Tay-Sachs disease in an animal model. Mol. Genet. Metab. 2008, 95, 59–65. [Google Scholar] [CrossRef]
- Torres, P.A.; Zeng, B.J.; Porter, B.F.; Alroy, J.; Horak, F.; Horak, J.; Kolodny, E.H. Tay-Sachs disease in Jacob sheep. Mol. Genet. Metab. 2010, 101, 357–363. [Google Scholar] [CrossRef]
- Tropak, M.B.; Bukovac, S.W.; Rigat, B.A.; Yonekawa, S.; Wakarchuk, W.; Mahuran, D.J. A sensitive fluorescence-based assay for monitoring GM2 ganglioside hydrolysis in live patient cells and their lysates. Glycobiology 2010, 20, 356–365. [Google Scholar] [CrossRef]
- Basirli, H.; Ates, N.; Seyrantepe, V. Imbalance in redox homeostasis is associated with neurodegeneration in the murine model of Tay-Sachs disease. Mol. Biol. Rep. 2025, 52, 282. [Google Scholar] [CrossRef]
- Ou, L.; Przybilla, M.J.; Tabaran, A.F.; Overn, P.; O’Sullivan, M.G.; Jiang, X.; Sidhu, R.; Kell, P.J.; Ory, D.S.; Whitley, C.B. A novel gene editing system to treat both Tay-Sachs and Sandhoff diseases. Gene Ther. 2020, 27, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Giugliani, R.; Vairo, F.; Kubaski, F.; Poswar, F.; Riegel, M.; Baldo, G.; Saute, J.A. Neurological manifestations of lysosomal disorders and emerging therapies targeting the CNS. Lancet Child. Adolesc. Health 2018, 2, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Garbade, S.F.; Zielonka, M.; Mechler, K.; Kolker, S.; Hoffmann, G.F.; Staufner, C.; Mengel, E.; Ries, M. FDA orphan drug designations for lysosomal storage disorders—A cross-sectional analysis. PLoS ONE 2020, 15, e0230898. [Google Scholar] [CrossRef]
- Ries, M. Enzyme replacement therapy and beyond-in memoriam Roscoe O. Brady, M.D. (1923-2016). J. Inherit. Metab. Dis. 2017, 40, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Motabar, O.; Ferrer, M.; Marugan, J.J.; Zheng, W.; Ottinger, E.A. Disease models for the development of therapies for lysosomal storage diseases. Ann. N. Y Acad. Sci. 2016, 1371, 15–29. [Google Scholar] [CrossRef]
- Johnson, W.G.; Desnick, R.J.; Long, D.M.; Sharp, H.L.; Krivit, W.; Brady, B.; Brady, R.O. Intravenous injection of purified hexosaminidase A into a patient with Tay-Sachs disease. Birth Defects Orig. Artic. Ser. 1973, 9, 120–124. [Google Scholar]
- Fitzpatrick, D.A.; FitzGerald, O.; McGeeney, K.F. Immunological aspects of urate oxidase therapy in hyperuricaemia. Ir. J. Med. Sci. 1977, 146, 155–161. [Google Scholar] [CrossRef]
- Beard, H.; Luck, A.J.; Hassiotis, S.; King, B.; Trim, P.J.; Snel, M.F.; Hopwood, J.J.; Hemsley, K.M. Determination of the role of injection site on the efficacy of intra-CSF enzyme replacement therapy in MPS IIIA mice. Mol. Genet. Metab. 2015, 115, 33–40. [Google Scholar] [CrossRef]
- Schulz, A.; Ajayi, T.; Specchio, N.; de Los Reyes, E.; Gissen, P.; Ballon, D.; Dyke, J.P.; Cahan, H.; Slasor, P.; Jacoby, D.; et al. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. N. Engl. J. Med. 2018, 378, 1898–1907. [Google Scholar] [CrossRef]
- Matsuoka, K.; Tsuji, D.; Aikawa, S.; Matsuzawa, F.; Sakuraba, H.; Itoh, K. Introduction of an N-glycan sequon into HEXA enhances human beta-hexosaminidase cellular uptake in a model of Sandhoff disease. Mol. Ther. 2010, 18, 1519–1526. [Google Scholar] [CrossRef]
- Tsuji, D.; Akeboshi, H.; Matsuoka, K.; Yasuoka, H.; Miyasaki, E.; Kasahara, Y.; Kawashima, I.; Chiba, Y.; Jigami, Y.; Taki, T.; et al. Highly phosphomannosylated enzyme replacement therapy for GM2 gangliosidosis. Ann. Neurol. 2011, 69, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Tavasoli, A.R.; Parvaneh, N.; Ashrafi, M.R.; Rezaei, Z.; Zschocke, J.; Rostami, P. Clinical presentation and outcome in infantile Sandhoff disease: A case series of 25 patients from Iranian neurometabolic bioregistry with five novel mutations. Orphanet J. Rare Dis. 2018, 13, 130. [Google Scholar] [CrossRef]
- Espejo-Mojica, A.J.; Almeciga-Diaz, C.J.; Rodriguez, A.; Mosquera, A.; Diaz, D.; Beltran, L.; Diaz, S.; Pimentel, N.; Moreno, J.; Sanchez, J.; et al. Human recombinant lysosomal enzymes produced in microorganisms. Mol. Genet. Metab. 2015, 116, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Akeboshi, H.; Chiba, Y.; Kasahara, Y.; Takashiba, M.; Takaoka, Y.; Ohsawa, M.; Tajima, Y.; Kawashima, I.; Tsuji, D.; Itoh, K.; et al. Production of recombinant beta-hexosaminidase A, a potential enzyme for replacement therapy for Tay-Sachs and Sandhoff diseases, in the methylotrophic yeast Ogataea minuta. Appl. Environ. Microbiol. 2007, 73, 4805–4812. [Google Scholar] [CrossRef]
- Braulke, T.; Bonifacino, J.S. Sorting of lysosomal proteins. Biochim. Biophys. Acta 2009, 1793, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef]
- Biffi, A. Hematopoietic Stem Cell Gene Therapy for Storage Disease: Current and New Indications. Mol. Ther. 2017, 25, 1155–1162. [Google Scholar] [CrossRef]
- Sawamoto, K.; Chen, H.H.; Almeciga-Diaz, C.J.; Mason, R.W.; Tomatsu, S. Gene therapy for Mucopolysaccharidoses. Mol. Genet. Metab. 2018, 123, 59–68. [Google Scholar] [CrossRef]
- Jacobs, J.F.; Willemsen, M.A.; Groot-Loonen, J.J.; Wevers, R.A.; Hoogerbrugge, P.M. Allogeneic BMT followed by substrate reduction therapy in a child with subacute Tay-Sachs disease. Bone Marrow Transplant. 2005, 36, 925–926. [Google Scholar] [CrossRef]
- Prasad, V.K.; Mendizabal, A.; Parikh, S.H.; Szabolcs, P.; Driscoll, T.A.; Page, K.; Lakshminarayanan, S.; Allison, J.; Wood, S.; Semmel, D.; et al. Unrelated donor umbilical cord blood transplantation for inherited metabolic disorders in 159 pediatric patients from a single center: Influence of cellular composition of the graft on transplantation outcomes. Blood 2008, 112, 2979–2989. [Google Scholar] [CrossRef]
- Ornaghi, F.; Sala, D.; Tedeschi, F.; Maffia, M.C.; Bazzucchi, M.; Morena, F.; Valsecchi, M.; Aureli, M.; Martino, S.; Gritti, A. Novel bicistronic lentiviral vectors correct beta-Hexosaminidase deficiency in neural and hematopoietic stem cells and progeny: Implications for in vivo and ex vivo gene therapy of GM2 gangliosidosis. Neurobiol. Dis. 2020, 134, 104667. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Ejima, D.; Kita, Y.; Tsumoto, K. Small molecule pharmacological chaperones: From thermodynamic stabilization to pharmaceutical drugs. Biochim. Biophys. Acta 2006, 1764, 1677–1687. [Google Scholar] [CrossRef]
- Cohen, F.E.; Kelly, J.W. Therapeutic approaches to protein-misfolding diseases. Nature 2003, 426, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Boyd, R.E.; Lee, G.; Rybczynski, P.; Benjamin, E.R.; Khanna, R.; Wustman, B.A.; Valenzano, K.J. Pharmacological chaperones as therapeutics for lysosomal storage diseases. J. Med. Chem. 2013, 56, 2705–2725. [Google Scholar] [CrossRef]
- Losada Diaz, J.C.; Cepeda Del Castillo, J.; Rodriguez-Lopez, E.A.; Almeciga-Diaz, C.J. Advances in the Development of Pharmacological Chaperones for the Mucopolysaccharidoses. Int. J. Mol. Sci. 2019, 21, 232. [Google Scholar] [CrossRef] [PubMed]
- Liguori, L.; Monticelli, M.; Allocca, M.; Hay Mele, B.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Pharmacological Chaperones: A Therapeutic Approach for Diseases Caused by Destabilizing Missense Mutations. Int. J. Mol. Sci. 2020, 21, 489. [Google Scholar] [CrossRef]
- Cortez, L.; Sim, V. The therapeutic potential of chemical chaperones in protein folding diseases. Prion 2014, 8, 197–202. [Google Scholar] [CrossRef]
- Bateman, K.S.; Cherney, M.M.; Mahuran, D.J.; Tropak, M.; James, M.N. Crystal structure of beta-hexosaminidase B in complex with pyrimethamine, a potential pharmacological chaperone. J. Med. Chem. 2011, 54, 1421–1429. [Google Scholar] [CrossRef]
- Maegawa, G.H.; Tropak, M.; Buttner, J.; Stockley, T.; Kok, F.; Clarke, J.T.; Mahuran, D.J. Pyrimethamine as a potential pharmacological chaperone for late-onset forms of GM2 gangliosidosis. J. Biol. Chem. 2007, 282, 9150–9161. [Google Scholar] [CrossRef]
- Clarke, J.T.; Mahuran, D.J.; Sathe, S.; Kolodny, E.H.; Rigat, B.A.; Raiman, J.A.; Tropak, M.B. An open-label Phase I/II clinical trial of pyrimethamine for the treatment of patients affected with chronic GM2 gangliosidosis (Tay-Sachs or Sandhoff variants). Mol. Genet. Metab. 2011, 102, 6–12. [Google Scholar] [CrossRef]
- Osher, E.; Fattal-Valevski, A.; Sagie, L.; Urshanski, N.; Sagiv, N.; Peleg, L.; Lerman-Sagie, T.; Zimran, A.; Elstein, D.; Navon, R.; et al. Effect of cyclic, low dose pyrimethamine treatment in patients with Late Onset Tay Sachs: An open label, extended pilot study. Orphanet J. Rare Dis. 2015, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jian, J.; Hettinghouse, A.; Zhao, X.; Setchell, K.D.R.; Sun, Y.; Liu, C.J. Progranulin associates with hexosaminidase A and ameliorates GM2 ganglioside accumulation and lysosomal storage in Tay-Sachs disease. J. Mol. Med. 2018, 96, 1359–1373. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, M.; Butters, T.D.; Cortina-Borja, M.; Hunnam, V.; Proia, R.L.; Perry, V.H.; Dwek, R.A.; Platt, F.M. Delayed symptom onset and increased life expectancy in Sandhoff disease mice treated with N-butyldeoxynojirimycin. Proc. Natl. Acad. Sci. USA 1999, 96, 6388–6393. [Google Scholar] [CrossRef]
- Platt, F.M.; Neises, G.R.; Reinkensmeier, G.; Townsend, M.J.; Perry, V.H.; Proia, R.L.; Winchester, B.; Dwek, R.A.; Butters, T.D. Prevention of lysosomal storage in Tay-Sachs mice treated with N-butyldeoxynojirimycin. Science 1997, 276, 428–431. [Google Scholar] [CrossRef]
- Masciullo, M.; Santoro, M.; Modoni, A.; Ricci, E.; Guitton, J.; Tonali, P.; Silvestri, G. Substrate reduction therapy with miglustat in chronic GM2 gangliosidosis type Sandhoff: Results of a 3-year follow-up. J. Inherit. Metab. Dis. 2010, 33 (Suppl. 3), S355–S361. [Google Scholar] [CrossRef]
- Ashe, K.M.; Bangari, D.; Li, L.; Cabrera-Salazar, M.A.; Bercury, S.D.; Nietupski, J.B.; Cooper, C.G.; Aerts, J.M.; Lee, E.R.; Copeland, D.P.; et al. Iminosugar-based inhibitors of glucosylceramide synthase increase brain glycosphingolipids and survival in a mouse model of Sandhoff disease. PLoS ONE 2011, 6, e21758. [Google Scholar] [CrossRef]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Ou, H.; Cui, R.; Jiang, N.; Zhang, M.; Li, X.; Ma, J.; Zhang, J.; Ma, D. Delivery of Glucosylceramidase Beta Gene Using AAV9 Vector Therapy as a Treatment Strategy in Mouse Models of Gaucher Disease. Hum. Gene Ther. 2019, 30, 155–167. [Google Scholar] [CrossRef]
- Latour, Y.L.; Yoon, R.; Thomas, S.E.; Grant, C.; Li, C.; Sena-Esteves, M.; Allende, M.L.; Proia, R.L.; Tifft, C.J. Human GLB1 knockout cerebral organoids: A model system for testing AAV9-mediated GLB1 gene therapy for reducing GM1 ganglioside storage in GM1 gangliosidosis. Mol. Genet. Metab. Rep. 2019, 21, 100513. [Google Scholar] [CrossRef]
- Fumagalli, F.; Calbi, V.; Natali Sora, M.G.; Sessa, M.; Baldoli, C.; Rancoita, P.M.V.; Ciotti, F.; Sarzana, M.; Fraschini, M.; Zambon, A.A.; et al. Lentiviral haematopoietic stem-cell gene therapy for early-onset metachromatic leukodystrophy: Long-term results from a non-randomised, open-label, phase 1/2 trial and expanded access. Lancet 2022, 399, 372–383. [Google Scholar] [CrossRef]
- Tardieu, M.; Zerah, M.; Gougeon, M.L.; Ausseil, J.; de Bournonville, S.; Husson, B.; Zafeiriou, D.; Parenti, G.; Bourget, P.; Poirier, B.; et al. Intracerebral gene therapy in children with mucopolysaccharidosis type IIIB syndrome: An uncontrolled phase 1/2 clinical trial. Lancet Neurol. 2017, 16, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Tardieu, M.; Zerah, M.; Husson, B.; de Bournonville, S.; Deiva, K.; Adamsbaum, C.; Vincent, F.; Hocquemiller, M.; Broissand, C.; Furlan, V.; et al. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: Results of a phase I/II trial. Hum. Gene Ther. 2014, 25, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Worgall, S.; Sondhi, D.; Hackett, N.R.; Kosofsky, B.; Kekatpure, M.V.; Neyzi, N.; Dyke, J.P.; Ballon, D.; Heier, L.; Greenwald, B.M.; et al. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Hum. Gene Ther. 2008, 19, 463–474. [Google Scholar] [CrossRef]
- Golebiowski, D.; van der Bom, I.M.J.; Kwon, C.S.; Miller, A.D.; Petrosky, K.; Bradbury, A.M.; Maitland, S.; Kuhn, A.L.; Bishop, N.; Curran, E.; et al. Direct Intracranial Injection of AAVrh8 Encoding Monkey beta-N-Acetylhexosaminidase Causes Neurotoxicity in the Primate Brain. Hum. Gene Ther. 2017, 28, 510–522. [Google Scholar] [CrossRef]
- Osmon, K.J.; Woodley, E.; Thompson, P.; Ong, K.; Karumuthil-Melethil, S.; Keimel, J.G.; Mark, B.L.; Mahuran, D.; Gray, S.J.; Walia, J.S. Systemic Gene Transfer of a Hexosaminidase Variant Using an scAAV9.47 Vector Corrects GM2 Gangliosidosis in Sandhoff Mice. Hum. Gene Ther. 2016, 27, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Flotte, T.R.; Cataltepe, O.; Puri, A.; Batista, A.R.; Moser, R.; McKenna-Yasek, D.; Douthwright, C.; Gernoux, G.; Blackwood, M.; Mueller, C.; et al. AAV gene therapy for Tay-Sachs disease. Nat. Med. 2022, 28, 251–259. [Google Scholar] [CrossRef]
- Flotte, T.R.; Gessler, D.J. Gene Therapy for Rare Neurological Disorders. Clin. Pharmacol. Ther. 2022, 111, 743–757. [Google Scholar] [CrossRef]
- Benner, E.J.; Mosley, R.L.; Destache, C.J.; Lewis, T.B.; Jackson-Lewis, V.; Gorantla, S.; Nemachek, C.; Green, S.R.; Przedborski, S.; Gendelman, H.E. Therapeutic immunization protects dopaminergic neurons in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 9435–9440. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Ghosh, A.; Roy, A.; Matras, J.; Brahmachari, S.; Gendelman, H.E.; Pahan, K. Simvastatin inhibits the activation of p21ras and prevents the loss of dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neurosci. 2009, 29, 13543–13556. [Google Scholar] [CrossRef]
- Jana, M.; Pahan, K. Gemfibrozil, a lipid lowering drug, inhibits the activation of primary human microglia via peroxisome proliferator-activated receptor beta. Neurochem. Res. 2012, 37, 1718–1729. [Google Scholar] [CrossRef] [PubMed]
- Pahan, K.; Jana, M.; Liu, X.; Taylor, B.S.; Wood, C.; Fischer, S.M. Gemfibrozil, a lipid-lowering drug, inhibits the induction of nitric-oxide synthase in human astrocytes. J. Biol. Chem. 2002, 277, 45984–45991. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Storer, P.D.; Chavis, J.A.; Racke, M.K.; Drew, P.D. Agonists for the peroxisome proliferator-activated receptor-alpha and the retinoid X receptor inhibit inflammatory responses of microglia. J. Neurosci. Res. 2005, 81, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Raha, S.; Dutta, D.; Paidi, R.K.; Pahan, K. Lipid-Lowering Drug Gemfibrozil Protects Mice from Tay-Sachs Disease via Peroxisome Proliferator-Activated Receptor alpha. Cells 2023, 12, 2791. [Google Scholar] [CrossRef]
- Inci, O.K.; Seyrantepe, V. Combined treatment of Ketogenic diet and propagermanium reduces neuroinflammation in Tay-Sachs disease mouse model. Metab. Brain Dis. 2025, 40, 133. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Sánchez, M.; Ramírez-Expósito, M.J.; Martínez-Martos, J.M. Advances in Diagnosis, Pathological Mechanisms, Clinical Impact, and Future Therapeutic Perspectives in Tay–Sachs Disease. Neurol. Int. 2025, 17, 98. https://doi.org/10.3390/neurolint17070098
González-Sánchez M, Ramírez-Expósito MJ, Martínez-Martos JM. Advances in Diagnosis, Pathological Mechanisms, Clinical Impact, and Future Therapeutic Perspectives in Tay–Sachs Disease. Neurology International. 2025; 17(7):98. https://doi.org/10.3390/neurolint17070098
Chicago/Turabian StyleGonzález-Sánchez, María, María Jesús Ramírez-Expósito, and José Manuel Martínez-Martos. 2025. "Advances in Diagnosis, Pathological Mechanisms, Clinical Impact, and Future Therapeutic Perspectives in Tay–Sachs Disease" Neurology International 17, no. 7: 98. https://doi.org/10.3390/neurolint17070098
APA StyleGonzález-Sánchez, M., Ramírez-Expósito, M. J., & Martínez-Martos, J. M. (2025). Advances in Diagnosis, Pathological Mechanisms, Clinical Impact, and Future Therapeutic Perspectives in Tay–Sachs Disease. Neurology International, 17(7), 98. https://doi.org/10.3390/neurolint17070098