
Molecular Pathogenic Mechanisms of Hypomyelinating Leukodystrophies (HLDs)



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. HLD1 (OMIM ID 312080)
3. HLD2 (OMIM ID 608803)
4. HLD3 (OMIM ID 260600) and HLD17 (OMIM ID 618006)
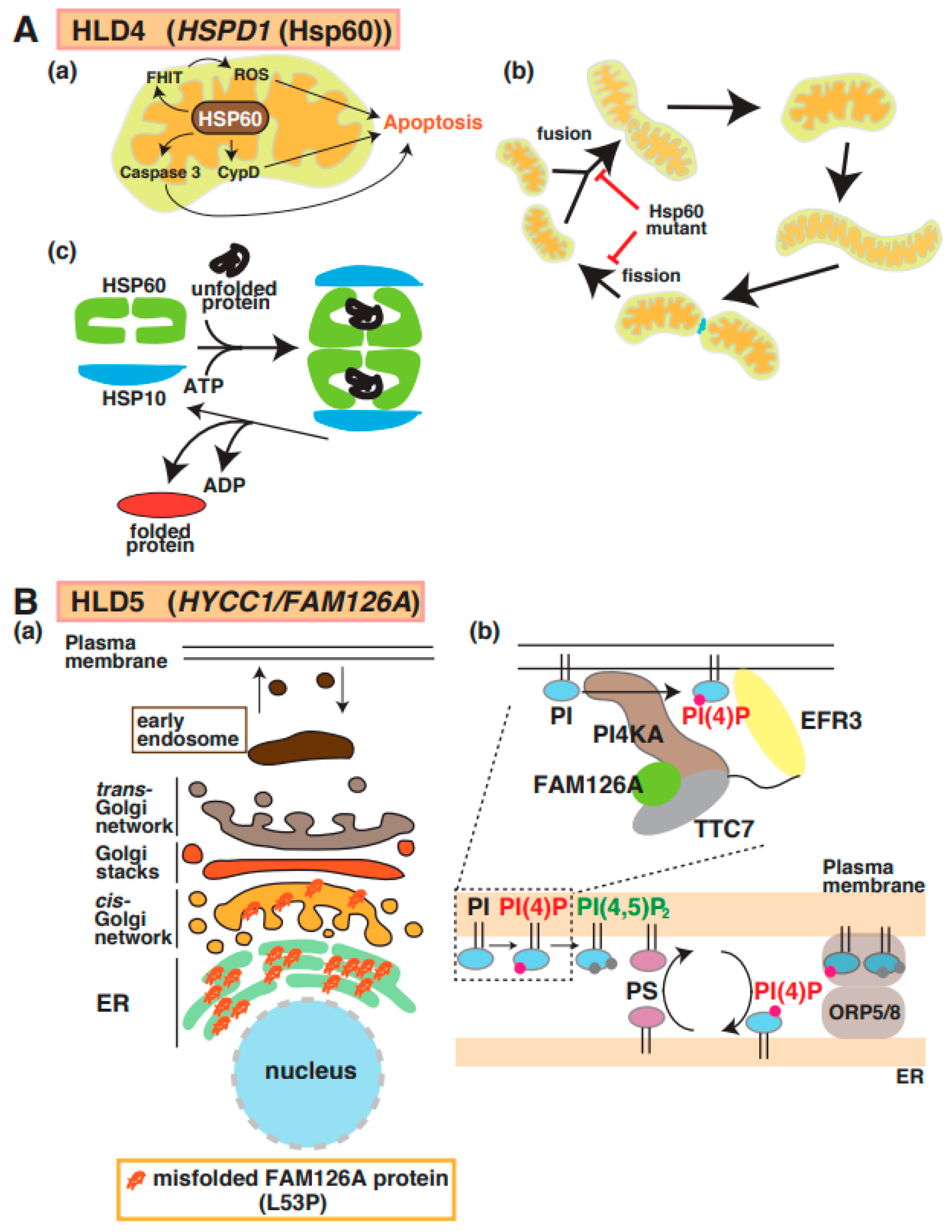
5. HLD4 (OMIM ID 612233)
6. HLD5 (OMIM ID 610532)
7. HLD6 (OMIM ID 612438)
8. HLD7 (OMIM ID 607694), HLD8 (OMIM ID 614381), HLD11 (OMIM ID 616494), and HLD21 (OMIM ID 619310)
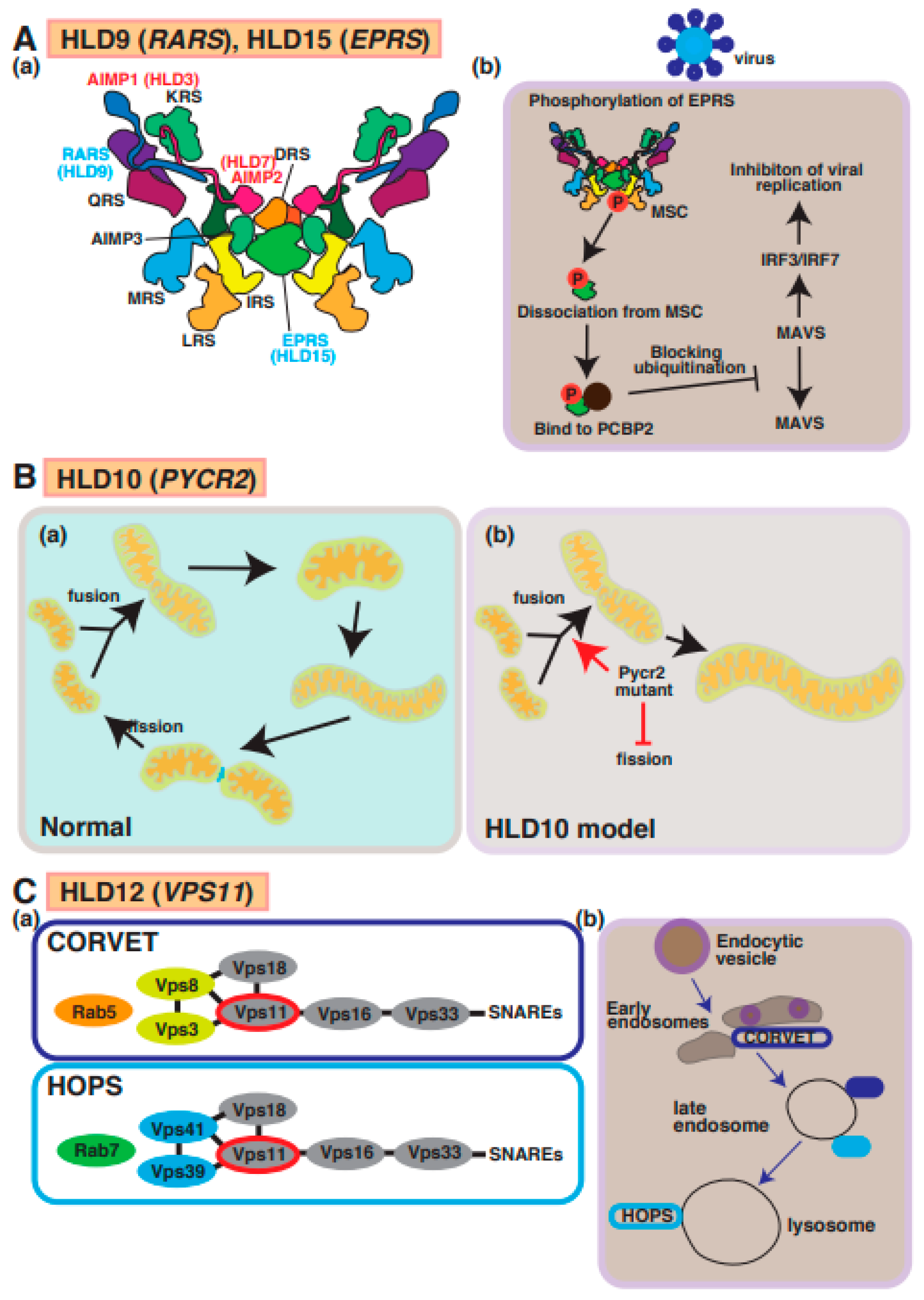
9. HLD9 (OMIM ID 616140) and HLD15 (OMIM ID 617951)
10. HLD10 (OMIM ID 616420)
11. HLD12 (OMIM ID 616683)
12. HLD13 (OMIM ID 616881)
13. HLD14 (OMIM ID 617899)
14. HLD16 (OMIM ID 617964)
15. HLD18 (OMIM ID 618404)
16. HLD19 (OMIM ID 618688)
17. HLD20 (OMIM ID 619071)
18. HLD22 (OMIM ID 619328)
19. HLD23 (OMIM ID 619688)
20. HLD24 (OMIM ID 619851)
21. Clinical Trial for Leukodystrophies
22. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bonetto, G.; Belin, D.; Káradóttir, R.T. Myelin: A gatekeeper of activity-dependent circuit plasticity? Science 2021, 374, eaba6905. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Nave, K.A. Oligodendrocytes: Myelination and axonal support. Cold Spring Harb. Perspect. Biol. 2015, 8, a020479. [Google Scholar] [CrossRef] [PubMed]
- Osso, L.A.; Rankin, K.A.; Chan, J.R. Experience-dependent myelination following stress is mediated by the neuropeptide dynorphin. Neuron 2021, 109, 3619–3632. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Manel, N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat. Rev. Immunol. 2015, 15, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Engelen, M.; Kemp, S.; Eichler, F. Endocrine dysfunction in adrenoleukodystrophy. Handb. Clin. Neurol. 2021, 182, 257–267. [Google Scholar]
- Messing, A. Alexander disease. Handb. Clin. Neurol. 2018, 148, 693–700. [Google Scholar]
- Hoshino, H.; Kubota, M. Canavan disease: Clinical features and recent advances in research. Pediatr. Int. 2014, 56, 477–483. [Google Scholar] [CrossRef]
- Nie, S.; Chen, G.; Cao, X.; Zhang, Y. Cerebrotendinous xanthomatosis: A comprehensive review of pathogenesis, clinical manifestations, diagnosis, and management. Orphanet. J. Rare. Dis. 2014, 9, 179. [Google Scholar] [CrossRef]
- Husain, A.M.; Altuwaijri, M.; Aldosari, M. Krabbe disease: Neurophysiologic studies and MRI correlations. Neurology 2004, 63, 617–620. [Google Scholar] [CrossRef]
- Kohlschütter, A. Lysosomal leukodystrophies: Krabbe disease and metachromatic leukodystrophy. Handb. Clin. Neurol. 2013, 113, 1611–1618. [Google Scholar]
- Vanier, M.T. Niemann-Pick diseases. Handb. Clin. Neurol. 2013, 113, 1717–1721. [Google Scholar] [PubMed]
- Torii, T.; Miyamoto, Y.; Yamauchi, J.; Tanoue, A. Pelizaeus-Merzbacher disease: Cellular pathogenesis and pharmacologic therapy. Pediatr. Int. 2014, 56, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, G.; Mattern, C.; Begou, M.; Boespflug-Tanguy, O.; Massaad, C.; Massaad-Massade, L. Mutation of Proteolipid Protein 1 Gene: From Severe Hypomyelinating Leukodystrophy to Inherited Spastic Paraplegia. Biomedicines 2022, 10, 1709. [Google Scholar] [CrossRef]
- Cailloux, F.; Gauthier-Barichard, F.; Mimault, C.; Isabelle, V.; Coutois, V.; Giraud, G.; Dastugue, B.; Boespflug-Tanguy, O. Genotype-phenotype correlation in inherited brain myelination defects due to proteolipid protein gene mutations. Eur. J. Hum. Genet. 2000, 8, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K. Cellular pathology of Pelizaeus-Merzbacher disease involving chaperones associated with endoplasmic reticulum stress. Front. Mol. Biosci. 2017, 4, 7. [Google Scholar] [CrossRef]
- Simons, M.; Kramer, E.M.; Macchi, P.; Rathke-Hartlieb, S.; Trotter, J.; Nave, K.A.; Schulz, J.B. Overexpression of the myelin proteolipid protein leads to accumulation of cholesterol and proteolipid protein in endosomes/lysosomes: Implications for Pelizaeus-Merzbacher disease. J. Cell Biol. 2002, 157, 327–336. [Google Scholar] [CrossRef]
- Numasawa-Kuroiwa, Y.; Okada, Y.; Shibata, S.; Kishi, N.; Akamatsu, W.; Shoji, M.; Nakanishi, A.; Oyama, M.; Osaka, H.; Inoue, K.; et al. Involvement of ER stress in dysmyelination of Perizaeus-Merzbacher disease with PLP1 missense mutations shown by iPS-derived oligodendrocytes. Stem Cell Rep. 2014, 2, 648–661. [Google Scholar] [CrossRef]
- Readhead, C.; Schneider, A.; Griffiths, I.; Nave, K.A. Premature arrest of myelin formation in transgenic mice with increased proteolipid protein gene dosage. Neuron 1994, 12, 583–595. [Google Scholar] [CrossRef]
- Kagawa, T.; Ikenaka, K.; Inoue, Y.; Kuriyama, S.; Tsujii, T.; Nakao, J.; Nakajima, K.; Aruga, J.; Okano, H.; Mikoshiba, K. Glial cell degeneration and hypomyelination caused by overexpression of myelin proteolipid protein gene. Neuron 1994, 13, 427–442. [Google Scholar] [CrossRef]
- Elitt, M.S.; Barbar, L.; Shick, H.E.; Powers, B.E.; Maeno-Hikichi, Y.; Madhavan, M.; Allan, K.C.; Nawash, B.S.; Gevorgyan, A.S.; Hung, S.; et al. Suppression of proteolipid protein rescues Pelizaeus-Merzbacher disease. Nature 2020, 585, 397–403. [Google Scholar] [CrossRef]
- Li, H.; Okada, H.; Suzuki, S.; Sakai, K.; Izumi, H.; Matsushima, Y.; Ichinohe, N.; Goto, Y.; Okada, T.; Inoue, K. Gene suppressing therapy for Pelizaeus-Merzbacher disease using artificial microRNA. JCI Insight 2019, 4, e125052. [Google Scholar] [CrossRef] [PubMed]
- Fasciani, I.; Pluta, P.; González-Nieto, D.; Martínez-Montero, P.; Molano, J.; Paíno, C.L.; Millet, O.; Barrio, L.C. Directional coupling of oligodendrocyte connexin-47 and astrocyte connexin-43 gap junctions. Glia 2018, 66, 2340–2352. [Google Scholar] [CrossRef] [PubMed]
- Uhlenberg, B.; Schuelke, M.; Rüschendorf, F.; Ruf, N.; Kaindl, A.M.; Henneke, M.; Thiele, H.; Stoltenburg-Didinger, G.; Aksu, F.; Topaloğlu, H.; et al. Mutations in the gene encoding gap junction protein α12 (connexin 46.6) cause Pelizaeus-Merzbacher–like disease. Am. J. Hum. Genet. 2004, 75, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Abrams, C.K.; Scherer, S.S.; Flores-Obando, R.; Freidin, M.M.; Wong, S.; Lamantea, E.; Farina, L.; Scaioli, V.; Pareyson, D.; Salsano, E. A new mutation in GJC2 associated with subclinical leukodystrophy. J. Neurol. 2014, 261, 1929–1938. [Google Scholar] [CrossRef]
- Orthmann-Murphy, J.L.; Enriquez, A.D.; Abrams, C.K.; Scherer, S.S. Loss-of-function GJA12/Connexin47 mutations cause Pelizaeus–Merzbacher-like disease. Mol. Cell. Neurosci. 2007, 34, 629–641. [Google Scholar] [CrossRef]
- Orthmann-Murphy, J.L.; Salsano, E.; Abrams, C.K.; Bizzi, A.; Uziel, G.; Freidin, M.M.; Lamantea, E.; Zeviani, M.; Scherer, S.S.; Pareyson, D. Hereditary spastic paraplegia is a novel phenotype for GJA12/GJC2 mutations. Brain 2009, 132, 426–438. [Google Scholar] [CrossRef]
- Feinstein, M.; Markus, B.; Noyman, I.; Shalev, H.; Flusser, H.; Shelef, I.; Liani-Leibson, K.; Shorer, Z.; Cohen, I.; Khateeb, S.; et al. Pelizaeus-Merzbacher-like disease caused by AIMP1/p43 homozygous mutation. Am. J. Hum. Genet. 2010, 87, 820–828. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Tanaka, M.; Okura, N.; Fukui, Y.; Noguchi, K.; Hayashi, Y.; Torii, T.; Ooizumi, H.; Ohbuchi, K.; Mizoguchi, K.; et al. Rare neurologic disease-associated mutations of AIMP1 are related with inhibitory neuronal differentiation which is reversed by ibuprofen. Medicines 2020, 7, 25. [Google Scholar] [CrossRef]
- Fu, Y.; Kim, Y.; Jin, K.S.; Kim, H.S.; Kim, J.H.; Wang, D.; Park, M.; Jo, C.H.; Kwon, N.H.; Kim, D.; et al. Structure of the ArgRS-GlnRS-AIMP1 complex and its implications for mammalian translation. Proc. Natl. Acad. Sci. USA 2014, 111, 15084–15089. [Google Scholar] [CrossRef]
- Halpert, M.M.; Konduri, V.; Liang, D.; Vazquez-Perez, J.; Hofferek, C.; Weldon, S.A.; Baig, Y.; Vedula, I.; Levitt, J.M.; Decker, W.K. MHC class Ⅰ and Ⅱ peptide homology regulates the cellular immune response. FASEB J. 2021, 34, 8082–8101. [Google Scholar] [CrossRef]
- Shukla, A.; Das Bhowmik, A.; Hebbar, M.; Rajagopal, K.V.; Girisha, K.M.; Gupta, N.; Dalal, A. Homozygosity for a nonsense variant in AIMP2 is associated with a progressive neurodevelopmental disorder with microcephaly, seizures, and spastic quadriparesis. J. Hum. Genet. 2018, 63, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, A.; Sawaguchi, S.; Memezawa, S.; Seki, Y.; Morimoto, T.; Oizumi, H.; Ohbuchi, K.; Yamamoto, M.; Mizoguchi, K.; Miyamoto, Y.; et al. Knockdown of Golgi stress-responsive caspase-2 ameliorates HLD17-associated AIMP2 mutant-mediated inhibition of oligodendroglial cell morphological differentiation. Neurochem. Res. 2022, 47, 2617–2631. [Google Scholar] [CrossRef] [PubMed]
- Magen, D.; Georgopoulos, C.; Bross, P.; Ang, D.; Segev, Y.; Goldsher, D.; Nemirovski, A.; Shahar, E.; Ravid, S.; Luder, A.; et al. Mitochondrial hsp60 chaperonopathy causes an autosomal-recessive neurodegenerative disorder linked to brain hypomyelination and leukodystrophy. Am. J. Hum. Genet. 2008, 83, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Scalia, F.; Marino Gammazza, A.; Conway de Macario, E.; Macario, A.J.L.; Cappello, F. Myelin pathology: Involvement of molecular chaperones and the promise of chaperonotherapy. Brain Sci. 2019, 9, 297. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Eguchi, T.; Kawahara, K.; Hasegawa, N.; Nakamura, K.; Funakoshi-Tago, M.; Tanoue, A.; Tamura, H.; Yamauchi, J. Hypomyelinating leukodystrophy-associated missense mutation in HSPD1 blunts mitochondrial dynamics. Biochem. Biophys. Res. Commun. 2015, 462, 275–281. [Google Scholar] [CrossRef]
- Zara, F.; Biancheri, R.; Bruno, C.; Bordo, L.; Assereto, S.; Gazzerro, E.; Sotgia, F.; Wang, X.B.; Gianotti, S.; Stringara, S.; et al. Deficiency of hyccin, a newly identified membrane protein, causes hypomyelination and congenital cataract. Nat. Genet. 2006, 38, 1111–1113. [Google Scholar] [CrossRef]
- Baskin, J.M.; Wu, X.; Christiano, R.; Oh, M.S.; Schauder, C.M.; Gazzerro, E.; Messa, M.; Baldassari, S.; Assereto, S.; Biancheri, R.; et al. The leukodystrophy protein FAM126A (hyccin) regulates PtdIns(4)P synthesis at the plasma membrane. Nat. Cell Biol. 2016, 18, 132–138. [Google Scholar] [CrossRef]
- Poitelon, Y.; Kopec, A.M.; Belin, S. Myelin Fat Facts: An Overview of Lipids and Fatty Acid Metabolism. Cells 2020, 9, 812. [Google Scholar] [CrossRef]
- Kister, A.; Kister, I. Overview of myelin, major myelin lipids, and myelin-associated proteins. Front. Chem. 2023, 10, 1041961. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Torii, T.; Eguchi, T.; Nakamura, K.; Tanoue, A.; Yamauchi, J. Hypomyelinating leukodystrophy-associated missense mutant of FAM126A/hyccin/DRCTNNB1A aggregates in the endoplasmic reticulum. J. Clin. Neurosci. 2014, 21, 1033–1039. [Google Scholar] [CrossRef]
- Curiel, J.; Rodríguez Bey, G.; Takanohashi, A.; Bugiani, M.; Fu, X.; Wolf, N.I.; Nmezi, B.; Schiffmann, R.; Bugaighis, M.; Pierson, T.; et al. TUBB4A mutations result in specific neuronal and oligodendrocytic defects that closely match clinically distinct phenotypes. Hum. Mol. Genet. 2017, 26, 4506–4518. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, K.; Wilcox, R.A.; Winkler, S.; Ramirez, A.; Rakovic, A.; Park, J.S.; Arns, B.; Lohnau, T.; Groen, J.; Kasten, M.; et al. Whispering dysphonia (DYT4 dystonia) is caused by a mutation in the TUBB4 gene. Ann. Neurol. 2013, 73, 537–545. [Google Scholar] [CrossRef]
- Duncan, I.D.; Bugiani, M.; Radcliff, A.B.; Moran, J.J.; Lopez-Anido, C.; Duong, P.; August, B.K.; Wolf, N.I.; van der Knaap, M.S.; Svaren, J. A mutation in the Tubb4a gene leads to microtubule accumulation with hypomyelination and demyelination. Ann. Neurol. 2017, 81, 690–702. [Google Scholar] [CrossRef]
- Song, J.; O’connor, L.T.; Yu, W.; Baas, P.W.; Duncan, I.D. Microtubule alterations in cultured taiep rat oligodendrocytes lead to deficits in myelin membrane formation. J. Neurocytol. 1999, 28, 671–683. [Google Scholar] [CrossRef]
- Bally, J.F.; Camargos, S.; Oliveira Dos Santos, C.; Kern, D.S.; Lee, T.; Pereira da Silva-Junior, F.; Puga, R.D.; Cardoso, F.; Barbosa, E.R.; Yadav, R.; et al. DYT-TUBB4A (DYT4 dystonia): New clinical and genetic observations. Neurology 2021, 96, e1887–e1897. [Google Scholar] [CrossRef]
- Vulinovic, F.; Krajka, V.; Hausrat, T.J.; Seibler, P.; Alvarez-Fischer, D.; Madoev, H.; Park, J.S.; Kumar, K.R.; Sue, C.M.; Lohmann, K.; et al. Motor protein binding and mitochondrial transport are altered by pathogenic TUBB4A variants. Hum. Mutat. 2018, 39, 1901–1915. [Google Scholar] [CrossRef]
- Paule, M.R.; White, R.J. Survey and summary: Transcription by RNA polymerase I and III. Nucleic Acids Res. 2000, 28, 1283–1298. [Google Scholar] [CrossRef]
- Ramsay, E.P.; Abascal-Palacios, G.; Daiß, J.L.; King, H.; Gouge, J.; Pilsl, M.; Beuron, F.; Morris, E.; Gunkel, P.; Engel, C.; et al. Structure of human RNA polymerase III. Nat. Commun. 2020, 11, 6409. [Google Scholar] [CrossRef]
- Sawaguchi, S.; Tago, K.; Oizumi, H.; Ohbuchi, K.; Yamamoto, M.; Mizoguchi, K.; Miyamoto, Y.; Yamauchi, J. Hypomyelinating leukodystrophy 7 (HLD7)-associated mutation of POLR3A is related to defective oligodendroglial cell differentiation, which is ameliorated by ibuprofen. Neurol. Int. 2021, 14, 11–33. [Google Scholar] [CrossRef] [PubMed]
- Wolf, N.I.; Vanderver, A.; van Spaendonk, R.M.; Schiffmann, R.; Brais, B.; Bugiani, M.; Sistermans, E.; Catsman-Berrevoets, C.; Kros, J.M.; Pinto, P.S.; et al. Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology 2014, 83, 1898–1905. [Google Scholar] [CrossRef] [PubMed]
- Wolf, N.I.; Salomons, G.S.; Rodenburg, R.J.; Pouwels, P.J.; Schieving, J.H.; Derks, T.G.; Fock, J.M.; Rump, P.; van Beek, D.M.; van der Knaap, M.S.; et al. Mutations in RARS cause hypomyelination. Ann. Neurol. 2014, 76, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Nafisinia, M.; Sobreira, N.; Riley, L.; Gold, W.; Uhlenberg, B.; Weiß, C.; Boehm, C.; Prelog, K.; Ouvrier, R.; Christodoulou, J. Mutations in RARS cause a hypomyelination disorder akin to Pelizaeus-Merzbacher disease. Eur. J. Hum. Genet. 2017, 25, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Mendes, M.I.; Gutierrez Salazar, M.; Guerrero, K.; Thiffault, I.; Salomons, G.S.; Gauquelin, L.; Tran, L.T.; Forget, D.; Gauthier, M.S.; Waisfisz, Q.; et al. Bi-allelic mutations in EPRS, encoding the glutamyl-prolyl-aminoacyl-tRNA synthetase, cause a hypomyelinating leukodystrophy. Am. J. Hum. Genet. 2018, 102, 676–684. [Google Scholar] [CrossRef]
- Nakayama, T.; Al-Maawali, A.; El-Quessny, M.; Rajab, A.; Khalil, S.; Stoler, J.M.; Tan, W.H.; Nasir, R.; Schmitz-Abe, K.; Hill, R.S.; et al. Mutations in PYCR2, encoding pyrroline-5-carboxylate reductase 2, cause microcephaly and hypomyelination. Am. J. Hum. Genet. 2015, 96, 709–719. [Google Scholar] [CrossRef]
- Zaki, M.S.; Bhat, G.; Sultan, T.; Issa, M.; Jung, H.J.; Dikoglu, E.; Selim, L.; Mahmoud, I.G.; Abdel-Hamid, M.S.; Abdel-Salam, G.; et al. PYCR2 mutations cause a lethal syndrome of microcephaly and failure to thrive. Ann. Neurol. 2016, 80, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Donti, T.; Xia, F.; Niu, Z.; AI Shamsi, A.; Hertecant, J.; Al-Jasmi, F.; Gibson, J.B.; Nagakura, H.; Zhang, J.; et al. Homozygous variants in pyrroline-5-carboxylate reductase 2 (PYCR2) in patients with progressive microcephaly and hypomyelinating leukodystrophy. Am. J. Med. Genet. A 2017, 173, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Escande-Beillard, N.; Loh, A.; Saleem, S.N.; Kanata, K.; Hashimoto, Y.; Altunoglu, U.; Metoska, A.; Grandjean, J.; Ng, F.M.; Pomp, O.; et al. Loss of PYCR2 Causes Neurodegeneration by Increasing Cerebral Glycine Levels via SHMT2. Neuron 2020, 107, 82–94.e6. [Google Scholar] [CrossRef]
- Torii, T.; Shirai, R.; Kiminami, R.; Nishino, S.; Sato, T.; Sawaguchi, S.; Fukushima, N.; Seki, Y.; Miyamoto, Y.; Yamauchi, J. Hypomyelinating leukodystrophy 10 (HLD10)-associated mutations of PYCR2 form large size mitochondria, inhibiting oligodendroglial cell morphological differentiation. Neurol. Int. 2022, 14, 1062–1080. [Google Scholar] [CrossRef]
- Zheng, H.; Gupta, V.; Patterson-Fortin, J.; Bhattacharya, S.; Katlinski, K.; Wu, J.; Varghese, B.; Carbone, C.J.; Aressy, B.; Fuchs, S.Y.; et al. A BRISC-SHMT complex deubiquitinates IFNAR1 and regulates interferon responses. Cell Rep. 2013, 5, 180–193. [Google Scholar] [CrossRef]
- van der Kant, R.; Jonker, C.T.; Wijdeven, R.H.; Bakker, J.; Janssen, L.; Klumperman, J.; Neefjes, J. Characterization of the mammalian CORVET and HOPS complexes and their modular restructuring for endosome specificity. J. Biol. Chem. 2015, 290, 30280–30290. [Google Scholar] [CrossRef]
- Skoff, R.P.; Bessert, D.; Banerjee, S.; Luo, X.; Thummel, R. Characterization of expression of vacuolar protein sorting 11 (Vps11) in mammalian oligodendrocytes. ASN Neuro 2021, 13, 17590914211009851. [Google Scholar] [CrossRef] [PubMed]
- Edvardson, S.; Gerhard, F.; Jalas, C.; Lachmann, J.; Golan, D.; Saada, A.; Shaag, A.; Ungermann, C.; Elpeleg, O. Hypomyelination and developmental delay associated with VPS11 mutation in Ashkenazi-Jewish patients. J. Med. Genet. 2015, 52, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lachance, V.; Schaffner, A.; Li, X.; Fedick, A.; Kaye, L.E.; Liao, J.; Rosenfeld, J.; Yachelevich, N.; Chu, M.L.; et al. A founder mutation in VPS11 causes an autosomal recessive leukoencephalopathy linked to autophagic defects. PLoS Genet. 2016, 12, e1005848. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, N.; Miyamoto, Y.; Hattori, K.; Ito, A.; Harada, H.; Oizumi, H.; Ohbuchi, K.; Mizoguchi, K.; Yamauchi, J. PP1C and PP2A are p70S6K phosphatases whose inhibition ameliorates HLD12-associated inhibition of oligodendroglial cell morphological differentiation. Biomedicines 2020, 8, 89. [Google Scholar] [CrossRef]
- Edvardson, S.; Kose, S.; Jalas, C.; Fattal-Valevski, A.; Watanabe, A.; Ogawa, Y.; Mamada, H.; Fedick, A.M.; Ben-Shachar, S.; Treff, N.R.; et al. Leukoencephalopathy and early death associated with an Ashkenazi-Jewish founder mutation in the Hikeshi gene. J. Med. Genet. 2016, 53, 132–137. [Google Scholar] [CrossRef]
- Hattori, K.; Tago, K.; Memezawa, S.; Ochiai, A.; Sawaguchi, S.; Kato, Y.; Sato, T.; Tomizuka, K.; Ooizumi, H.; Ohbuchi, K.; et al. The infantile leukoencephalopathy-associated mutation of C11ORF73/HIKESHI proteins generates de novo interactive activity with Filamin A, inhibiting oligodendroglial cell morphological differentiation. Medicines 2021, 8, 9. [Google Scholar] [CrossRef]
- Alcover-Sanchez, B.; Garcia-Martin, G.; Wandosell, F.; Cubelos, B. R-Ras GTPases signaling role in myelin neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 5911. [Google Scholar] [CrossRef]
- Nahorski, M.S.; Maddirevula, S.; Ishimura, R.; Alsahli, S.; Brady, A.F.; Begemann, A.; Mizushima, T.; Guzmán-Vega, F.J.; Obata, M.; Ichimura, Y.; et al. Biallelic UFM1 and UFC1 mutations expand the essential role of ufmylation in brain development. Brain 2018, 141, 1934–1945. [Google Scholar] [CrossRef]
- Banerjee, S.; Kumar, M.; Wiener, R. Decrypting UFMylation: How proteins are modified with UFM1. Biomolecules 2020, 10, 1442. [Google Scholar] [CrossRef]
- Ishimura, R.; El-Gowily, A.H.; Noshiro, D.; Komatsu-Hirota, S.; Ono, Y.; Shindo, M.; Hatta, T.; Abe, M.; Uemura, T.; Lee-Okada, H.C.; et al. The UFM1 system regulates ER-phagy through the ufmylation of CYB5R3. Nat. Commun. 2023, 13, 7857. [Google Scholar] [CrossRef]
- Simons, C.; Dyment, D.; Bent, S.J.; Crawford, J.; D’Hooghe, M.; Kohlschütter, A.; Venkateswaran, S.; Helman, G.; Poll-The, B.T.; Makowski, C.C.; et al. A recurrent de novo mutation in TMEM106B causes hypomyelinating leukodystrophy. Brain 2017, 140, 3105–3111. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Sheng, R.R.; Solé-Domènech, S.; Ullah, M.; Zhou, X.; Mendoza, C.S.; Enriquez, L.C.M.; Katz, I.I.; Paushter, D.H.; Sullivan, P.M.; et al. A role of the frontotemporal lobar degeneration risk factor TMEM106B in myelination. Brain 2020, 143, 2255–2271. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Nicholson, A.M.; Ren, Y.; Brooks, M.; Jiang, P.; Zuberi, A.; Phuoc, H.N.; Perkerson, R.B.; Matchett, B.; Parsons, T.M.; et al. Loss of TMEM106B leads to myelination deficits: Implications for frontotemporal dementia treatment strategies. Brain 2020, 143, 1905–1919. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Lacrampe, A.; Hu, F. Physiological and pathological functions of TMEM106B: A gene associated with brain aging and multiple brain disorders. Acta Neuropathol. 2021, 141, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Karsai, G.; Kraft, F.; Haag, N.; Korenke, G.C.; Hänisch, B.; Othman, A.; Suriyanarayanan, S.; Steiner, R.; Knopp, C.; Mull, M.; et al. DEGS1-associated aberrant sphingolipid metabolism impairs nervous system function in humans. J. Clin. Investig. 2019, 129, 1229–1239. [Google Scholar] [CrossRef]
- Planas-Serra, L.; Launay, N.; Goicoechea, L.; Heron, B.; Jou, C.; Juliá-Palacios, N.; Ruiz, M.; Fourcade, S.; Casasnovas, C.; De La Torre, C.; et al. Sphingolipid desaturase DEGS1 is essential for mitochondria-associated membrane integrity. J. Clin. Investig. 2023, 133, e162957. [Google Scholar] [CrossRef]
- Fukumura, S.; Hiraide, T.; Yamamoto, A.; Tsuchida, K.; Aoto, K.; Nakashima, M.; Saitsu, H. A novel de novo TMEM63A variant in a patient with severe hypomyelination and global developmental delay. Brain Dev. 2022, 44, 178–183. [Google Scholar] [CrossRef]
- Zhang, T.M.; Liao, L.; Yang, S.Y.; Huang, M.Y.; Zhang, Y.L.; Deng, L.; Hu, S.Y.; Yang, F.; Zhang, F.L.; Shao, Z.M.; et al. TOLLIP-mediated autophagic degradation pathway links the VCP-TMEM63A-DERL1 signaling axis to triple-negative breast cancer progression. Autophagy 2023, 19, 805–821. [Google Scholar] [CrossRef]
- Acheta, J.; Bhatia, U.; Haley, J.; Hong, J.; Rich, K.; Close, R.; Bechler, M.E.; Belin, S.; Poitelon, Y. Piezo channels contribute to the regulation of myelination in Schwann cells. Glia 2022, 70, 2276–2289. [Google Scholar] [CrossRef]
- Lee, J.; Gravel, M.; Zhang, R.; Thibault, P.; Braun, P.E. Process outgrowth in oligodendrocytes is mediated by CNP, a novel microtubule assembly myelin protein. J. Cell Biol. 2005, 170, 661–673. [Google Scholar] [CrossRef]
- Al-Abdi, L.; Al Murshedi, F.; Elmanzalawy, A.; Al Habsi, A.; Helaby, R.; Ganesh, A.; Ibrahim, N.; Patel, N.; Alkuraya, F.S. CNP deficiency causes severe hypomyelinating leukodystrophy in humans. Hum. Genet. 2020, 139, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Sasaki, H.; Fujimoto, K.; Furuse, M.; Tsukita, S. Claudin-11/OSP-based tight junctions of myelin sheaths in brain and Sertoli cells in testis. J. Cell Biol. 1999, 145, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Gow, A.; Southwood, C.M.; Li, J.S.; Pariali, M.; Riordan, G.P.; Brodie, S.E.; Danias, J.; Bronstein, J.M.; Kachar, B.; Lazzarini, R.A. CNS myelin and sertoli cell tight junction strands are absent in Osp/claudin-11 null mice. Cell 1999, 99, 649–959. [Google Scholar] [CrossRef]
- Li, C.F.; Chen, J.Y.; Ho, Y.H.; Hsu, W.H.; Wu, L.C.; Lan, H.Y.; Hsu, D.S.; Tai, S.K.; Chang, Y.C.; Yang, M.H. Snail-induced claudin-11 prompts collective migration for tumour progression. Nat. Cell Biol. 2019, 21, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Riedhammer, K.M.; Stockler, S.; Ploski, R.; Wenzel, M.; Adis-Dutschmann, B.; Ahting, U.; Alhaddad, B.; Blaschek, A.; Haack, T.B.; Kopajtich, R.; et al. De novo stop-loss variants in CLDN11 cause hypomyelinating leukodystrophy. Brain 2021, 144, 411–419. [Google Scholar] [CrossRef]
- Kong, Q.; Zeng, W.; Wu, J.; Hu, W.; Li, C.; Mao, B. RNF220, an E3 ubiquitin ligase that targets Sin3B for ubiquitination. Biochem. Biophys. Res. Commun. 2010, 393, 708–713. [Google Scholar] [CrossRef]
- Guo, X.; Ma, P.; Li, Y.; Yang, Y.; Wang, C.; Xu, T.; Wang, H.; Li, C.; Mao, B.; Qi, X. RNF220 mediates K63-linked polyubiquitination of STAT1 and promotes host defense. Cell Death. Differ. 2021, 28, 640–656. [Google Scholar] [CrossRef]
- Ma, P.; Wan, L.P.; Li, Y.; He, C.H.; Song, N.N.; Zhao, S.; Wang, H.; Ding, Y.Q.; Mao, B.; Sheng, N. RNF220 is an E3 ubiquitin ligase for AMPA receptors to regulate synaptic transmission. Sci. Adv. 2022, 8, eabq4736. [Google Scholar] [CrossRef]
- Sferra, A.; Fortugno, P.; Motta, M.; Aiello, C.; Petrini, S.; Ciolfi, A.; Cipressa, F.; Moroni, I.; Leuzzi, V.; Pieroni, L.; et al. Biallelic mutations in RNF220 cause laminopathies featuring leukodystrophy, ataxia and deafness. Brain 2021, 144, 3020–3035. [Google Scholar] [CrossRef]
- Segawa, K.; Kikuchi, A.; Noji, T.; Sugiura, Y.; Hiraga, K.; Suzuki, C.; Haginoya, K.; Kobayashi, Y.; Matsunaga, M.; Ochiai, Y.; et al. A sublethal ATP11A mutation associated with neurological deterioration causes aberrant phosphatidylcholine flipping in plasma membranes. J. Clin. Investig. 2021, 131, e148005. [Google Scholar] [CrossRef]
- Uchida, N.; Chen, K.; Dohse, M.; Hansen, K.D.; Dean, J.; Buser, J.R.; Riddle, A.; Beardsley, D.J.; Wan, Y.; Gong, X.; et al. Human neural stem cells induce functional myelination in mice with severe dysmyelination. Sci. Transl. Med. 2012, 4, 155ra136. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Henry, R.G.; Kang, S.M.; Strober, J.; Lim, D.A.; Ryan, T.; Perry, R.; Farrell, J.; Ulman, M.; Rajalingam, R.; et al. Neural stem cell engraftment and myelination in the human brain. Sci. Transl. Med. 2012, 4, 155ra137. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Henry, R.G.; Kang, S.M.; Strober, J.; Lim, D.A.; Ryan, T.; Perry, R.; Farrell, J.; Ulman, M.; Rajalingam, R.; et al. Long-Term Safety, Immunologic Response, and Imaging Outcomes following Neural Stem Cell Transplantation for Pelizaeus-Merzbacher Disease. Stem Cell Rep. 2019, 13, 254–261. [Google Scholar] [CrossRef]
- Xue, Y.Y.; Cheng, H.L.; Dong, H.L.; Yin, H.M.; Yuan, Y.; Meng, L.C.; Wu, Z.Y.; Yu, H. A de novo variant of POLR3B causes demyelinating Charcot-Marie-Tooth disease in a Chinese patient: A case report. BMC Neurol. 2021, 21, 402. [Google Scholar] [CrossRef]
- Ando, M.; Higuchi, Y.; Yuan, J.H.; Yoshimura, A.; Kitao, R.; Morimoto, T.; Taniguchi, T.; Takeuchi, M.; Takei, J.; Hiramatsu, Y.; et al. Novel de novo POLR3B mutations responsible for demyelinating Charcot-Marie-Tooth disease in Japan. Ann. Clin. Transl. Neurol. 2022, 9, 747–755. [Google Scholar] [CrossRef]
- Figlia, G.; Gerber, D.; Suter, U. Myelination and mTOR. Glia 2018, 66, 693–707. [Google Scholar] [CrossRef] [PubMed]
- Wolf, N.I.; Ffrench-Constant, C.; van der Knaap, M.S. Hypomyelinating leukodystrophies—Unravelling myelin biology. Nat. Rev. Neurol. 2021, 17, 88–103. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torii, T.; Yamauchi, J. Molecular Pathogenic Mechanisms of Hypomyelinating Leukodystrophies (HLDs). Neurol. Int. 2023, 15, 1155-1173. https://doi.org/10.3390/neurolint15030072
Torii T, Yamauchi J. Molecular Pathogenic Mechanisms of Hypomyelinating Leukodystrophies (HLDs). Neurology International. 2023; 15(3):1155-1173. https://doi.org/10.3390/neurolint15030072
Chicago/Turabian StyleTorii, Tomohiro, and Junji Yamauchi. 2023. "Molecular Pathogenic Mechanisms of Hypomyelinating Leukodystrophies (HLDs)" Neurology International 15, no. 3: 1155-1173. https://doi.org/10.3390/neurolint15030072
APA StyleTorii, T., & Yamauchi, J. (2023). Molecular Pathogenic Mechanisms of Hypomyelinating Leukodystrophies (HLDs). Neurology International, 15(3), 1155-1173. https://doi.org/10.3390/neurolint15030072