
FTD/ALS Type 7-Associated Thr104Asn Mutation of CHMP2B Blunts Neuronal Process Elongation, and Is Recovered by Knockdown of Arf4, the Golgi Stress Regulator

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Key Antibodies and siRNA Sequences
2.2. Reverse Transcription-Linked Polymerase Chain Reactions (RT-PCR)
2.3. Cell Line and Stable Clone Cultures and Differentiation
2.4. siRNA and Plasmid Transfection
2.5. Polyacrylamide Electrophoresis and Immunoblotting Techniques
2.6. Capturing Fluorescence Images
2.7. Statistical Analyses
2.8. Ethical Approval
3. Results
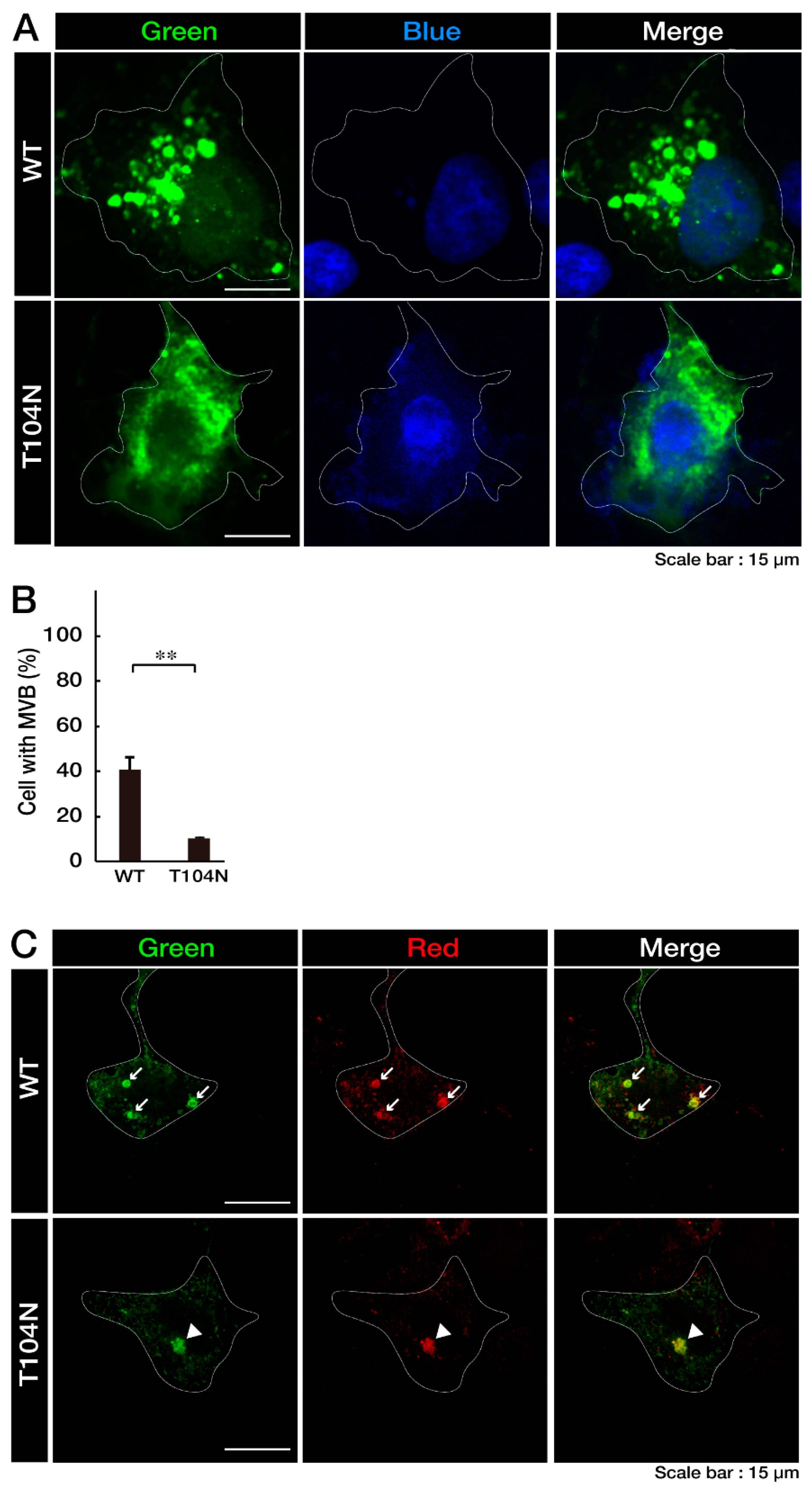
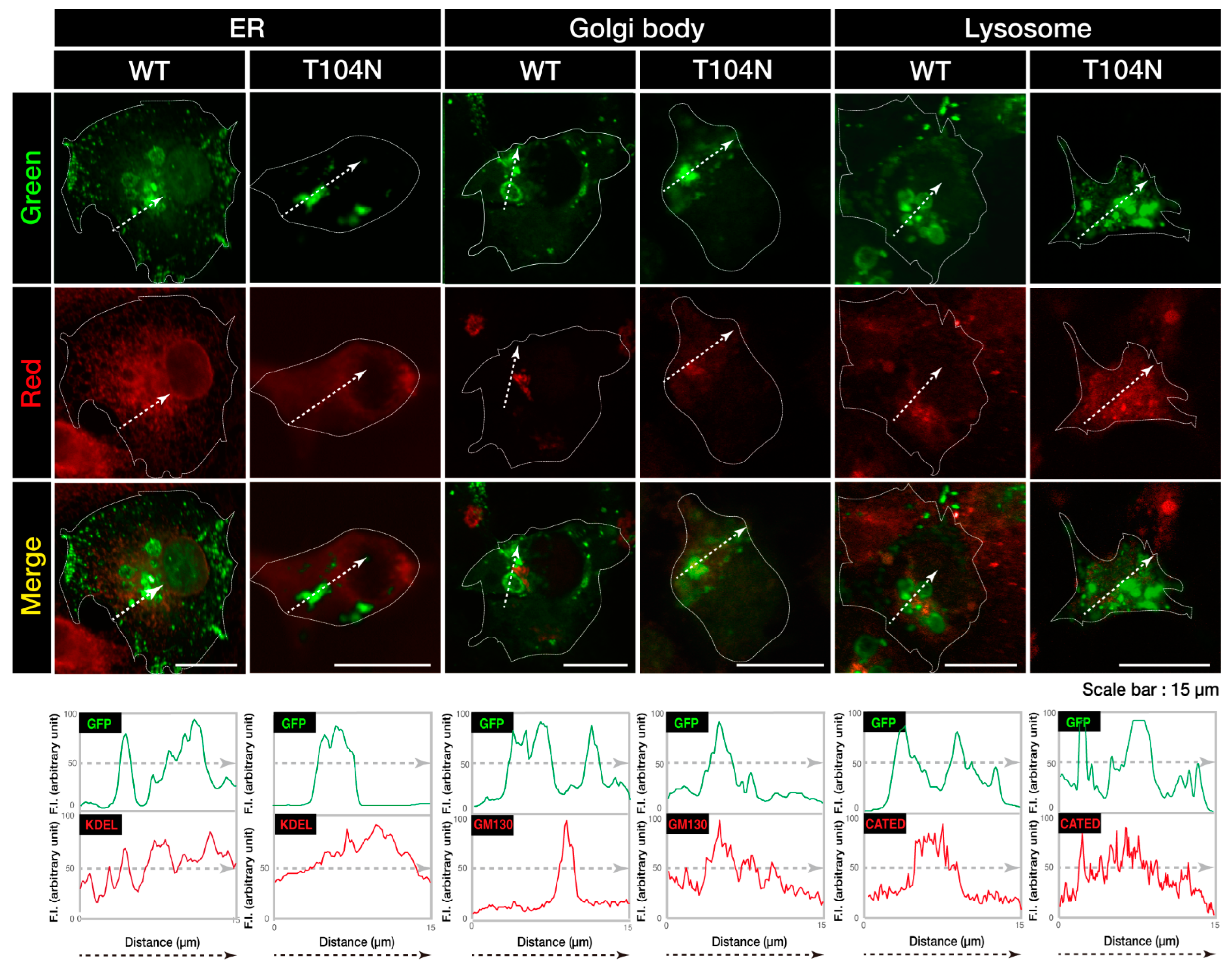
3.1. Wild-Type CHMP2B Is Contained in MVB-Like Structures Whereas T104N-Mutant CHMP2B Forms Aggregate-Like Structures
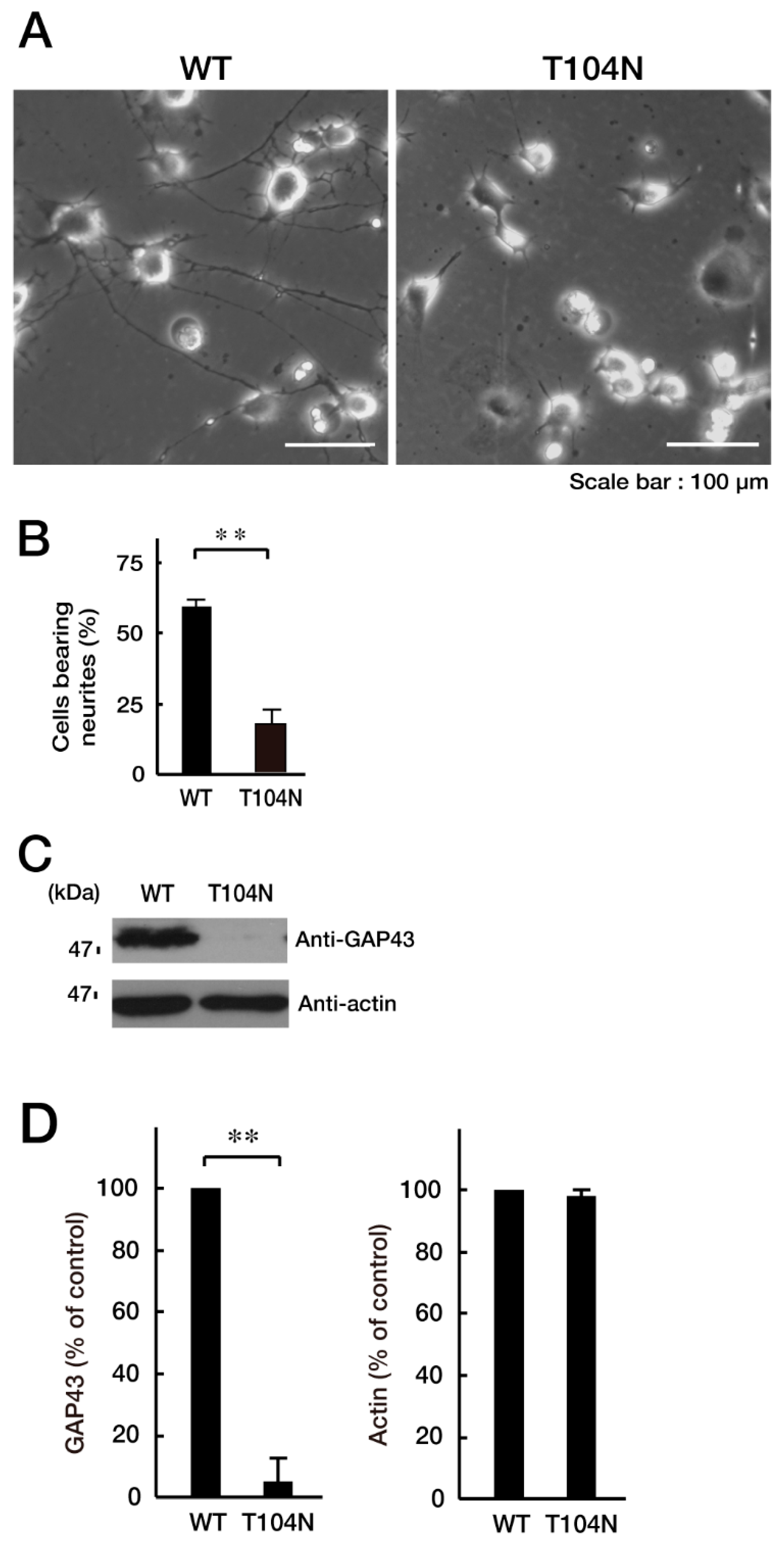
3.2. CHMP2B with the T104N Mutation Inhibits Neuronal Morphological Differentiation
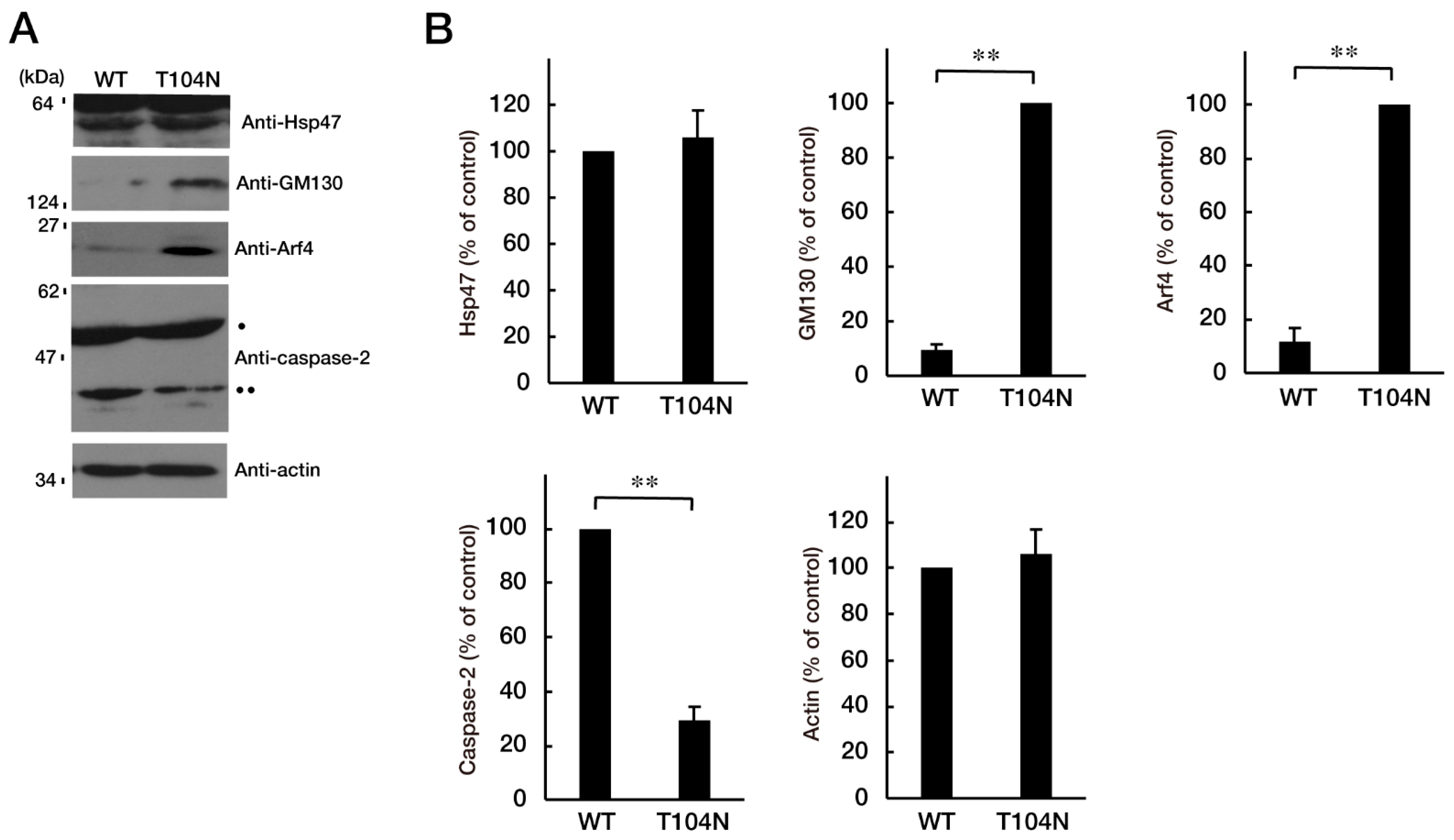
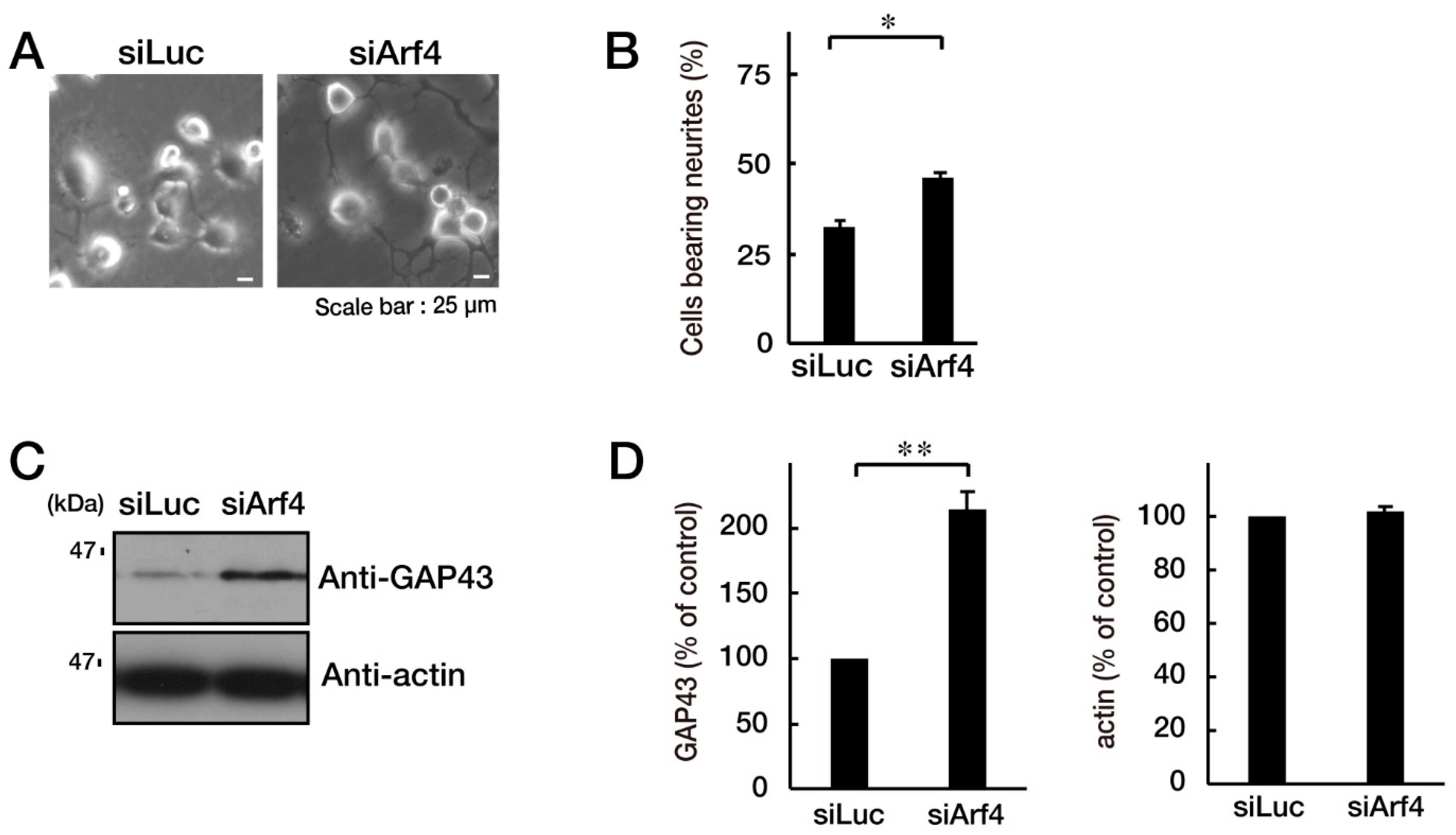
3.3. Attenuating Golgi Stress Recovers an Inhibitory Morphological Differentiation Phenotype
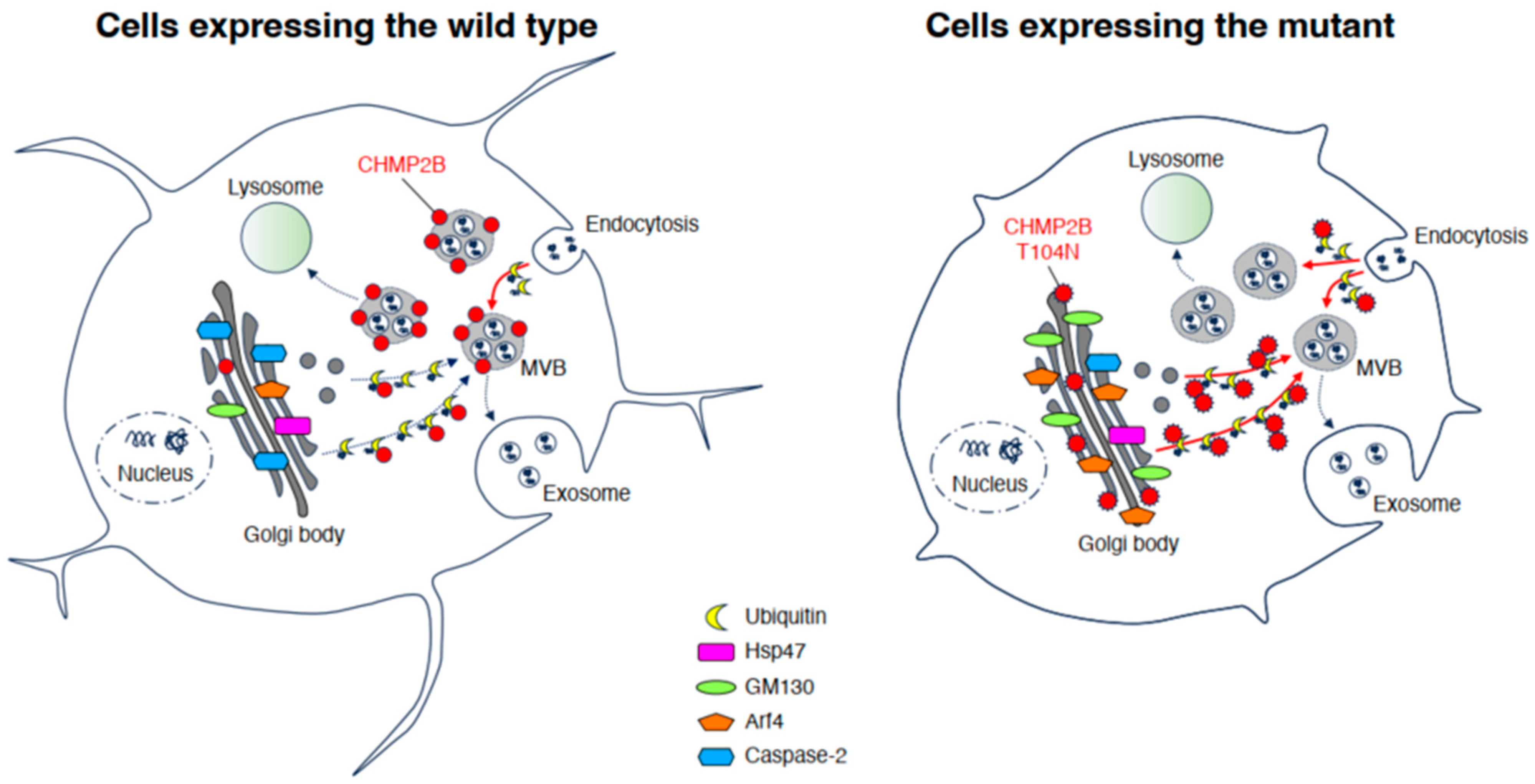
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Craig, A.M.; Banker, G. Neuronal polarity. Annu. Rev. Neurosci. 1994, 17, 267–310. [Google Scholar] [CrossRef] [PubMed]
- da Silva, J.S.; Dotti, C.G. Breaking the neuronal sphere: Regulation of the actin cytoskeleton in neuritogenesis. Nat. Rev. Neurosci. 2002, 3, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Arimura, N.; Kaibuchi, K. Neuronal polarity: From extracellular signals to intracellular mechanisms. Nat. Rev. Neurosci. 2007, 8, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Poo, M.M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef]
- Bray, D. Surface movements during the growth of single explanted neurons. Proc. Natl. Acad. Sci. USA 1970, 65, 905–910. [Google Scholar] [CrossRef]
- Rigby, M.J.; Gomez, T.M.; Puglielli, L. Glial cell-axonal growth cone interactions in neurodevelopment and regeneration. Front. Neurosci. 2020, 14, 203. [Google Scholar] [CrossRef]
- Ugbode, C.; West, R.J.H. Lessons learned from CHMP2B, implications for frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 2021, 147, 105144. [Google Scholar] [CrossRef]
- Root, J.; Merino, P.; Nuckols, A.; Johnson, M.; Kukar, T. Lysosome dysfunction as a cause of neurodegenerative diseases: Lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 2021, 154, 105360. [Google Scholar] [CrossRef]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef]
- Sadoul, R.; Laporte, M.H.; Chassefeyre, R.; Chi, K.I.; Goldberg, Y.; Chatellard, C.; Hemming, F.J.; Fraboulet, S. The role of ESCRT during development and functioning of the nervous system. Semin. Cell Dev. Biol. 2018, 74, 40–49. [Google Scholar] [CrossRef]
- Skibinski, G.; Parkinson, N.J.; Brown, J.M.; Chakrabarti, L.; Lloyd, S.L.; Hummerich, H.; Nielsen, J.E.; Hodges, J.R.; Spillantini, M.G.; Thusgaard, T.; et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 2005, 37, 806–808. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.E.; Ferraiuolo, L.; Goodall, E.F.; Heath, P.R.; Higginbottom, A.; Mortiboys, H.; Hollinger, H.C.; Hartley, J.A.; Brockington, A.; Burness, C.E.; et al. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS ONE 2010, 5, e9872. [Google Scholar] [CrossRef] [PubMed]
- Hirose, M.; Ishizaki, T.; Watanabe, N.; Uehata, M.; Kranenburg, O.; Moolenaar, W.H.; Matsumura, F.; Maekawa, M.; Bito, H.; Narumiya, S. Molecular dissection of the Rho-associated protein kinase (p160ROCK)-regulated neurite remodeling in neuroblastoma N1E-115 cells. J. Cell Biol. 1998, 141, 1625–1636. [Google Scholar] [CrossRef]
- Memezawa, S.; Sato, T.; Ochiai, A.; Fukawa, M.; Sawaguchi, S.; Sango, K.; Miyamoto, Y.; Yamauchi, J. The antiepileptic valproic acid ameliorates Charcot-Marie-Tooth 2W (CMT2W) disease-associated HARS1 mutation-induced inhibition of neuronal cell morphological differentiation through c-Jun N-terminal kinase. Neurochem. Res. 2022, 47, 2684–2702. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, N.; Miyamoto, Y.; Hattori, K.; Ito, A.; Harada, H.; Oizumi, H.; Ohbuchi, K.; Mizoguchi, K.; Yamauchi, J. PP1C and PP2A are p70S6K phosphatases whose inhibition ameliorates HLD12-associated inhibition of oligodendroglial cell morphological differentiation. Biomedicines 2020, 8, 89. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Yamauchi, J.; Sanbe, A.; Tanoue, A. Dock6, a Dock-C subfamily guanine nucleotide exchanger, has the dual specificity for Rac1 and Cdc42 and regulates neurite outgrowth. Exp. Cell Res. 2007, 313, 791–804. [Google Scholar] [CrossRef]
- Machamer, C.M. The Golgi complex in stress and death. Front. Neurosci. 2015, 9, 421. [Google Scholar] [CrossRef]
- Taniguchi, M.; Yoshida, H. TFE3, HSP47, and CREB3 pathways of the mammalian Golgi stress response. Cell Struct. Funct. 2017, 42, 27–36. [Google Scholar] [CrossRef]
- Sasaki, K.; Yoshida, H. Golgi stress response and organelle zones. FEBS Lett. 2019, S593, 2330–2340. [Google Scholar] [CrossRef]
- Olsson, M.; Forsberg, J.; Zhivotovsky, B. Caspase-2: The reinvented enzyme. Oncogene 2015, 34, 1877–1882. [Google Scholar] [CrossRef]
- Reiling, J.H.; Olive, A.J.; Sanyal, S.; Carette, J.E.; Brummelkamp, T.R.; Ploegh, H.L.; Starnbach, M.N.; Sabatini, D.M. A CREB3–ARF4 signalling pathway mediates the response to Golgi stress and susceptibility to pathogens. Nat. Cell Biol. 2013, 15, 1473–1485. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Nadanaka, S.; Tanakura, S.; Sawaguchi, S.; Midori, S.; Kawai, Y.; Yamaguchi, S.; Shimada, Y.; Nakamura, Y.; Matsumura, Y.; et al. TFE3 is a bHLH-ZIP-type transcription factor that regulates the mammalian Golgi stress response. Cell Struct. Funct. 2015, 40, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Miyata, S.; Mizuno, T.; Koyama, Y.; Katayama, T.; Tohyama, M. The endoplasmic reticulum-resident chaperone heat shock protein 47 protects the Golgi apparatus from the effects of O-glycosylation inhibition. PLoS ONE 2013, 8, e69732. [Google Scholar] [CrossRef]
- Kim, W.K.; Choi, W.; Deshar, B.; Kang, S.; Kim, J. Golgi stress response: New insights into the pathogenesis and therapeutic targets of human diseases. Mol. Cells 2023, 46, 191–199. [Google Scholar] [CrossRef]
- Avila, J.; Lucas, J.J.; Perez, M.; Hernandez, F. Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.; Park, S.B.; Vucic, S.; Yiannikas, C.; Spies, J.; Howells, J.; Huynh, W.; Matamala, J.M.; Krishnan, A.V.; Pollard, J.D.; et al. Differentiating lower motor neuron syndromes. J. Neurol. Neurosurg. Psychiatry 2017, 88, 474–483. [Google Scholar] [CrossRef]
- Scott-Solomon, E.; Boehm, E.; Kuruvilla, R. The sympathetic nervous system in development and disease. Nat. Rev. Neurosci. 2021, 22, 685–702. [Google Scholar] [CrossRef]
- Deng, X.; Sun, X.; Yue, W.; Duan, Y.; Hu, R.; Zhang, K.; Ni, J.; Cui, J.; Wang, Q.; Chen, Y.; et al. CHMP2B regulates TDP-43 phosphorylation and cytotoxicity independent of autophagy via CK1. J. Cell Biol. 2022, 221, e202103033. [Google Scholar] [CrossRef]
- Luan, W.; Wright, A.L.; Brown-Wright, H.; Le, S.; San Gil, R.; Madrid San Martin, L.; Ling, K.; Jafar-Nejad, P.; Rigo, F.; Walker, A.K. Early activation of cellular stress and death pathways caused by cytoplasmic TDP-43 in the rNLS8 mouse model of ALS and FTD. Mol. Psychiatry, 2023; in press. [Google Scholar] [CrossRef]
- Tan, A.; Prasad, R.; Jho, E.H. TFEB regulates pluripotency transcriptional network in mouse embryonic stem cells independent of autophagy-lysosomal biogenesis. Cell Death Dis. 2021, 12, 343. [Google Scholar] [CrossRef]
- Jackson, C.L. Activators and effectors of the small G protein Arf1 in regulation of Golgi dynamics during the cell division cycle. Front. Cell Dev. Biol. 2018, 6, 29. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Torii, T.; Tago, K.; Tanoue, A.; Takashima, S.; Yamauchi, J. BIG1/Arfgef1 and Arf1 regulate the initiation of myelination by Schwann cells in mice. Sci. Adv. 2018, 4, eaar4471. [Google Scholar] [CrossRef]
- Ezratty, E.J.; Pasolli, H.A.; Fuchs, E. A Presenilin-2-ARF4 trafficking axis modulates Notch signaling during epidermal differentiation. J. Cell Biol. 2016, 214, 89–101. [Google Scholar] [CrossRef]
- Wang, J.; Fresquez, T.; Kandachar, V.; Deretic, D. The Arf GEF GBF1 and Arf4 synergize with the sensory receptor cargo, rhodopsin, to regulate ciliary membrane trafficking. J. Cell Sci. 2017, 130, 3975–3987. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, L.; Bai, B.; Li, D.; Xiao, P.; Li, Q.; Zhang, Z.; Wang, H.; Li, L.; Jiang, Q. Quantitative proteomics reveals association of neuron projection development genes ARF4, KIF5B, and RAB8A with hirschsprung disease. Mol. Cell Proteom. 2021, 20, 100007. [Google Scholar] [CrossRef] [PubMed]
- Pennauer, M.; Buczak, K.; Prescianotto-Baschong, C.; Spiess, M. Shared and specific functions of Arfs 1-5 at the Golgi revealed by systematic knockouts. J. Cell Biol. 2022, 221, e202106100. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Todd, T.; Wu, Y.; Jones, C.; Tong, L.; Jansen-West, K.; Daughrity, L.; Park, J.; Koike, Y.; Kurti, A.; et al. Two FTD-ALS genes converge on the endosomal pathway to induce TDP-43 pathology and degeneration. Science 2022, 378, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Baradaran-Heravi, Y.; Broeckhoven, C.; Zee, J. Stress granule mediated protein aggregation and underlying gene defects in the FTD-ALS spectrum. Neurobiol. Dis. 2020, 134, 104639. [Google Scholar] [CrossRef]
- Wood, H. FTD-ALS risk factors converge on the endolysosomal pathway. Nat. Rev. Neurol. 2022, 18, 699. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent or Material | Company or Source | Cat. No. | Lot. No. | Concentration Used |
|---|---|---|---|---|
| Antibody | ||||
| Anti-heat shock protein (HSP) 47 | Santa Cruz Biotechnology | sc-5293 | I2118 | Immunoblotting (IB), 1/200 |
| Anti-Arf4 | Proteintech | 11673-1-AP | 00048284 | IB, 1/1000 |
| Anti-caspase-2 | Abcam | ab179520 | GR209449-2 | IB, 1/1000 |
| Anti-actin | MBL | M177-3 | 007 | IB, 1/5000 |
| Ant-growth-associated protein 43 (GAP43) | Santa Cruz Biotechnology | sc-17790 | J0920 | IB, 1/5000 |
| Anti-Lys-Asp-Glu-Leu (KDEL) | MBL | M181-3 | 004 | Immunofluorescence (IF), 1/200 |
| Anti-130 kDa Golgi membrane protein (GM130) | BD Biosciences | 610823 | 8352796 | IB, 1/500 and IF, 1/200 |
| Anti-cathepsin D | Abcam | ab75852 | GR260148-33 | IF, 1/200 |
| Anti-IgG (H+L chain) (Rabbit) pAb-HRP | MBL | 458 | 353 | IB, 1/5000 |
| Anti-IgG (H+L chain) (Mouse) pAb-HRP | MBL | 330 | 365 | IB, 1/5000 |
| Goat pAb to Ms IgG (Alexa Fluor 488 conjugate) | abcam | ab150113 | GR173498-1 | IF, 1/500 |
| Alexa Fluor TM 594 goat anti-mouse IgG (H+L) | invitrogen | A11005 | 226-8383 | IF, 1/500 |
| Alexa Fluor TM 488 goat anti-rabbit IgG (H+L) | invitrogen | A11008 | 075-1094 | IF, 1/500 |
| Alexa Fluor TM 594 goat anti-rabbit IgG (H+L) | invitrogen | A11012 | 201-8240 | IF, 1/500 |
| Recombinant DNA | ||||
| pEGFP-C1-human CHMP2B | Generated in this study | Not applicable | 1.25 μg of DNA per 6 cm dish | |
| pEGFP-C1-human CHMP2B with the T104N mutation | Generated in this study | Not applicable | 1.25 μg of DNA per 6 cm dish |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shirai, R.; Cho, M.; Isogai, M.; Fukatsu, S.; Okabe, M.; Okawa, M.; Miyamoto, Y.; Torii, T.; Yamauchi, J. FTD/ALS Type 7-Associated Thr104Asn Mutation of CHMP2B Blunts Neuronal Process Elongation, and Is Recovered by Knockdown of Arf4, the Golgi Stress Regulator. Neurol. Int. 2023, 15, 980-993. https://doi.org/10.3390/neurolint15030063
Shirai R, Cho M, Isogai M, Fukatsu S, Okabe M, Okawa M, Miyamoto Y, Torii T, Yamauchi J. FTD/ALS Type 7-Associated Thr104Asn Mutation of CHMP2B Blunts Neuronal Process Elongation, and Is Recovered by Knockdown of Arf4, the Golgi Stress Regulator. Neurology International. 2023; 15(3):980-993. https://doi.org/10.3390/neurolint15030063
Chicago/Turabian StyleShirai, Remina, Mizuka Cho, Mikinori Isogai, Shoya Fukatsu, Miyu Okabe, Maho Okawa, Yuki Miyamoto, Tomohiro Torii, and Junji Yamauchi. 2023. "FTD/ALS Type 7-Associated Thr104Asn Mutation of CHMP2B Blunts Neuronal Process Elongation, and Is Recovered by Knockdown of Arf4, the Golgi Stress Regulator" Neurology International 15, no. 3: 980-993. https://doi.org/10.3390/neurolint15030063
APA StyleShirai, R., Cho, M., Isogai, M., Fukatsu, S., Okabe, M., Okawa, M., Miyamoto, Y., Torii, T., & Yamauchi, J. (2023). FTD/ALS Type 7-Associated Thr104Asn Mutation of CHMP2B Blunts Neuronal Process Elongation, and Is Recovered by Knockdown of Arf4, the Golgi Stress Regulator. Neurology International, 15(3), 980-993. https://doi.org/10.3390/neurolint15030063