

Soluble TREM2 Concentrations in the Cerebrospinal Fluid Correlate with the Severity of Neurofibrillary Degeneration, Cognitive Impairment, and Inflammasome Activation in Alzheimer’s Disease



Abstract
1. Introduction
2. Materials and Methods
2.1. Cerebrospinal Fluid and Blood Collection
2.2. ELISA Procedure
- TREM2 (Human TREM2 ELISA Kit, Abcam, Cambridge, UK);
- Aβ1-42 (Innotest β-amyloid1-42, Fujirebio, Gent, Belgium);
- total tau (Innotest hTau Ag, Fujirebio, Gent, Belgium);
- p-tau181 (Innotest Phospho-Tau(181P), Fujirebio, Gent, Belgium);
- ASC (Human PYCARD/ASC/TMS1 Sandwich ELISA, LSBio, Seattle, WA, USA).
2.3. Statistical Analysis
3. Results
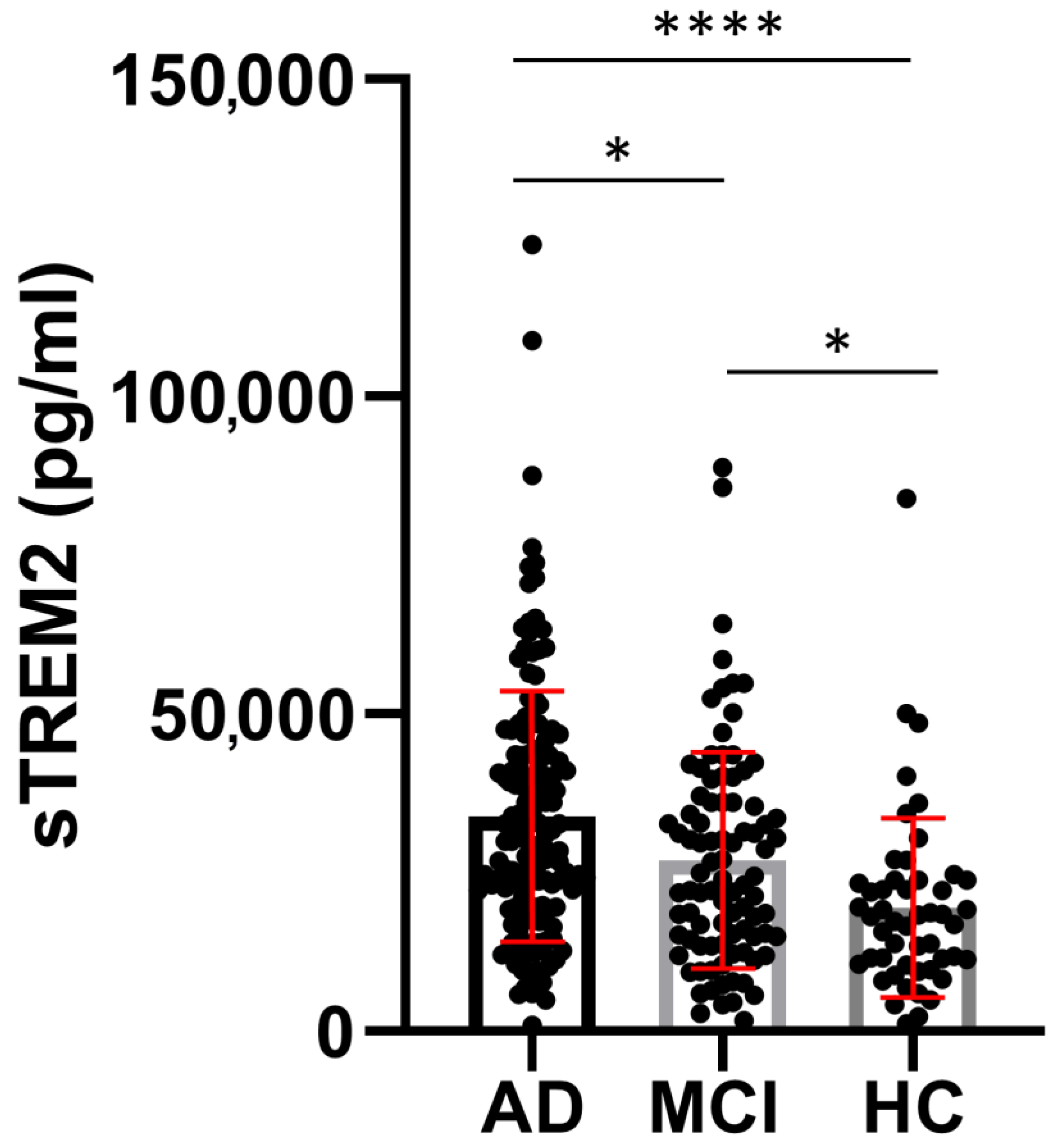
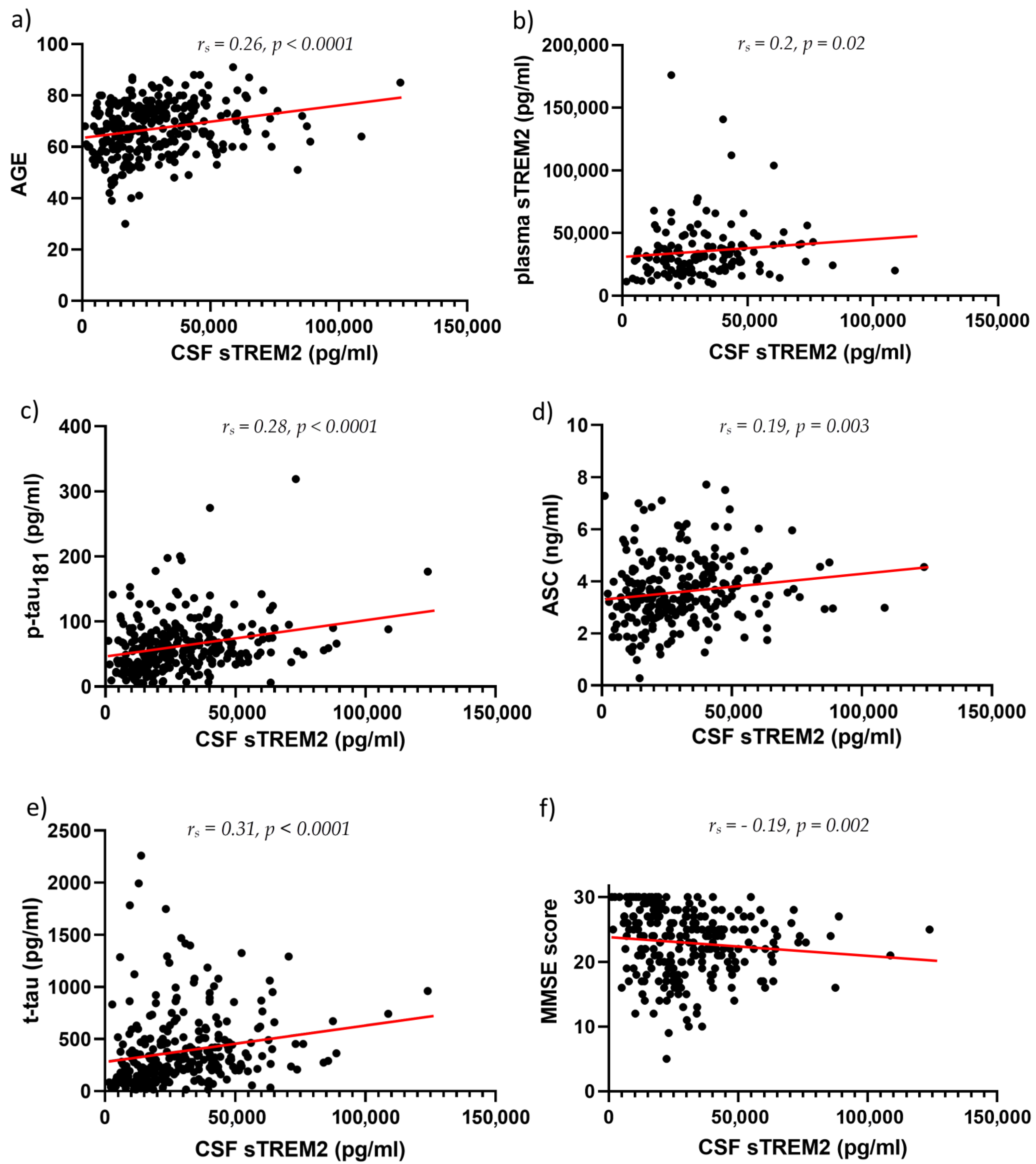
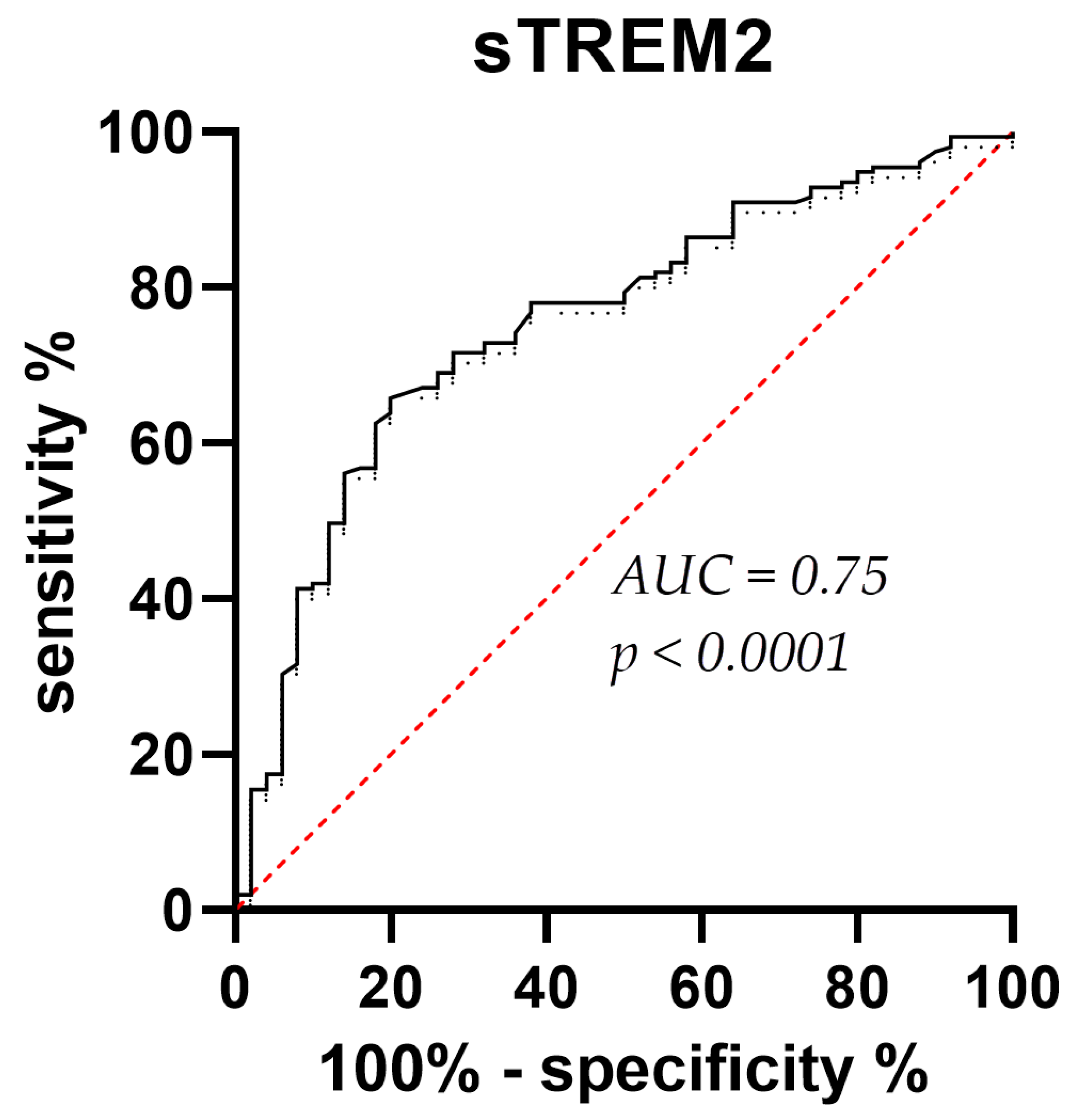
3.1. sTREM2 Levels in CSF and Plasma Samples
3.2. ASC Levels in CSF and Plasma Samples
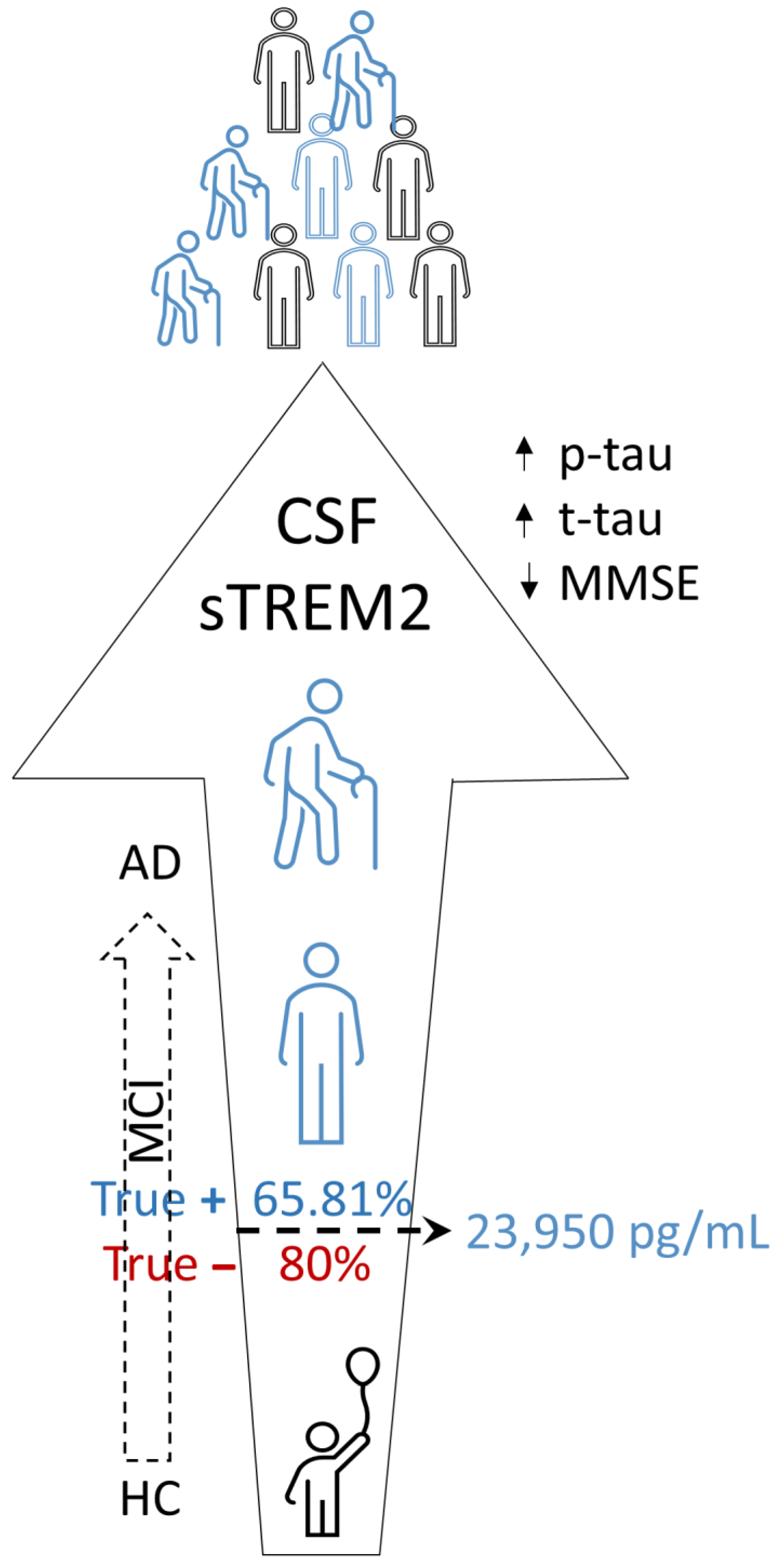
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhong, L.; Chen, X.F. The emerging roles and therapeutic potential of soluble TREM2 in Alzheimer’s disease. Front. Aging Neurosci. 2019, 11, 328. [Google Scholar] [CrossRef] [PubMed]
- Jay, T.R.; von Saucken, V.E.; Landreth, G.E. TREM2 in neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Wang, M.; Yin, Y.; Tang, Y. The specific mechanism of TREM2 regulation of synaptic clearance in Alzheimer’s disease. Front. Immunol. 2022, 13, 845897. [Google Scholar] [CrossRef]
- Li, R.Y.; Qin, Q.; Yang, H.C.; Wang, Y.Y.; Mi, Y.X.; Yin, Y.S.; Wang, M.; Yu, C.J.; Tang, Y. TREM2 in the pathogenesis of AD: A lipid metabolism regulator and potential metabolic therapeutic target. Mol. Neurodegener. 2022, 17, 40. [Google Scholar] [CrossRef]
- Filipello, F.; You, S.F.; Mirfakhar, F.S.; Mahali, S.; Bollman, B.; Acquarone, M.; Korvatska, O.; Marsh, J.A.; Sivaraman, A.; Martinez, R.; et al. Defects in lysosomal function and lipid metabolism in human microglia harboring a TREM2 loss of function mutation. Acta Neuropathol. 2023, 145, 749–772. [Google Scholar] [CrossRef]
- Šimić, G.; Španić, E.; Langer Horvat, L.; Hof, P.R. Blood-brain barrier and innate immunity in the pathogenesis of Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 2019, 168, 99–145. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Ulland, T.K.; Song, W.M.; Huang, S.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 2017, 170, 649–663.e13. [Google Scholar] [CrossRef]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef]
- Yang, J.; Fu, Z.; Zhang, X.; Xiong, M.; Meng, L.; Zhang, Z. TREM2 ectodomain and its soluble form in Alzheimer’s disease. J. Neuroinflammation 2020, 17, 204. [Google Scholar] [CrossRef] [PubMed]
- Ewers, M.; Biechele, G.; Suárez-Calvet, M.; Sacher, C.; Blume, T.; Morenas-Rodriguez, E.; Deming, Y.; Piccio, L.; Cruchaga, C.; Kleinberger, G.; et al. Higher CSF sTREM2 and microglia activation are associated with slower rates of β-amyloid accumulation. EMBO Mol. Med. 2020, 12, e12308. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 2020, 217, e20200785. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Lewis, C.A.; Ulrich, J.D.; Holtzman, D.M. Chronic TREM2 activation exacerbates Aβ-associated tau seeding and spreading. J. Exp. Med. 2023, 220, e20220654. [Google Scholar] [CrossRef]
- Zhong, L.; Xu, Y.; Zhuo, R.; Wang, T.; Wang, K.; Huang, R.; Wang, D.; Yue Gao, Y.; Zhu, Y.; Sheng, X.; et al. Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 1365. [Google Scholar] [CrossRef]
- Peng, X.; Guo, H.; Zhang, X.; Yang, Z.; Ruganzu, J.B.; Yang, Z.; Wu, X.; Bi, W.; Ji, S.; Yang, W. TREM2 inhibits tau hyperphosphorylation and neuronal apoptosis via the PI3K/Akt/GSK-3β signaling pathway in vivo and in vitro. Mol. Neurobiol. 2023, 60, 2470–2485. [Google Scholar] [CrossRef]
- Pereira, J.B.; Janelidze, S.; Strandberg, O.; Whelan, C.D.; Zetterberg, H.; Blennow, K.; Palmqvist, S.; Stomrud, E.; Mattsson-Carlgren, N.; Hansson, O. Microglial activation protects against accumulation of tau aggregates in nondemented individuals with underlying Alzheimer’s disease pathology. Nat. Aging 2022, 2, 1138–1144. [Google Scholar] [CrossRef]
- Lee, C.Y.D.; Daggett, A.; Gu, X.; Jiang, L.L.; Langfelder, P.; Li, X.; Wang, N.; Zhao, Y.; Park, C.S.; Cooper, Y.; et al. Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer’s disease models. Neuron 2018, 97, 1032–1048.e5. [Google Scholar] [CrossRef]
- Cosker, K.; Mallach, A.; Limaye, J.; Piers, T.M.; Staddon, J.; Neame, S.J.; Hardy, J.; Pocock, J.M. Microglial signalling pathway deficits associated with the patient-derived R47H TREM2 variants linked to AD indicate inability to activate inflammasome. Sci. Rep. 2021, 11, 13316. [Google Scholar] [CrossRef]
- Yang, S.; Yang, Y.; Wang, F.; Luo, Q.Y.; Zhang, Y.; Zheng, F.; Shu, Q.; Chen, Q.; Fang, X. TREM2 dictates antibacterial defense and viability of bone marrow-derived macrophages during bacterial infection. Am. J. Respir. Cell Mol. Biol. 2021, 65, 176–188. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, C.; Zhu, Y.; Fan, H.; Liu, Q.; Liu, Y.; Chen, K.; Wu, Y.; Liang, S.; Li, M.; et al. TREM2/β-catenin attenuates NLRP3 inflammasome-mediated macrophage pyroptosis to promote bacterial clearance of pyogenic bacteria. Cell Death Dis. 2022, 13, 771. [Google Scholar] [CrossRef] [PubMed]
- Jay, T.R.; Miller, C.M.; Cheng, P.J.; Graham, L.C.; Bemiller, S.; Broihier, M.L.; Xu, G.; Margevicius, D.; Karlo, J.C.; Sousa, G.L.; et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 2015, 212, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Long, W.; Gao, M.; Jiao, F.; Chen, Z.; Liu, M.; Yu, L. TREM2 regulates high glucose-induced microglial inflammation via the NLRP3 signaling pathway. Brain Sci. 2021, 11, 896. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Liu, F.; Li, H.; Wang, K.; Cao, X.; Xu, X.; Zhou, Y.; Zou, J.; Zhang, X.; Cui, X. TREM2 ameliorates anesthesia and surgery-induced cognitive impairment by regulating mitophagy and NLRP3 inflammasome in aged C57/BL6 mice. Neurotoxicology 2022, 90, 216–227. [Google Scholar] [CrossRef]
- Li, H.; Liu, F.; Jiang, W.; Wang, K.; Cao, X.; Zou, J.; Zhou, Y.; Li, Z.; Liu, S.; Cui, X.; et al. TREM2 ameliorates lipopolysaccharide-induced oxidative stress response and neuroinflammation by promoting sirtuin3 in BV2 cells. Neurotox. Res. 2022, 40, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Del-Aguila, J.L.; Benitez, B.A.; Li, Z.; Dube, U.; Mihindukulasuriya, K.A.; Budde, J.P.; Farias, F.H.G.; Fernández, M.V.; Ibanez, L.; Jiang, S.; et al. TREM2 brain transcript-specific studies in AD and TREM2 mutation carriers. Mol. Neurodegener. 2019, 14, 18. [Google Scholar] [CrossRef]
- Filipello, F.; Goldsbury, C.; You, S.F.; Locca, A.; Karch, C.M.; Piccio, L. Soluble TREM2: Innocent bystander or active player in neurological diseases? Neurobiol. Dis. 2022, 165, 105630. [Google Scholar] [CrossRef]
- Zhong, L.; Chen, X.F.; Wang, T.; Wang, Z.; Liao, C.; Wang, Z.; Huang, R.; Wang, D.; Li, X.; Wu, L.; et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 2017, 214, 597–607. [Google Scholar] [CrossRef]
- Qiao, W.; Chen, Y.; Zhong, J.; Madden, B.J.; Charlesworth, C.M.; Martens, Y.A.; Liu, C.C.; Knight, J.; Ikezu, T.C.; Kurti, A.; et al. Trem2 H157Y increases soluble TREM2 production and reduces amyloid pathology. Mol. Neurodegener. 2023, 18, 8. [Google Scholar] [CrossRef]
- Gratuze, M.; Leyns, C.E.; Sauerbeck, A.D.; St-Pierre, M.K.; Xiong, M.; Kim, N.; Serrano, J.R.; Tremblay, M.È.; Kummer, T.T.; Colonna, M.; et al. Impact of TREM2R47H variant on tau pathology-induced gliosis and neurodegeneration. J. Clin. Investig. 2020, 130, 4954–4968. [Google Scholar] [CrossRef]
- Albanus, R.; Jain, N.; Novotny, B.; Brase, L.; Rodriguez, L.; Mansel, C.; Kipnis, M.; O’Brien, S.; Pasillas, M.P.; Lee, C.; et al. TREM2-independent microgliosis promotes tau-mediated neurodegeneration in the presence of ApoE4. Neuron 2023, 111, 202–219.e7. [Google Scholar] [CrossRef]
- Vautheny, A.; Duwat, C.; Aurégan, G.; Joséphine, C.; Hérard, A.S.; Jan, C.; Mitja, J.; Gipchtein, P.; Gaillard, M.C.; Buée, L.; et al. THY-Tau22 mouse model accumulates more tauopathy at late stage of the disease in response to microglia deactivation through TREM2 deficiency. Neurobiol. Dis. 2021, 155, 105398. [Google Scholar] [CrossRef]
- Brown, G.C.; St George-Hyslop, P. Does soluble TREM2 protect against Alzheimer’s disease? Front. Aging Neurosci. 2022, 13, 834697. [Google Scholar] [CrossRef]
- Ulrich, J.D.; Ulland, T.K.; Colonna, M.; Holtzman, D.M. Elucidating the role of TREM2 in Alzheimer’s disease. Neuron 2017, 94, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Calvet, M.; Kleinberger, G.; Araque Caballero, M.Á.; Brendel, M.; Rominger, A.; Alcolea, D.; Fortea, J.; Lleó, A.; Blesa, R.; Gispert, J.D.; et al. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol. Med. 2016, 8, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Calvet, M.; Morenas-Rodríguez, E.; Kleinberger, G.; Schlepckow, K.; Araque Caballero, M.A.; Franzmeier, N.; Capell, A.; Fellerer, K.; Nuscher, B.; Eren, E.; et al. Early increase of CSF sTREM2 in Alzheimer’s disease is associated with tau related-neurodegeneration but not with amyloid-β pathology. Mol. Neurodegener. 2019, 14, 1. [Google Scholar] [CrossRef]
- Ma, L.Z.; Tan, L.; Bi, Y.L.; Shen, X.N.; Xu, W.; Ma, Y.H.; Li, H.Q.; Dong, Q.; Yu, J.T. Dynamic changes of CSF sTREM2 in preclinical Alzheimer’s disease: The CABLE study. Mol. Neurodegener. 2020, 15, 25. [Google Scholar] [CrossRef]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement. 2012, 8, 1–13. [Google Scholar] [CrossRef]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 4th ed.; Text Revision (DSM-IV-TR); American Psychiatric Association: Washington, DC, USA, 2000. [Google Scholar]
- Petersen, R.C.; Smith, G.E.; Waring, S.C.; Ivnik, R.J.; Tangalos, E.G.; Kokmen, E. Mild cognitive impairment: Clinical characterization and outcome. Arch. Neurol. 1999, 56, 303–308. [Google Scholar] [CrossRef]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Heslegrave, A.; Heywood, W.; Paterson, R.; Magdalinou, N.; Svensson, J.; Johansson, P.; Öhrfelt, A.; Blennow, K.; Hardy, J.; Schott, J.; et al. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 3. [Google Scholar] [CrossRef]
- Bonomi, C.G.; Assogna, M.; Di Donna, M.G.; Bernocchi, F.; De Lucia, V.; Nuccetelli, M.; Fiorelli, D.; Loizzo, S.; Mercuri, N.B.; Koch, G.; et al. Cerebrospinal fluid sTREM-2, GFAP, and β-S100 in symptomatic sporadic Alzheimer’s disease: Microglial, astrocytic, and APOE contributions along the Alzheimer’s disease continuum. J. Alzheimers Dis. 2023, 92, 1385–1397. [Google Scholar] [CrossRef]
- Zhao, A.; Jiao, Y.; Ye, G.; Kang, W.; Tan, L.; Li, Y.; Deng, Y.; Liu, J. Alzheimer’s disease neuroimaging initiative (ADNI). Soluble TREM2 levels associate with conversion from mild cognitive impairment to Alzheimer’s disease. J. Clin. Investig. 2022, 132, e158708. [Google Scholar] [CrossRef]
- Huang, S.; Wang, Y.J.; Guo, J. Biofluid biomarkers of Alzheimer’s disease: Progress, problems, and perspectives. Neurosci. Bull. 2022, 38, 677–691. [Google Scholar] [CrossRef]
- Salih, D.A.; Bayram, S.; Guelfi, S.; Reynolds, R.H.; Shoai, M.; Ryten, M.; Brenton, J.W.; Zhang, D.; Matarin, M.; Botia, J.A.; et al. Genetic variability in response to amyloid beta deposition influences Alzheimer’s disease risk. Brain Commun. 2019, 1, fcz022. [Google Scholar] [CrossRef]
- Andrews, S.J.; Renton, A.E.; Fulton-Howard, B.; Podlesny-Drabiniok, A.; Marcora, E.; Goate, A.M. The complex genetic architecture of Alzheimer’s disease: Novel insights and future directions. EBioMedicine 2023, 90, 104511. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Firulyova, M.; Manis, M.; Herz, J.; Smirnov, I.; Aladyeva, E.; Wang, C.; Bao, X.; Finn, M.B.; Hu, H.; et al. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature 2023, 615, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-R.; Lyu, D.-Y.; Liu, F.-Q. Alzheimer’s Disease Imaging Initiative. Cerebrospinal fluid sTREM2 in Alzheimer’s disease is associated with both amyloid and tau pathologies but not with cognitive status. J. Alzheimers Dis. 2022, 90, 1123–1138. [Google Scholar] [CrossRef] [PubMed]
- Morenas-Rodríguez, E.; Li, Y.; Nuscher, B.; Franzmeier, N.; Xiong, C.; Suárez-Calvet, M.; Fagan, A.M.; Schultz, S.; Gordon, B.A.; Benzinger, T.L.S.; et al. Soluble TREM2 in CSF and its association with other biomarkers and cognition in autosomal-dominant Alzheimer’s disease: A longitudinal observational study. Lancet Neurol. 2022, 21, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Winfree, R.L.; Dumitrescu, L.; Blennow, K.; Zetterberg, H.; Gifford, K.A.; Pechman, K.R.; Jefferson, A.L.; Hohman, T.J. Alzheimer’s Disease Neuroimaging Initiative. Biological correlates of elevated soluble TREM2 in cerebrospinal fluid. Neurobiol. Aging 2022, 118, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Attems, J.; Lintner, F.; Jellinger, K.A. Amyloid beta peptide 1-42 highly correlates with capillary cerebral amyloid angiopathy and Alzheimer disease pathology. Acta Neuropathol. 2004, 107, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Attems, J.; Jellinger, K.A. Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology—A pilot study. Acta Neuropathol. 2004, 107, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, D.V.; Zaretskaia, M.V.; Molkov, Y.I. Alzheimer’s Disease Neuroimaging Initiative. Patients with Alzheimer’s disease have an increased removal rate of soluble beta-amyloid-42. PLoS ONE 2022, 17, e0276933. [Google Scholar] [CrossRef]
- Španić, E.; Langer Horvat, L.; Hof, P.R.; Šimić, G. Role of microglial cells in Alzheimer’s disease tau propagation. Front. Aging Neurosci. 2019, 11, 271. [Google Scholar] [CrossRef]
- Španić, E.; Langer Horvat, L.; Ilić, K.; Hof, P.R.; Šimić, G. NLRP1 Inflammasome activation in the hippocampal formation in Alzheimer’s disease: Correlation with neuropathological changes and unbiasedly estimated neuronal loss. Cells 2022, 11, 2223. [Google Scholar] [CrossRef]
- Tsai, H.H.; Chen, Y.F.; Yen, R.F.; Lo, Y.L.; Yang, K.C.; Jeng, J.S.; Tsai, L.K.; Chang, C.F. Plasma soluble TREM2 is associated with white matter lesions independent of amyloid and tau. Brain 2021, 144, 3371–3380. [Google Scholar] [CrossRef]
- Scott, X.O.; Stephens, M.E.; Desir, M.C.; Dietrich, W.D.; Keane, R.W.; de Rivero Vaccari, J.P. The inflammasome adaptor protein ASC in mild cognitive impairment and Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 4674. [Google Scholar] [CrossRef]
- Zilka, N.; Stozicka, Z.; Kovac, A.; Pilipcines, E.; Bugos, O.; Novak, M. Human misfolded truncated tau protein promotes activation of microglia and leukocyte infiltration in the transgenic rat model of tauopathy. J. Neuroimmunol. 2009, 209, 16–25. [Google Scholar] [CrossRef]
- Zilka, N.; Kazmerova, Z.; Jadhav, S.; Neradil, P.; Madari, A.; Obetkova, D.; Bugos, O.; Novak, M. Who fans the flames of Alzheimer’s disease brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. J. Neuroinflammation 2012, 9, 47. [Google Scholar] [CrossRef]
- Stancu, I.-C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brône, B.; Huaux, F.; Octave, J.-N.; et al. Aggregated tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded tau pathology in vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Varma, V.R.; Varma, S.; Casanova, R.; Dammer, E.; Pletnikova, O.; Chia, C.W.; Egan, J.M.; Ferrucci, L.; Troncoso, J.; et al. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Lee, H.; Seo, J. Impact of genetic risk factors for Alzheimer’s disease on brain glucose metabolism. Mol. Neurobiol. 2021, 58, 2608–2619. [Google Scholar] [CrossRef]
- Knezović, A.; Osmanović-Barilar, J.; Ćurlin, M.; Hof, P.R.; Šimić, G.; Riederer, P.; Šalković-Petrišić, M. Staging of cognitive deficits and neuropathological and ultrastructural changes in streptozotocin-induced rat model of Alzheimer’s disease. J. Neural Transm. 2015, 122, 577–592. [Google Scholar] [CrossRef]
- Choi, H.; Choi, Y.; Lee, E.J.; Kim, H.; Lee, Y.; Kwon, S.; Hwang, D.W.; Lee, D.S. Hippocampal glucose uptake as a surrogate of metabolic change of microglia in Alzheimer’s disease. J. Neuroinflammation 2021, 18, 190. [Google Scholar] [CrossRef]
- Piccio, L.; Buonsanti, C.; Cella, M.; Tassi, I.; Schmidt, R.E.; Fenoglio, C.; Rinker, J., 2nd; Naismith, R.T.; Panina-Bordignon, P.; Passini, N.; et al. Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain 2008, 131, 3081–3091. [Google Scholar] [CrossRef]
- Piccio, L.; Deming, Y.; Del-Águila, J.L.; Ghezzi, L.; Holtzman, D.M.; Fagan, A.M.; Fenoglio, C.; Galimberti, D.; Borroni, B.; Cruchaga, C. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 2016, 131, 925–933. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, E.H.; Kim, H.J.; Jo, S.; Lee, S.; Seo, S.W.; Park, H.H.; Koh, S.H.; Lee, J.H. The relationship of soluble TREM2 to other biomarkers of sporadic Alzheimer’s disease. Sci. Rep. 2021, 11, 13050. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Age (Years) Mean ± SD (n) | MMSE (Points) Mean ± SD (n) | sTREM2 (pg/mL) Mean ± SD (n) | ASC (ng/mL) Mean ± SD (n) | Aβ1-42 (pg/mL) Mean ± SD (n) | p-tau181 (pg/mL) Mean ± SD (n) | t-tau (pg/mL) Mean ± SD (n) |
|---|---|---|---|---|---|---|---|
| AD | 70.58 ± 8.670 (154) a,b | 20.30 ± 4.395 (142) a,b | 33,854 ± 19,631 (155) a,b | 3.817± 1.348 (126) | 549.5 ± 303.9 (152) | 77.06 ± 45.25 (147) a,b | 522.5 ± 400.9 (146) a,b |
| MCI | 66.42 ± 8.832 (90) a,c | 25.64 ± 2.903 (74) a,c | 26,940 ± 17,043 (90) a,c | 3.432 ± 1.121 (78) | 665.9 ± 374.8 (86) c | 53.32 ± 29.90 (87) a,c | 257.5 ± 177.1 (86) a |
| HC | 58.80 ± 10.68 (49) b,c | 29.06 ± 2.199 (32) b,c | 18,527 ± 13,566 (50) b,c | 3.369 ± 1.272 (39) | 552.8 ± 521.7 (49) c | 35.18 ± 22.70 (49) b,c | 222.7 ± 284.0 (48) b |
| Kruskal–Wallis test | H test = 45.13 p < 0.0001 | H test = 124.4 p < 0.0001 | H test = 35.38 p < 0.0001 | H test = 6.44 p = 0.04 | H test = 8.873 p = 0.01 | H test = 57.40 p < 0.0001 | H test = 58.02 p < 0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Španić Popovački, E.; Babić Leko, M.; Langer Horvat, L.; Brgić, K.; Vogrinc, Ž.; Boban, M.; Klepac, N.; Borovečki, F.; Šimić, G. Soluble TREM2 Concentrations in the Cerebrospinal Fluid Correlate with the Severity of Neurofibrillary Degeneration, Cognitive Impairment, and Inflammasome Activation in Alzheimer’s Disease. Neurol. Int. 2023, 15, 842-856. https://doi.org/10.3390/neurolint15030053
Španić Popovački E, Babić Leko M, Langer Horvat L, Brgić K, Vogrinc Ž, Boban M, Klepac N, Borovečki F, Šimić G. Soluble TREM2 Concentrations in the Cerebrospinal Fluid Correlate with the Severity of Neurofibrillary Degeneration, Cognitive Impairment, and Inflammasome Activation in Alzheimer’s Disease. Neurology International. 2023; 15(3):842-856. https://doi.org/10.3390/neurolint15030053
Chicago/Turabian StyleŠpanić Popovački, Ena, Mirjana Babić Leko, Lea Langer Horvat, Klara Brgić, Željka Vogrinc, Marina Boban, Nataša Klepac, Fran Borovečki, and Goran Šimić. 2023. "Soluble TREM2 Concentrations in the Cerebrospinal Fluid Correlate with the Severity of Neurofibrillary Degeneration, Cognitive Impairment, and Inflammasome Activation in Alzheimer’s Disease" Neurology International 15, no. 3: 842-856. https://doi.org/10.3390/neurolint15030053
APA StyleŠpanić Popovački, E., Babić Leko, M., Langer Horvat, L., Brgić, K., Vogrinc, Ž., Boban, M., Klepac, N., Borovečki, F., & Šimić, G. (2023). Soluble TREM2 Concentrations in the Cerebrospinal Fluid Correlate with the Severity of Neurofibrillary Degeneration, Cognitive Impairment, and Inflammasome Activation in Alzheimer’s Disease. Neurology International, 15(3), 842-856. https://doi.org/10.3390/neurolint15030053