
Confusing Onset of MOGAD in the Form of Focal Seizures


Abstract
1. Introduction
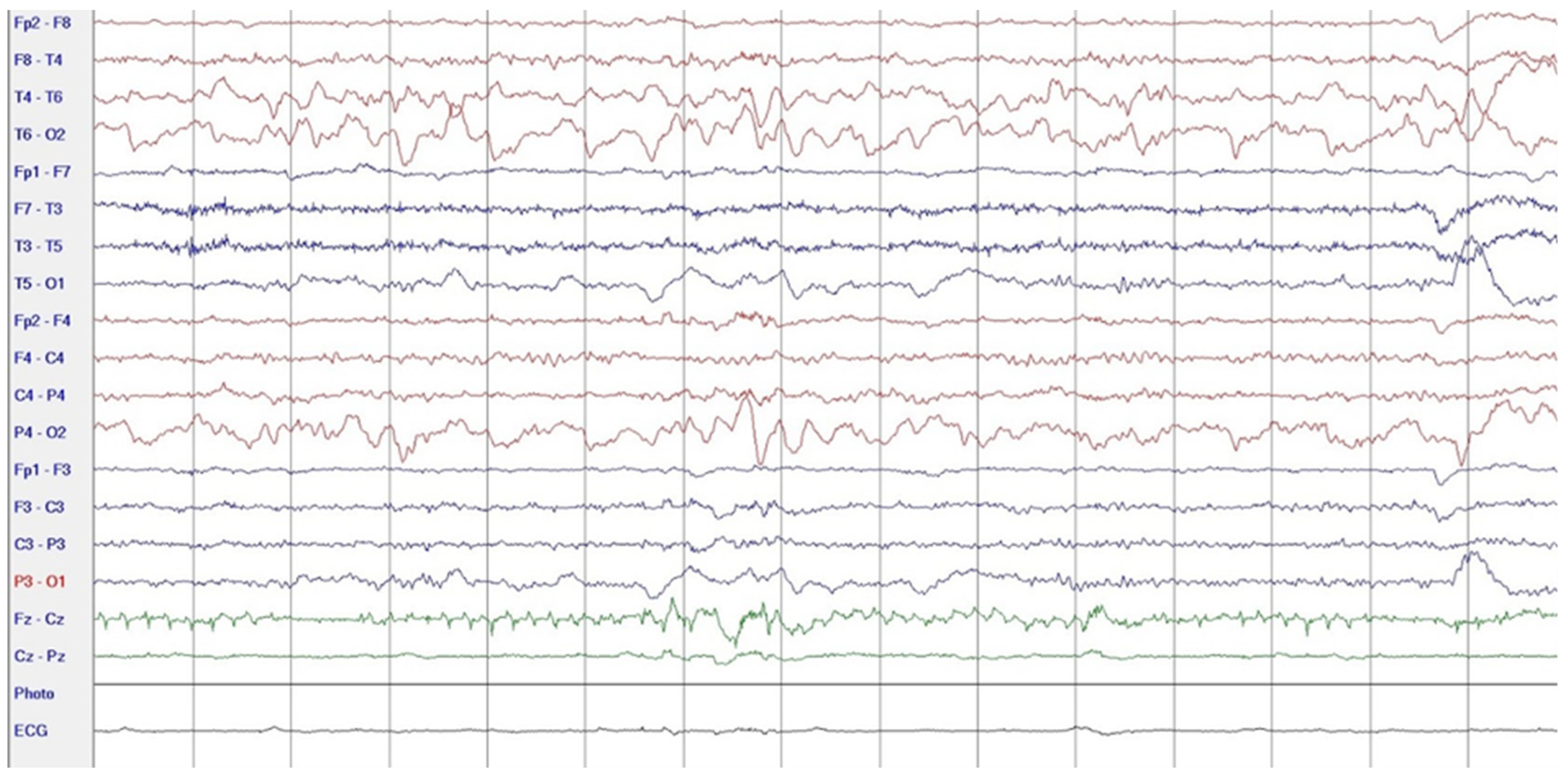
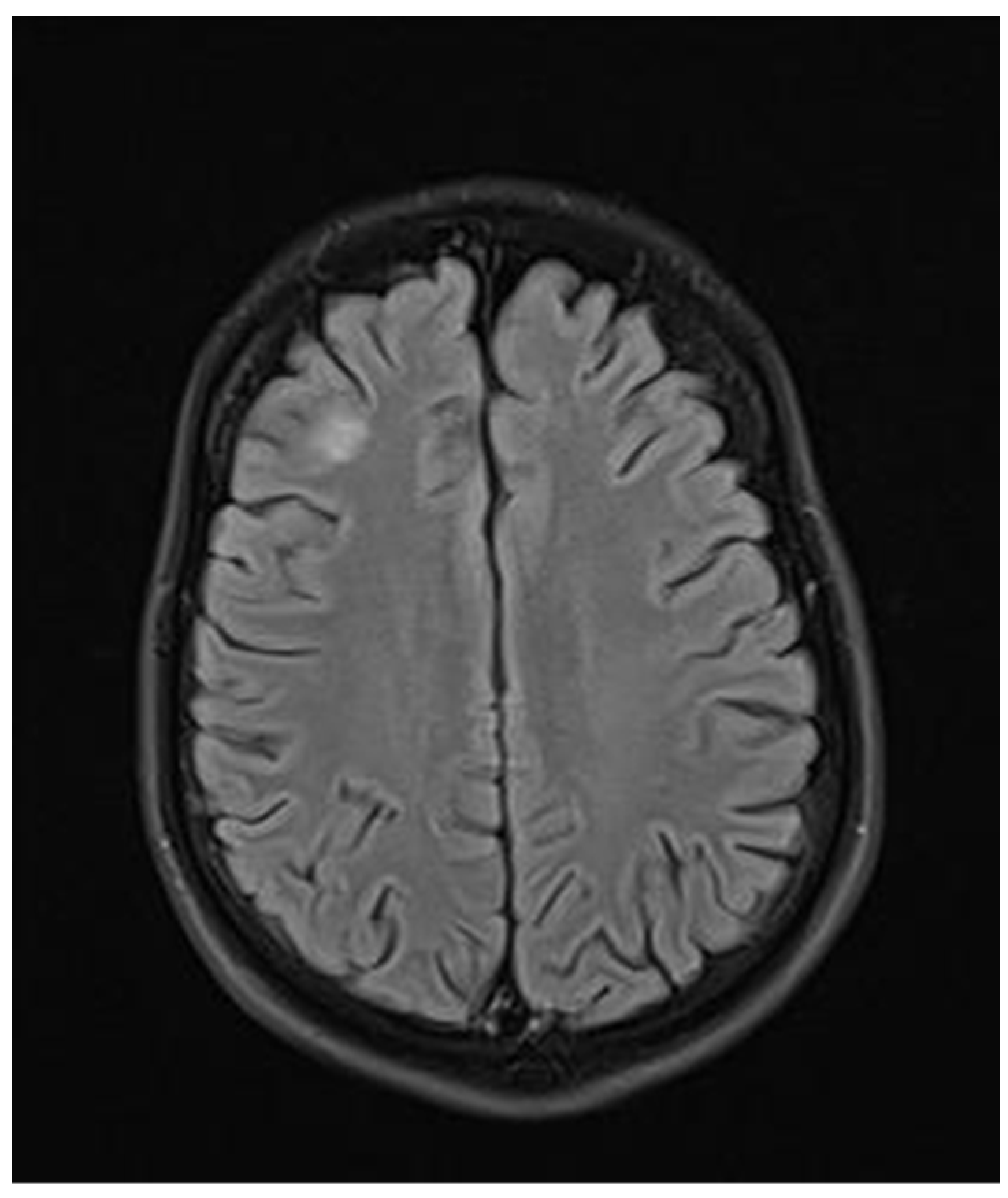
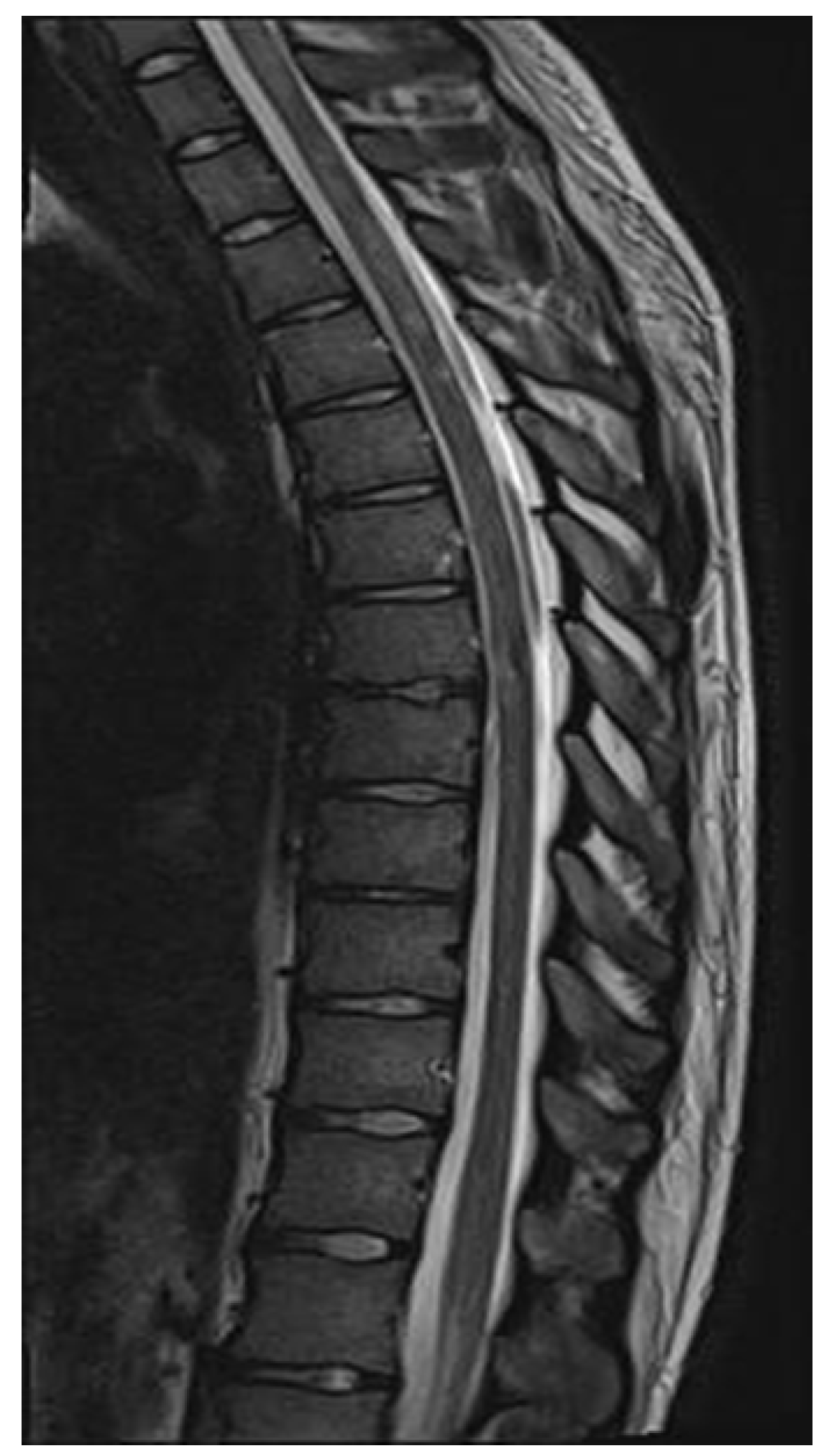
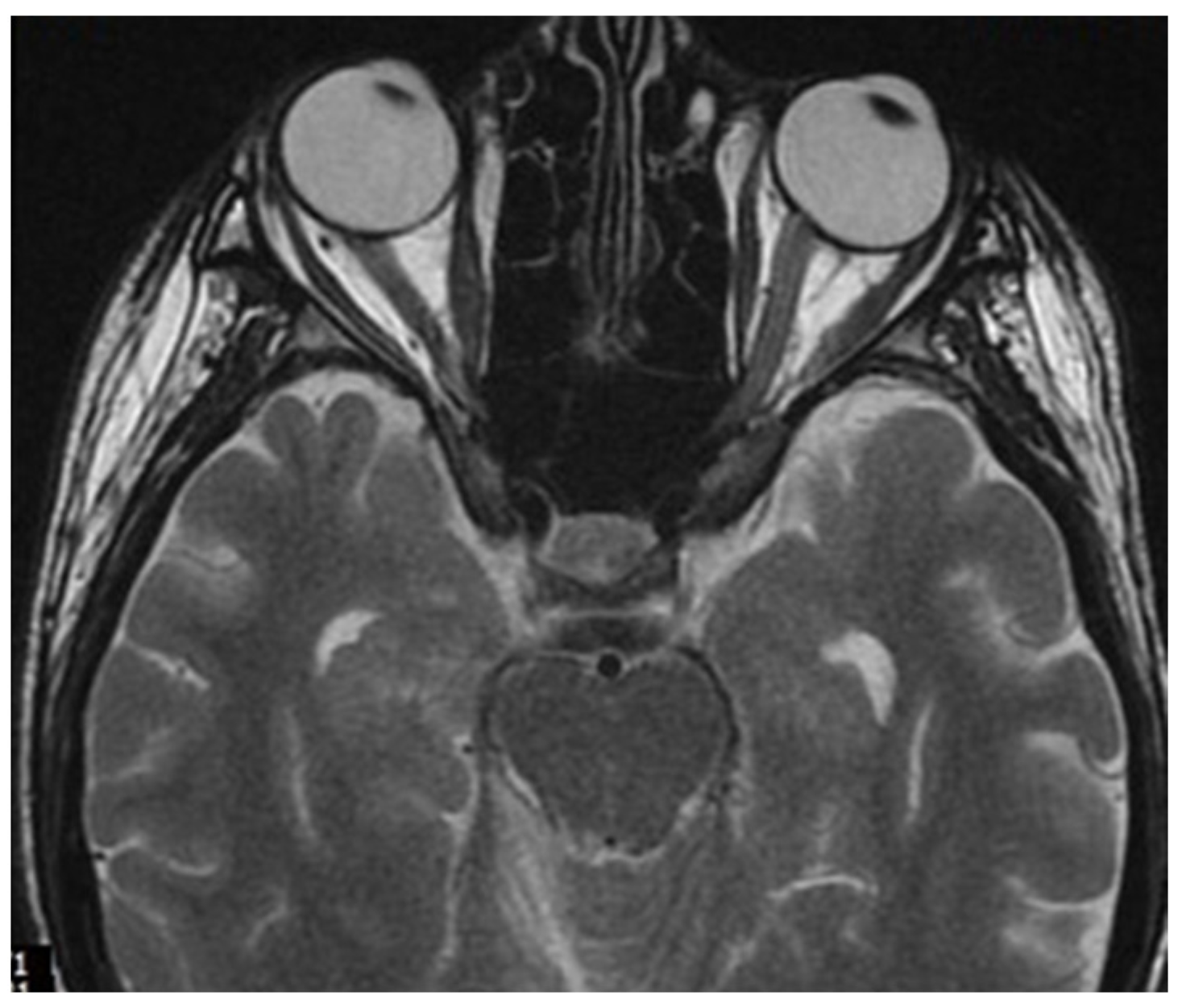
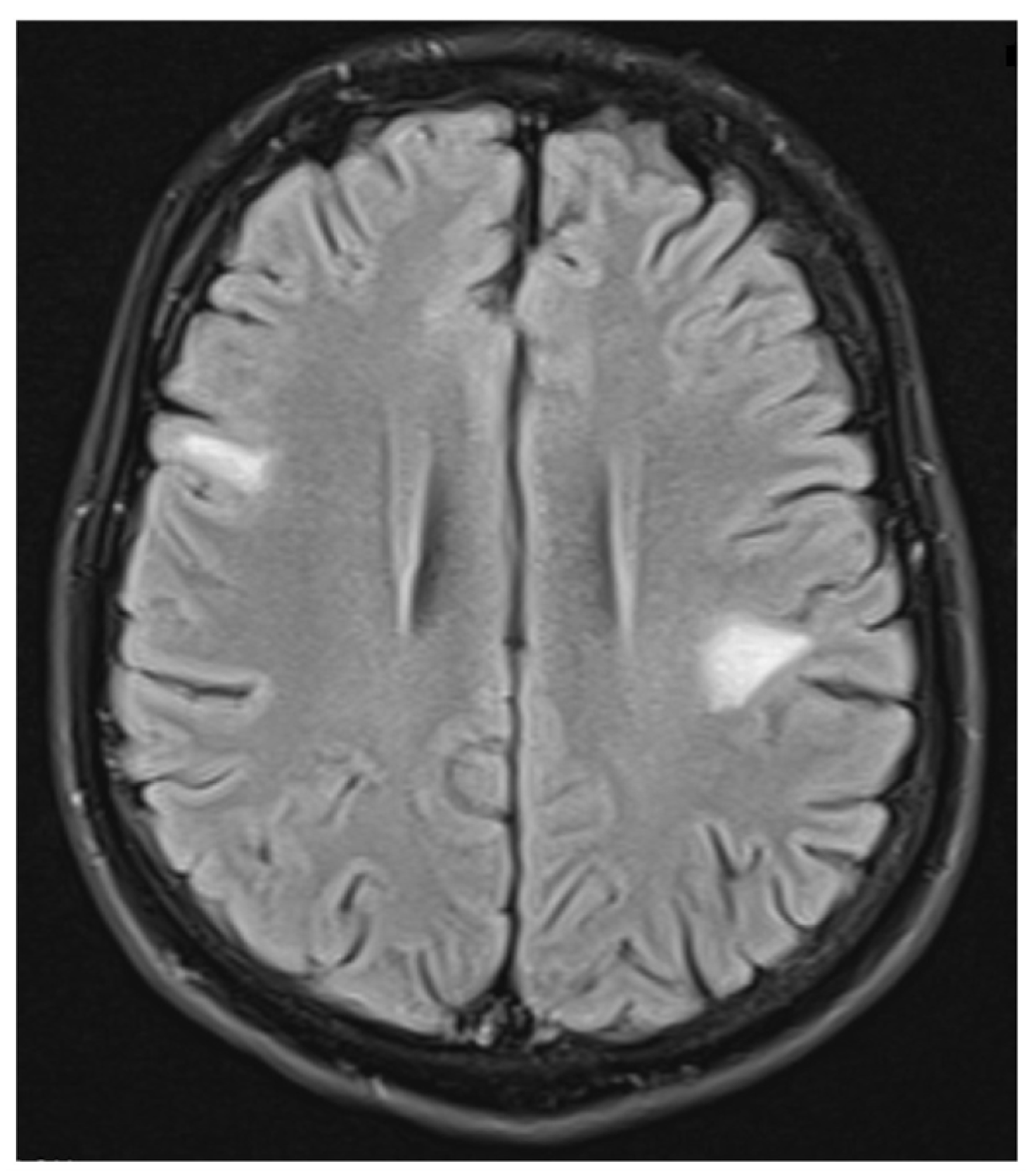
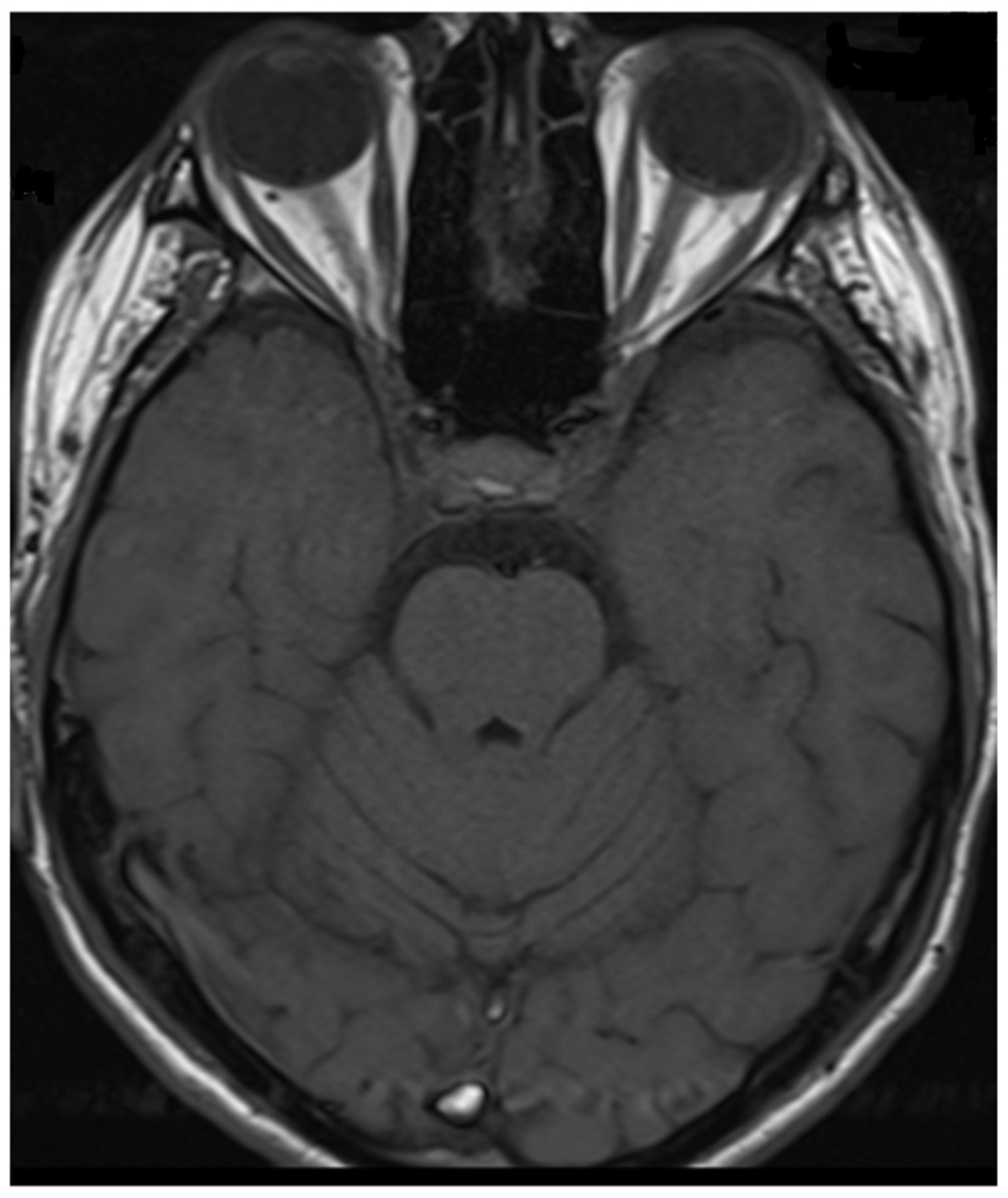
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MOGAD | demyelinating syndrome with the presence of antibodies against myelin oligodendrocyte glycoprotein |
| NMOSD | neuromyelitis optica spectrum disorders |
| MS | Multiple sclerosis |
| ON | optic neuritis |
| TM | transverse myelitis |
| ADEM | acute disseminated encephalomyelitis |
References
- Sechi, E.; Cacciaguerra, L.; Chen, J.J.; Mariotto, S.; Fadda, G.; Dinoto, A.; Lopez-Chiriboga, A.S.; Pittock, S.J.; Flanagan, E.P. Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease (MOGAD): A Review of Clinical and MRI Features, Diagnosis, and Management. Front. Neurol. 2022, 13, 885218. [Google Scholar] [CrossRef]
- Bruijstens, A.L.; Lechner, C.; Flet-Berliac, L.; Deiva, K.; Neuteboom, R.F.; Hemingway, C.; Wassmer, E.; E.U. Pediatric MOG Consortium; Baumann, M.; Bartels, F.; et al. E.U. pediatric MOG consortium consensus: Part 1—Classification of clinical phenotypes of pediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur. J. Paediatr. Neurol. 2020, 29, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Al-Ani, A.; Chen, J.J.; Costello, F. Myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD): Current understanding and challenges. J. Neurol. 2023, 270, 4132–4150. [Google Scholar] [CrossRef] [PubMed]
- Frade, H.C.; Elnaeem, A.; Banerjee, P.; Sharma, T.; Wu, L.; Dabi, A. Aggressive Course of Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease (MOGAD): An Illustration of Two Cases and Review of Literature. Cureus 2024, 16, e68563. [Google Scholar] [CrossRef]
- Baumann, M.; Bartels, F.; Finke, C.; Adamsbaum, C.; Hacohen, Y.; Rostásy, K.; E.U. Pediatric MOG Consortium. E.U. pediatric MOG consortium consensus: Part 2—Neuroimaging features of pediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur. J. Paediatr. Neurol. 2020, 29, 14–21. [Google Scholar] [CrossRef]
- Bruijstens, A.L.; Wendel, E.M.; Lechner, C.; Bartels, F.; Finke, C.; Breu, M.; Flet-Berliac, L.; de Chalus, A.; Adamsbaum, C.; Capobianco, M.; et al. E.U. pediatric MOG consortium consensus: Part 5—Treatment of pediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur. J. Paediatr. Neurol. 2020, 29, 41–53. [Google Scholar] [CrossRef]
- Kroenke, E.; Ankar, A.; Malani Shukla, N. Refractory MOG-Associated Demyelinating Disease in a Pediatric Patient. Child Neurol. Open 2022, 9, 2329048X221079093. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Li, A.; Liu, L.; Wang, J.; Fan, D. Acute Myelitis, Recurrent Optic Neuritis, and Seizures Over 17 Years. Front. Neurol. 2020, 11, 541146. [Google Scholar] [CrossRef]
- Ramanathan, S.; O’Grady, G.L.; Malone, S.; Spooner, C.G.; Brown, D.A.; Gill, D.; Brilot, F.; Dale, R.C. Isolated seizures during the first episode of relapsing myelin oligodendrocyte glycoprotein antibody-associated demyelination in children. Dev. Med. Child Neurol. 2019, 61, 610–614. [Google Scholar] [CrossRef]
- Tenembaum, S.; Yeh, E.A.; Guthy-Jackson Foundation International Clinical Consortium (GJCF-ICC). Pediatric NMOSD:A Review and Position Statement on Approach to Work-Up and Diagnosis. Front. Pediatr. 2020, 8, 339. [Google Scholar] [CrossRef]
- Hino-Fukuyo, N.; Kawai, E.; Itoh, S.; Oba, S.; Sato, Y.; Abe, S.; Ichikawa, Y.; Kitazawa, H.; Atobe, Y.; Fujimori, J.; et al. Myelin oligodendrocyte glycoprotein antibody-associated disease in a patient with symptoms of aseptic meningitis who achieved spontaneous remission: A case report and review of the literature. Brain Dev. 2023, 45, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Long, W.; Wen, J.; Su, Q.; Liao, J.; Hu, Z. Myelin oligodendrocyte glycoprotein antibody-associated aseptic meningitis without neurological parenchymal lesions: A novel phenotype. Mult. Scler. Relat. Disord. 2022, 68, 104126. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Maekawa, K.; Matsuo, K.; Yamasaki, M.; Shibata, M.; Takahashi, T.; Naito, Y. Aseptic Meningitis as an Initial Manifestation of Anti-myelin Oligodendrocyte Glycoprotein Antibody-associated Disease. Intern. Med. 2019, 58, 3319–3321. [Google Scholar] [CrossRef]
- Gombolay, G.Y.; Gadde, J.A. Aseptic meningitis and leptomeningeal enhancement associated with anti-MOG antibodies: A review. J. Neuroimmunol. 2021, 358, 577653. [Google Scholar] [CrossRef]
- Wang, X.Y.; Jiang, Y.; Wu, P.; Ma, J.N.; Yuan, P.; Li, X.J.; Jiang, L. Less common phenotypes of myelin oligodendrocyte glycoprotein antibody-related diseases in children deserve more attention. Pediatr. Res. 2024, 96, 731–739. [Google Scholar] [CrossRef]
- Karavassilis, M.E.; Chernov, D.; Dakhlia, S.; Saravanan, P. Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease (MOGAD) Initially Presenting as Septic Meningoencephalitis in a 16-Year-Old Male. Eur. J. Case Rep. Intern. Med. 2024, 11, 004596. [Google Scholar] [CrossRef] [PubMed]
- Banwell, B.; Bennett, J.L.; Marignier, R.; Kim, H.J.; Brilot, F.; Flanagan, E.P.; Ramanathan, S.; Waters, P.; Tenembaum, S.; Graves, J.S.; et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria. Lancet Neurol. 2023, 22, 268–282. [Google Scholar] [CrossRef] [PubMed]
- Varley, J.A.; Champsas, D.; Prossor, T.; Pontillo, G.; Abdel-Mannan, O.; Khaleeli, Z.; Petzold, A.; Toosy, A.T.; Trip, S.A.; Wilson, H.; et al. Validation of the 2023 International Diagnostic Criteria for MOGAD in a Selected Cohort of Adults and Children. Neurology 2024, 103, e209321. [Google Scholar] [CrossRef]
- Trewin, B.P.; Brilot, F.; Reddel, S.W.; Dale, R.C.; Ramanathan, S. MOGAD: A comprehensive review of clinicoradiological features, therapy and outcomes in 4699 patients globally. Autoimmun. Rev. 2025, 24, 103693. [Google Scholar] [CrossRef]
- Graus, F.; Dalmau, J. MOGAD comes of age with new criteria. Lancet Neurol. 2023, 22, 193–194. [Google Scholar] [CrossRef]
- Marignier, R. What s new in NMOSD and MOGAD? Rev. Neurol. 2024, 180, 957–962. [Google Scholar] [CrossRef]
- Nguyen, L.; Singh, S.; Feltrin, F.S.; Tardo, L.M.; Clarke, R.L.; Wang, C.X.; Greenberg, B.M. The positive predictive value of MOG-IgG testing based on the 2023 diagnostic criteria for MOGAD. Mult. Scler. J. Exp. Transl. Clin. 2024, 10, 20552173241274610. [Google Scholar] [CrossRef] [PubMed]
- Juryńczyk, M.; Jakuszyk, P.; Kurkowska-Jastrzębska, I.; Palace, J. Increasing role of imaging in differentiating MS from non-MS and defining indeterminate borderline cases. Neurol. Neurochir. Pol. 2022, 56, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, N.; Lerch, M.; Dale, R.C.; Ramanathan, S. MOG antibody-associated optic neuritis. Eye 2024, 38, 2289–2301. [Google Scholar] [CrossRef]
- Li, E.C.; Zheng, Y.; Cai, M.T.; Lai, Q.L.; Fang, G.L.; Du, B.Q.; Shen, C.H.; Zhang, Y.X.; Wu, L.J.; Ding, M.P. Seizures and epilepsy in multiple sclerosis, aquaporin four antibody-positive neuromyelitis optica spectrum disorder, and myelin oligodendrocyte glycoprotein antibody-associated disease. Epilepsia 2022, 63, 2173–2191. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Sun, S.; Cui, J.; Zhang, L.; Zhang, K.; Zhang, L. Seizures in myelin oligodendrocyte glycoprotein antibody-associated disorders and related immune factors. Seizure 2021, 92, 216–220. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, Y.; Ren, H.; Zhou, X.; Jin, L.; Huang, Y.; Lu, Q.; Yang, X.; Zhang, Y.; Zhu, Y.; et al. Acute epileptic seizures in myelin oligodendrocyte glycoprotein encephalomyelitis and neuromyelitis optica spectrum disorder: A comparative cohort study. Mult. Scler. Relat. Disord. 2019, 27, 281–288. [Google Scholar] [CrossRef]
- Foiadelli, T.; Gastaldi, M.; Scaranzin, S.; Franciotta, D.; Savasta, S. Seizures and myelin oligodendrocyte glycoprotein (MOG) antibodies: Two paradigmatic cases and a review of the literature. Mult. Scler. Relat. Disord. 2020, 41, 102011. [Google Scholar] [CrossRef] [PubMed]
- Höftberger, R.; Guo, Y.; Flanagan, E.P.; Lopez-Chiriboga, A.S.; Endmayr, V.; Hochmeister, S.; Joldic, D.; Pittock, S.J.; Tillema, J.M.; Gorman, M.; et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol. 2020, 139, 875–892. [Google Scholar] [CrossRef] [PubMed]
- de Curtis, M.; Garbelli, R.; Uva, L. A hypothesis for the role of axon demyelination in seizure generation. Epilepsia 2021, 62, 583–595. [Google Scholar] [CrossRef]
- Montalvo, M.; Khattak, J.F.; Redenbaugh, V.; Britton, J.; Sanchez, C.V.; Datta, A.; Tillema, J.M.; Chen, J.; McKeon, A.; Pittock, S.J.; et al. Acute symptomatic seizures secondary to myelin oligodendrocyte glycoprotein antibody-associated disease. Epilepsia 2022, 63, 3180–3191. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Demyelinating Syndrome | Supportive Radiological Features |
|---|---|
| Optic neuritis | bilateral simultaneous clinical involvement, longitudinal optic nerve involvement (>50% length of optic nerve), perineural optic sheath enhancement, optic disc edema |
| Myelitis | longitudinally extensive enhancement, central cord lesion, or H-sign, conus lesion |
| Brain, brainstem or cerebellar syndrome | multiple ill-defined T2-hyperintense lesions in supratentorial and often infratentorial white matter, deep gray matter involvement, ill-defined T2-hyperintensity involving pons, middle cerebellar peduncle, or medulla, cortical lesion with /without lesional and overlying meningeal enhancement |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jączak-Goździak, M.; Steinborn, B. Confusing Onset of MOGAD in the Form of Focal Seizures. Neurol. Int. 2025, 17, 37. https://doi.org/10.3390/neurolint17030037
Jączak-Goździak M, Steinborn B. Confusing Onset of MOGAD in the Form of Focal Seizures. Neurology International. 2025; 17(3):37. https://doi.org/10.3390/neurolint17030037
Chicago/Turabian StyleJączak-Goździak, Małgorzata, and Barbara Steinborn. 2025. "Confusing Onset of MOGAD in the Form of Focal Seizures" Neurology International 17, no. 3: 37. https://doi.org/10.3390/neurolint17030037
APA StyleJączak-Goździak, M., & Steinborn, B. (2025). Confusing Onset of MOGAD in the Form of Focal Seizures. Neurology International, 17(3), 37. https://doi.org/10.3390/neurolint17030037