1. Introduction
The prevalence of cardiovascular illnesses is rising worldwide and currently stands as the principal source of morbidity and mortality globally, responsible for over 32% of all fatalities [
1]. While the number of cardiovascular disease (CVD) mortalities climbed consistently by approximately 53.7%, from around 12.1 million in 1990 to 18.6 million in 2019, the total prevalence of CVDs nearly doubled, increasing by approximately 93.0%, from roughly 271 million in 1990 to approximately 523 million in 2019 [
2]. Despite concerted efforts spanning decades to address modifiable risk factors, there have been notable strides in reducing mortality rates and age-adjusted prevalence. However, this progress has also coincided with a staggering 193% surge in overall CVD prevalence globally over the last three decades.
Along with risk factors including obesity, smoking, diabetes, hypertension, hyperlipidemia, and immobility, the etiology of CVDs is complicated and involves both hereditary and environmental variables. Nearly all CVDs have an underlying pathogenesis and progression that is primarily of atherosclerotic origin. This causes coronary artery disease, cerebrovascular disease, thromboembolism, and peripheral vascular disease, which in turn causes myocardial infarction (MI), cardiac arrhythmias, or stroke. Novel strategies for illness prevention and early diagnosis are therefore needed.
The care of monogenic cardiovascular diseases, such as hypertrophic cardiomyopathy and primary arrhythmic disorders, now includes genetic testing. Recent developments in computational biology and sequencing technology have provided previously unattainable insight into the genetic foundations of common complicated illnesses including atrial fibrillation (AF) and coronary artery disease (CAD). Genetic testing and personalized treatment plans hold great promise for managing complex CVDs, but their broader application faces significant challenges. High costs, complex data interpretation, lack of standardized guidelines, healthcare system limitations, and privacy concerns hinder their widespread use. Integrating these innovations into clinical practice requires advanced tools, expertise, and substantial changes in healthcare systems. Overcoming these obstacles necessitates a concerted effort from various stakeholders to make genetic testing a standard diagnostic procedure. To fully utilize genetic techniques for CVD detection and prediction, creative prospective implementation studies and a quick improvement in our healthcare workforce’s skill set beyond traditional clinical genetic routes are required [
3].
2. Understanding Inherited Cardiomyopathies and Inherited Arrhythmic Syndromes: Types, Testing, and Treatment
Cardiomyopathies are a class of cardiac illnesses characterized by abnormalities in the structure and function of the heart muscle, which can result in diastolic and/or systolic dysfunction and an increased risk of malignant arrhythmias [
3]. Based on the predominant structural and hemodynamic characteristics, the World Health Organization (WHO) and the International Society and Federation of Cardiology (ISFC) introduced the first categorization of cardiomyopathies in 1980 [
4]. At the outset, the disease was officially classified into five forms as follows: restrictive cardiomyopathy (RCM), dilated cardiomyopathy (DCM), and hypertrophic cardiomyopathy (HCM), as well as arrhythmogenic ventricular cardiomyopathy (AVC). Originally, all cardiomyopathies were classified as idiopathic; however, as genetic testing became more sophisticated, these conditions were separated into acquired/secondary forms and hereditary/inherited types. Cardiomyopathies have been reclassified into five categories as follows: dilated cardiomyopathy, non-dilated cardiomyopathy, hypertrophic cardiomyopathy, restrictive cardiomyopathy, and arrhythmogenic ventricular cardiomyopathy, according to recently published recommendations [
5].
Individuals exhibiting features consistent with hypertrophic cardiomyopathy, dilated cardiomyopathy, or arrhythmogenic cardiomyopathy, particularly those eligible for cascade screening, are advised to undergo genetic testing as part of their diagnostic evaluation for hereditary cardiomyopathies [
6,
7,
8]. If the requirements for clinical diagnosis are not met, genetic testing is generally not advised. There could be importance in situations when a monogenic etiology is quite plausible given the frequency of variations with uncertain significance, particularly if testing is carried out in a specialized facility [
9,
10]. It is advised to screen first-degree biological relatives for these disorders using cascade genetic testing if a mutation causing the disease is found. Medicare-rebated genetic testing for hypertrophic cardiomyopathy, dilated cardiomyopathy, or arrhythmogenic cardiomyopathy may be requested by specialists or consulting physicians for patients whose clinical presentation, family history, or laboratory results are consistent with the inherited cardiomyopathy. If a P/LP mutation is discovered, specialists or consulting physicians could investigate first-degree biological relations specifically for genetic screening. Genetic testing on the proband’s partners may also be required to determine reproductive risk [
11]. Although there is a lack of data on the prevalence of inherited cardiomyopathies because of underdiagnosis, it is estimated that at least 0.6% of the world’s population suffers from this pathology, with racial and sexual orientation-related differences in clinical presentation [
6,
7,
8,
12].
Asymmetric ventricular enlargement and distinctive histological features including disarray, interstitial fibrosis, and myofibril hypertrophy are hallmarks of a major heart disorder known as hypertrophic cardiomyopathy. Approximately 70% of cases of hypertrophic cardiomyopathy are attributed to deleterious mutations in sarcomere genes, including cardiac β-myosin heavy chain—MYH7 and cardiac myosin binding protein—C-MYBPC3 variants [
13]. Different therapies are available for some of these ailments, including cardiac amyloidosis, Fabry disease, and glycogen storage disorders, which are like hypertrophic cardiomyopathy and must be separated from one another [
14]. For certain people who test negative for their genes, the subject of the genetic origin of hypertrophic cardiomyopathy remains unresolved after years of inquiry. Ventricular hypertrophy has been proposed to occasionally result from combinations of commonly occurring variants, even if this may reflect undiscovered monogenic kinds of disease.
2.1. Dilated Cardiomyopathy
Dilated cardiomyopathy (DCM) is a disorder defined by the absence of aberrant loading circumstances such as coronary artery disease, major valvopathies, or viral infections like COVID-19, ventricular dilatation, and systolic dysfunction with a multitude of genetic and acquired causes that can affect up to one in 250 people [
15,
16,
17]. These differences are becoming more and more blurry because of evidence of rare genetic abnormalities in individuals with environmental causes of DCM and vice versa. Uncommon genetic anomalies complicate distinguishing hereditary from environmental causes of dilated DCM due to several factors. Limited data on rare mutations and their roles in DCM indeed create uncertainty. Variable penetrance and expressivity mean that not all carriers exhibit symptoms, and severity can vary widely among patients. Gene–environment interactions further obscure the origins of the disease, as environmental factors can trigger symptoms in genetically predisposed individuals. Sporadic cases and phenotypic similarities between hereditary and environmentally induced DCM make clinical differentiation challenging. It is common for a genetic basis of the illness to remain hidden in the index case, and a positive family history may not emerge until later in life when more relatives are diagnosed [
18].
Though approximately 250 disease genes have been found, only ten to twenty genes now provide compelling proof of a dilated cardiomyopathy etiology [
18,
19,
20]. Fractional changes in the Titin gene, Titin-truncating variants (TTN, TTNtv), which generate the sarcomeric protein Titin, can account for up to 20% of cases of dilated cardiomyopathy. For the disorder, this is the most common genetic cause [
21]. It may be challenging to assess the therapeutic consequences of TTNtv because these changes can impact up to 3% of the general population. The high prevalence of TTNtv variants poses challenges in clinical management, genetic counseling, research, and health policy. Key issues include predicting disease risk, managing asymptomatic carriers, interpreting incidental findings, and developing care guidelines. Thirty research studies are currently being conducted, but there have been suggestions that a “second hit” might change the severity in TTNtv carriers [
22,
23].
2.2. Arrhythmogenic Cardiomyopathies
Arrhythmogenic cardiomyopathies are a group of highly arrhythmic cardiomyopathies with the gradual loss of myocytes as fibro-adipose tissue replaces the myocardium. Fatty-fibrous tissue envelops groups of myocytes, forming an electrical route for malignant ventricular arrhythmias [
3,
24]. Variations in LMNA, FLNC, RBM20, and SCN5A are associated with arrhythmic forms of dilated cardiomyopathy, such as arrhythmogenic right ventricular cardiomyopathy, which mainly impacts the right ventricle [
25]. Most of the time, pathogenic mutations in desmosomal genes cause arrhythmogenic right ventricular cardiomyopathy, which is inherited autosomally dominant [
26].
Positive family history is seen in up to 50% of patients with AVC. AVC typically exhibits incomplete penetrance autosomal dominant transmission, while two autosomal recessive forms—Naxos disease and Carvajal syndrome—have also been identified [
27]. Up to 60% of patients with ACM have genetic variations mostly in the genes that encode structural proteins known as desmosome proteins, such as plakoglobin, desmoplakin, and plakophillin [
3]. The Plakophilin 2 (PKP2) variants that are most frequently linked to the right-sided phenotype are truncating mutations [
28]. It has also been discovered that genes that do not encode desmosomes contain pathogenic mutations, but all at a lesser frequency.
2.3. Exploring Inherited Arrhythmic Syndromes
Inherited arrhythmic syndromes include monogenic illnesses such as catecholaminergic polymorphic ventricular tachycardia, familial forms of atrial fibrillation, long QT syndrome (LQTS), and Brugada syndrome. The prevalence of LQTS, one of the most common inherited cardiac conduction anomalies that can result in sudden cardiac death, is estimated to be 50 per 100,000 infants [
29]. In approximately 80% of patients with a prolonged corrected QT interval in the absence of structural cardiac illness or exposure to QT-prolonging drugs, there is a detectable LQTS-associated genetic variant, which mostly shows an inheritance with an autosomal dominant pattern. For those with known or suspected LQTS, genetic testing is recommended based on ECG, family history, and clinical symptoms. For asymptomatic people with a QTc of more than 500 ms in adults and greater than 480 ms in pre-puberty, genetic testing is also recommended if QT prolongation is not suspected [
6]. Most importantly, the ability of the data to inform treatment decisions and aid in independent risk prediction makes genetic testing for LQTS unique [
30]. It is recommended to perform variation-specific cascade genetic screening of first-degree relatives if a proband is determined to have a confident genetic variant. Three genes are undeniably associated with LQTS. Potassium channel protein-encoding genes KCNQ1 and KCNH2 have been connected to LQT1 and LQT2, respectively [
31,
32]. There is further complexity in the genetic link between LQT3 and SCN5A. The sodium channel subunit that is encoded by the gene SCN5A has been associated with disease, and around 10% of LQTS patients have gain-of-function mutations in this gene [
33]. Physicians who treat patients with symptoms or a family history that indicates a higher than 10% likelihood of a pathogenic variant may perform genetic testing for channelopathies or hereditary cardiac arrhythmic disorders. In the event of a P/LP variation, experts or consulting physicians may seek variant-specific testing in first-degree biological family members or reproductive partners [
11].
2.4. Vascular Aneurysm
Vascular aneurysms are pathological dilations of blood vessels caused by a weakening of the vessel wall, leading to a localized enlargement that can predispose the vessel to rupture. Vascular aneurysms are critical areas of focus in recent clinical research and guidelines. The European Society for Vascular Surgery (ESVS) updated its 2024 guidelines, which provide comprehensive recommendations on the management of these conditions [
31]. The most clinically significant aneurysms include cerebral aneurysms, thoracic aortic aneurysms (TAA), and abdominal aortic aneurysms (AAA). Cerebral aneurysms typically occur at arterial bifurcations within the Circle of Willis and can vary widely in size, often being asymptomatic until they rupture, leading to subarachnoid hemorrhage, which carries a high risk of morbidity and mortality. Factors such as hypertension, smoking, and genetic predisposition contribute to their development. Thoracic aortic aneurysms occur in the segment of the aorta and are often associated with degenerative changes in the aortic wall, such as cystic medial necrosis, and can be linked to conditions like Marfan syndrome, Ehlers–Danlos syndrome, and bicuspid aortic valve disease. TAAs can present asymptomatically or with symptoms related to compression of surrounding structures, such as chest pain or shortness of breath, similar to CVD. Abdominal aortic aneurysms are the most common type of aortic aneurysm, occurring below the renal arteries and above the iliac bifurcation. Risk factors include age, smoking, hypertension, and atherosclerosis. AAAs are often asymptomatic and discovered incidentally through imaging studies performed for other reasons. The risk of rupture increases with aneurysm size, and rupture is an extremely serious event with a high mortality rate. Advanced imaging techniques, such as magnetic resonance angiography (MRA) and computed tomography angiography (CTA), are crucial for the detection and management of these life-threatening conditions [
2,
31].
3. Unraveling the Complexity of Genetics in Coronary Arterial Disease and Atrial Fibrillation
Because of developments in sequencing, computer power, analytical methods, and international collaborations, it is now possible to identify the genetic diversity-related contributing factors to often occurring complex CVDs, such as CAD and atrial fibrillation. Using comprehensive case-control studies and genome-wide association studies (GWASs), which have reduced surveillance bias towards specific biological processes or genes of interest, common variants associated with disease have been discovered. The substantial overlap between cases and controls limits the predictive significance of a single nucleotide polymorphism (SNP) in a single patient. To increase the therapeutic utility of these data, polygenic risk scores (PRSs) were created by assessing the cumulative effect of each participant’s single nucleotide polymorphism (SNP) profile. People with exceptionally high scores are at risk for illness at rates several times higher than the population norm, and in other situations, their risk is like that of some monogenic disorders, per a recent study on several complex vascular diseases.
Traditional estimates place the heritability of 40–60% of CAD cases at play, a figure that is not fully explained by rare genetic variants associated with monogenic lipid disorders [
34,
35]. Over 200 significant GWAS loci have been found in European populations, and these loci have been included in many CAD-PRS techniques. Just around half of the SNPs that are included have previously been connected to established risk factors for CAD, such as blood pressure and lipid metabolism. Notably, a healthy lifestyle might mitigate the adverse consequences of a high CAD-PRS, as seen by the observed 46% relative risk reduction in CAD events. Those with top vs. bottom quintile scores showed a 4.17-fold higher relative risk of CAD in a second study that employed CAD-PRSs of 1.7 million genetic variants; this conclusion persisted even after adjusting for traditional risk markers [
36].
Although there is a possibility of familial atrial fibrillation, atrial fibrillation is more commonly associated with factors related to age, sex, genetics, concurrent diseases, and lifestyle. Nearly 200 chromosomal areas that influence atrial fibrillation susceptibility have been identified by GWASs, and several atrial fibrillation PRSs have been created. Numerous prospective cohort studies suggest that the atrial fibrillation PRS is indicative of incident atrial fibrillation. The atrial fibrillation polygenic risk score (AF-PRS) is thought to have prognostic value for the development of atrial fibrillation, according to a number of prospective cohort studies. Predictive accuracy is further improved by including the AF-PRS in models together with clinical variables such as anthropometrics, blood pressure, smoking status, blood pressure medication, diabetes, and history of myocardial infarction and heart failure [
37,
38,
39,
40,
41]. It is also possible to use the atrial fibrillation PRS to predict the chance of an atrial fibrillation recurrence after ablation therapy [
37].
4. Therapeutic Opportunities
4.1. Nucleic Acid-Based Therapies
Because of notable advancements in both efficacy and safety, nucleic acid-based therapies for cardiovascular diseases (CVDs) are advancing rapidly. This is especially true for therapeutics aimed at novel targets that conventional small molecule or antibody-based approaches may not be able to address optimally. Recent research in genetics and epigenetics has found many possible therapeutic targets for the management and prevention of CVDs. This review focuses on important approaches in nucleic acid-based therapeutics, such as gene therapies, epigenetic therapies, small interfering RNAs (siRNAs), RNA-targeted antisense oligonucleotides (ASOs), and microRNA (miRNA)-modulating medications. It also describes the current clinical translational status and potential applications of these approaches.
Notably, ongoing clinical studies are investigating the use of RNA-targeted nucleic acid-based therapeutics such as ASOs and siRNAs to reduce levels of transthyretin (TTR), PCSK9, apolipoprotein(a) [apo(a)], or apoCIII in patients with amyloidosis or advanced atherosclerotic CVD [
42,
43,
44,
45,
46]. These technologies have undergone extensive research and development to ensure safety and efficacy, enabling large-scale clinical trials to evaluate their effectiveness in achieving clinical endpoints.
It is important to mention that two intriguing therapeutic techniques with potentially significant effects have emerged from recent findings in biological cell-based systems, specifically RNA interference (RNAi) and CRISPR-associated protein 9 (Cas9) gene editing, together with their evolutionary progression. Furthermore, significant new target types have been identified by recent developments in the study of the human genome and epigenome, which might one day make it possible to target highly integrated processes like immune response regulation and cell differentiation.
Although RNA-targeted treatments such as small interfering RNAs (siRNAs) and antisense oligonucleotides (ASOs) have demonstrated significant clinical translational progress, including impressive long-term prospects, difficulties still exist in guaranteeing clinical safety and efficacy, particularly for more recent technological advancements, calling for careful consideration. In addition, the possibility of human genome editing presents ethical questions that need to be considered [
47].
These innovative technologies must finally be evaluated in light of key clinical effectiveness and safety characteristics, such as the absence of major adverse effects, high molecular target specificity, effective transport to target organs, and suitable stability for clinical use. The results will provide mechanistic insights into the synthesis, administration, principles of action, and salient characteristics of nucleic acid-based medications, such as siRNAs and ASOs. These methods not only provide fresh approaches to previously “undruggable” targets such as apolipoprotein(a), but they also hold the potential to greatly simplify treatment regimens in comparison with current treatments, like RNAi-based PCSK9 suppression that lasts for several months after a single subcutaneous injection of the medication [
48].
4.2. Unraveling Molecular Mechanisms and Implementing ASO Therapeutics
The development of fully synthetic “nucleic acid drugs”, which may preserve essential structural components of DNA or RNA while simultaneously undergoing substantial alterations (e.g., “xeno nucleic acids”, or XNAs), has been made possible by considerable advances in nucleic acid chemistry. Synthetic oligonucleotides that cause target gene silence have been developed into therapeutically safe and effective “antisense” medications thanks to recent advancements in nucleic acid chemistry methods. Usually comprising 13–20 nucleic acids, ASOs attach to target RNA by Watson–Crick hybridization. Nevertheless, bare, unaltered DNA or RNA utilized as medications is not effective and is easily degraded. Changes in the backbone (P-O to P-S moieties) and/or sugar moieties, especially at the 2′ position with different chemistries, have been made to circumvent this obstacle by increasing the target sequence’s affinity and resistance to degrading enzymes. This has resulted in increased potency, better tolerance, and increased clinical efficacy [
49].
New developments also involve conjugating N-acetylgalactosamine (GalNAc) to ASOs that bind to high-capacity asialoglycoprotein receptors in the liver, which allows ASOs to be specifically targeted and delivered to hepatocytes. This improves potency up to thirty times over unconjugated molecules [
49,
50]. With modified DNA wings on each side to improve binding stability, efficacy, and tolerability, ASOs are created as “gapmers”, with the core 10 bases acting as DNA mediating RNAse H1 cleavage of the sense strand. Single-stranded ASOs attach themselves directly to messenger RNA (mRNA) to create a duplex. This duplex is subsequently targeted by the ubiquitous RNAse H1 to cleave the target mRNA and prevent the synthesis of proteins [
48]. These medications have longer half-lives (3–4 weeks) because the antisense strand can attach to another target mRNA and exhibits relative resistance to cleavage [
49]. This makes it possible to administer less frequently and at lower levels to obtain the desired results [
51,
52,
53,
54].
The new lipid-lowering ASO treatments are either being developed and evaluated by the FDA or are now under authorization. Angiopoietin-like 3 (ANGPTL3), lipoprotein (a) [Lp(a)], and ApoC-III are the targets of ASOs. To date, none of the three has been able to successfully treat molecular targets using conventional pharmacological approaches [
55,
56].
4.2.1. ASOs Directed towards ApoC-III
The EU approved volanesorsen, a second-generation ASO without GalNAc, to treat familial chylomicronemia syndrome (FCS) [
55]. Because of lipoprotein-lipase (LPL) deficiency or associated enzyme deficiencies, people with FCS have higher plasma levels of dietary-derived chylomicrons. This can result in recurrent episodes of acute pancreatitis and an increased risk of atherosclerotic cardiovascular disease (ACVD) in later life. Research has demonstrated that apoC-III, which functions as a triglyceride-rich lipoprotein (TLR) clearance factor and an LPL inhibitor, enhances the therapeutic efficacy of volanesorsen in the treatment of FCS [
57]. In most individuals, volanesorsen has proven to be highly effective in lowering triglyceride levels by 78%, bringing them below the threshold for pancreatitis. Although therapy with volanesorsen or FCS can both cause thrombocytopenia, the risk of significant bleeding episodes has been reduced by routinely checking platelet levels.
Triglyceride levels ≥200 mg/dL in patients with pre-existing cardiovascular disease and a GalNac-conjugated ASO targeting ApoC-III with the same sequence as volanesorsen are now being developed for late-phase 2 patients. Broad improvements in the atherogenic lipid profile were shown in a phase 1 trial that involved volunteers with elevated triglyceride levels, including those receiving monthly dosing. Total cholesterol, ApoB, non-high-density lipoprotein cholesterol (HDL-C), very-low-density lipoprotein cholesterol (LDL-C), and increases in HDL-C were significantly reduced, and there was also a favorable safety and tolerability profile [
42]. In individuals with pre-existing cardiovascular disease and increased triglyceride levels, a phase 3 cardiovascular outcomes study will be designed based on the results of these trials.
4.2.2. ASOs Directed towards Apolipoprotein(a)
The primary target of antisense oligonucleotides (ASOs) is apolipoprotein(a) (ApoA), a vital part of lipoprotein(a) (Lp[a]), which is linked to cardiovascular disease. ASOs effectively lower Lp(a) plasma levels by blocking hepatocytes’ synthesis of apolipoprotein(a). Recent developments in ASO technology have greatly improved their potency and safety profile, especially when it comes to conjugating ASOs with a triantennary N-acetylgalactosamine (GalNAc3) moiety [
48]. Clinical studies using the GalNAc3-conjugated ASO medication APO(a)-LRx have produced encouraging findings, indicating a dose-dependent decrease in the levels of Lp(a) in circulation. This novel strategy might be used as a targeted therapy to treat people with high Lp(a) levels, opening up new treatment options for cardiovascular risk factors [
58]. Following the publication of the research design, the ASO treatment aimed at targeting apolipoprotein(a) (ApoA) has progressed to a pivotal phase 3 clinical trial focused on cardiovascular outcomes [
59].
In the experimental medication, IONIS-APO(a)-LRx, galNAc conjugation, and various structural alterations led to a >30-fold increase in efficacy and a >80% decrease in Lp(a) levels. Additionally, there were notable drops in proinflammatory oxidized phospholipids (OxPL), LDL-C, and apoB-100 along with the Lp(a) reductions. During the drug’s administration, circulating monocytes’ promigratory behavior towards endothelial cells was notably reduced; nevertheless, monocytes eventually recovered their promigratory phenotype. For practically all patients, the level of Lp(a) reduction achieved with a single monthly dosage should be sufficient to return Lp(a) concentrations to normal [
48].
The promising outcomes observed with IONIS-APO(a)-LRx underscore its potential as a valuable therapeutic option for reducing Lp(a) levels and associated cardiovascular risk factors. With its ability to effectively modulate lipid profiles and inflammatory markers, alongside the attenuation of promigratory behavior in monocytes, this medication holds great promise for improving cardiovascular health outcomes [
48,
59].
4.2.3. ASOs Directed towards ANGPTL3
ANGPTL3, a protein that is mostly produced in the liver, is a potential genetic target for lowering the risk of CVD since it inhibits both endothelial lipase and lipoprotein lipase (LPL). People who have full loss-of-function mutations in ANGPTL3 have familial hypolipidemia, which is defined by lifelong low levels of HDL-C, triglycerides, and LDL-C. Interestingly, ANGPTL3 loss-of-function mutations have also been connected to a lower risk of CVDs [
44] A GalNAc-modified ASO was reported to reproduce the lipid profile seen in individuals with familial mixed hypolipidemia in a phase 1 investigation on human volunteers with increased triglyceride levels. According to research on animals, this ASO improves insulin sensitivity, lowers the amount of triglycerides in the liver, and delays the development of atherosclerosis. Importantly, inhibition of ANGPTL3 also results in significant reductions in non-high-density lipoprotein cholesterol, very-low-density lipoprotein cholesterol (27.9–60.0%), ApoB, and ApoC-III. These findings suggest that ANGPTL3 inhibition may be an excellent therapeutic agent for patients who, regardless of obtaining otherwise optimal lipid-modifying therapy, have elevated remnant cholesterol [
59].
4.2.4. ASOs Targeting Transthyretin
Severe cardiomyopathy can result from transthyretin amyloidosis, which affects the heart in addition to the neurological system [
46,
60]. For inherited TTR amyloidosis polyneuropathy, the 2′-MOE-engineered ASO inotersen, which is intended to decrease the synthesis of both wild-type and mutant TTR in the liver, showed better disease progression and quality of life in a 15-month phase III research [
61]. In individuals with cardiac involvement, preliminary research indicates that inotersen may also improve cardiac function [
62,
63]. As of right now, inotersen is approved worldwide for treatment in TTR polyneuropathy. Phase I studies are now underway for a version of the drug that has been modified for use in polyneuropathy, as well as in hereditary and wild-type TTR cardiac amyloidosis.
5. Translational Trials for Heart Health: Pharmaceutical Approaches
5.1. siRNA Targeting PCSK9
To evaluate clinical effectiveness and safety, the ORION clinical trials program is currently unique in that it examines RNAi-mediated PCSK9 inhibition in a variety of CVD cohorts. The ORION-1 trial’s results were released [
64,
65,
66]. By specifically targeting PCSK9, patients treated with the siRNA medication Inclisiran had a significant decrease in LDL-C levels. Significantly, long-lasting drops in LDL-C and PCSK9 levels after a single medication dose over a 240-day period suggested that RNAi-mediated liver PCSK9 inhibition provides an alternative to circulating PCSK9 being targeted by monoclonal antibodies (mAbs), probably requiring fewer injections. More than 3000 patients participated in the phase 3 ORION-10 and ORION-11 studies, which validated the medication’s safety and effectiveness in decreasing LDL. The ORION-4 study is a phase III clinical trial with an emphasis on cardiovascular outcomes, which is now underway [
48].
5.2. siRNA Targeting Transthyretin
siRNAi is a therapeutic approach that targets TTR amyloidosis. In the APOLLO phase III clinical trial, the anti-TTR sRNAi drug patisiran corrected multiple clinical symptoms of hereditary TTR amyloidosis with polyneuropathy. Five of one patisiran was found to be superior to a placebo in the subanalysis of the study for left ventricular global longitudinal strain [
67,
68].
5.3. Targeting MicroRNA Therapies for Cardiovascular Conditions
By either preventing translation or encouraging the destruction of target mRNAs, tiny non-coding RNAs known as microRNAs (miRNAs), which have an approximate length of 22 nucleotides, function as powerful post-transcriptional regulators of gene expression [
69]. miRNAs recognize certain gene transcripts by matching the 3′-untranslated region of target mRNAs with positions 2–8 of the miRNA 5′-end. A single miRNA may control many genes, and its targeting can alter the transcriptional landscape significantly, affecting the phenotype of cells [
70]. Since mature miRNA sequences are short and largely conserved throughout mammalian species, miRNAs are a prospective target for therapeutic intervention in a variety of illnesses, including cardiovascular disease (CVD). In order to modify the amounts of miRNA, the following methods are used: (i) chemically modified anti-miR oligonucleotides or viral vector-based overexpression and (ii) synthetic double-stranded miRNAs [
71].
Research on gain- and loss-of-function in hindlimb ischemia experimental models has shown that reprogramming particular miRNAs, like miR-92a, miR-21, and miR-29b, can significantly alter transcriptional networks that control fibrosis, hypertrophy, myocyte growth, and angiogenic responses [
72]. The functional recovery of the ischemic limb and myocardium in mice is achieved by the intravenous injection of a particular antagomir that targets miR-92a, a member of the miR-17/92 cluster that orchestrates angiogenic genes (integrin α5) and restores blood vessel growth [
73]. Cardiomyocyte hypertrophy and interstitial fibrosis are reduced in vivo by inhibiting miR-21, a master regulator of MAP kinase signaling, which thereby prevents cardiac dysfunction brought on by transaortic constriction [
74]. Similar to this, TAC-induced cardiac fibrosis in mice is avoided when miR-29 is deleted from cardiac myocytes [
75].
Clinical Utilization of miRNA-Based Treatments
A large number of miRNA-based treatments are now in preclinical research, and more and more are making their way into clinical trials. When given as a 4-week monotherapy, the locked nucleic acid-targeting miR-122, miravirsen, showed dose-dependent antiviral efficacy against hepatitis C virus RNA in a phase 2 human investigation [
76]. Moreover, patients with refractory advanced solid tumors showed a decrease in tumor progression when MRX34, a double-stranded miRNA mimic of miR-34, was administered [
77]. Recently, Santaris Pharma and Servier, two major players in the pharmaceutical business, partnered with a clinical-stage biopharmaceutical startup, MiRagen Medicines, to promote miRNA-based medicines with potential uses in cardiovascular disease (CVD). This experiment will be a prelude to phase 2 clinical studies evaluating the therapeutic potential of MRG-110 in patients with ischemic cardiomyopathy, heart failure, and/or peripheral artery disease, taking into account the pro-angiogenic effects of miR-92a suppression. A few years back, a different study began accepting patients to determine if Remlarsen (MRG-201), a miR-29 mimic, can prevent or lessen the formation of keloid scars in those who have already had keloid scarring [
78,
79]. The trial’s predicted outcomes, which were anticipated in 2020, have great potential since they will open the door to further research into how Remlarsen affects myocardial fibrosis and extracellular matrix remodeling in heart failure patients. The relevance of these findings is further highlighted by the dearth of effective anti-fibrotic medicines for cardiovascular patients. Moreover, Regulus therapies are leading the way in the development of miRNA therapies with potential uses in CVDs, such as anti-miR-21, anti-miR-155, and anti-miR-33, which are used to treat inflammation, cardiometabolic disorders, and fibrotic illnesses, respectively [
48].
5.4. Therapeutic Approaches to Epigenetic Changes in Cardiovascular Health
Protein-coding genes have been identified as a result of DNA sequencing of the human genome. Nevertheless, the next ENCODE Project demonstrated that these genes account for a minuscule portion of total RNA transcripts generated within human cells [
80,
81,
82]. Furthermore, it was discovered that the non-coding genome is widely translated into a wide variety of RNA species, many of which have unknown roles.
Research on the human epigenome has shown further complexity [
83,
84,
85]. The human epigenome is impacted by a variety of non-genetic factors and includes reversible genome alterations that take place above the fixed DNA sequence level. The transcriptional activity of large genomic areas can be strongly impacted by these epigenetic changes.
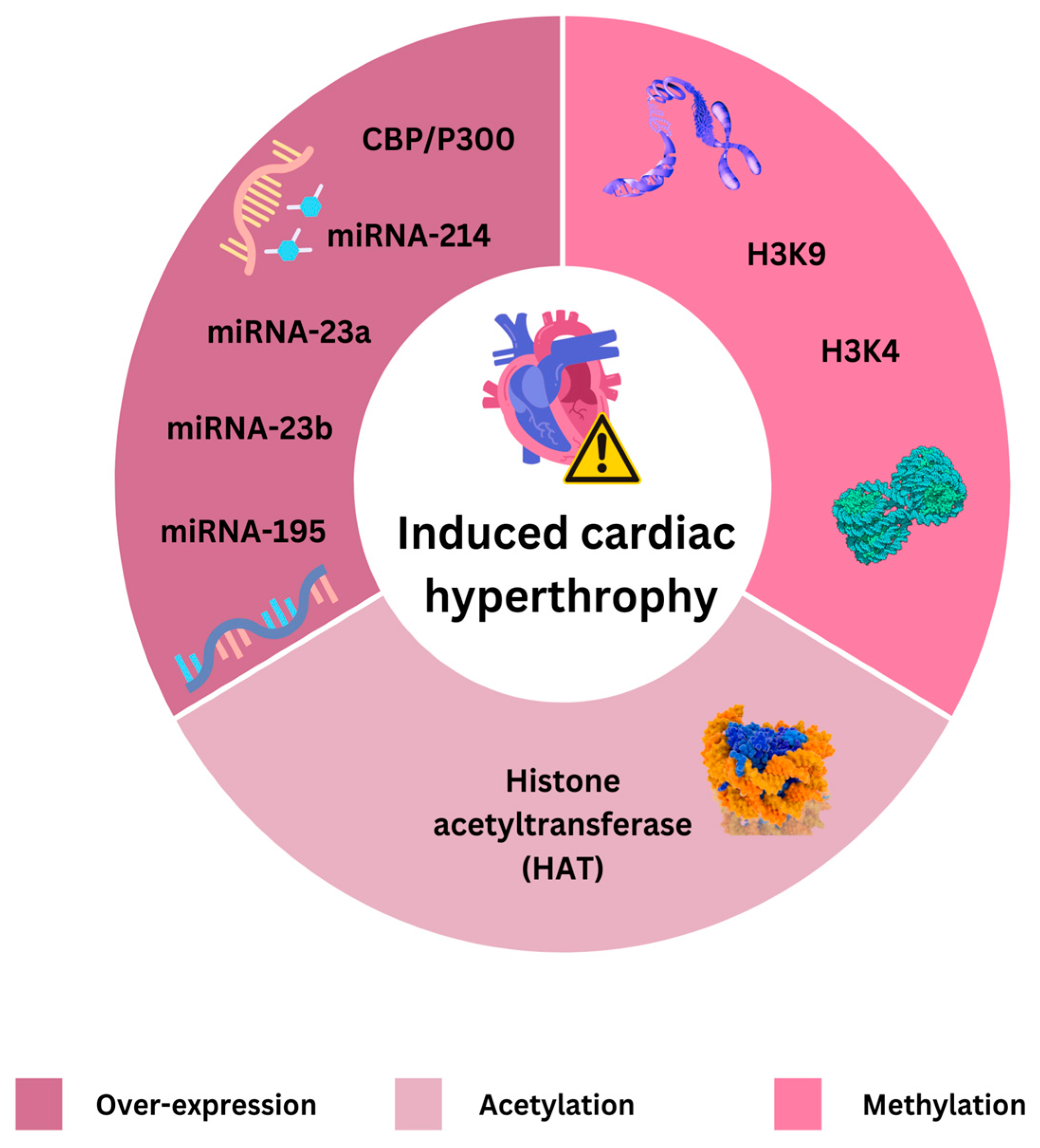
Histone modifications, RNA-based processes, and DNA methylation are all examples of epigenetic mechanisms. Research has revealed a variety of DNA methylation patterns in heart failure patients. For example, a recent study found areas of epigenetic vulnerability linked to heart failure [
86]. Differentiable methylation patterns were found across tissues using extensive epigenome-wide mapping of DNA methylation in left ventricular myocardial biopsies and entire peripheral blood, in addition to RNA deep sequencing and whole-genome sequencing. Methylation of CpG sites has been proposed as a potential epigenetic biomarker for heart failure.
Histone modifications, such as methylation and acetylation, are regulated by enzymes known as writers (e.g., histone acetyltransferases and histone methyltransferases (H3K4 and H3K9) and erasers (e.g., histone deacetylases—HDACs) and are recognized by proteins like BET proteins [
87,
88]. BET proteins act as epigenetic readers of lysine acetylation and represent a potential novel therapeutic target. Investigations into BET protein inhibitors, such as apabetalone, are underway [
89,
90]. Epigenetic mechanisms claimed to induce cardiac hypertrophy are presented on
Figure 1.
Novel Approaches to Epigenetic Therapies for Cardiovascular Disorders
According to recent experimental research, epigenetic modifications are important in the etiology of CVDs and could provide compelling treatment targets. Epigenetically reprogrammed endothelial progenitor cells (EPCs) were transplanted intramyocardially into a mouse model of myocardial infarction (MI), and this improved cardiac function [
91]. Similarly, pharmacological inhibition of histone deacetylases (HDACs) reduced cardiac hypertrophy in a transverse aortic constriction (TAC) model [
92]. An epigenetic process involving chromatin-modifying enzymes and microRNAs (miRNAs) was reported to induce persistent transcription of p66Shc in diabetic mice with heart failure, leading to the production of reactive oxygen species (ROS) [
93].
Furthermore, recent research found that failed mice hearts have lower levels of the stress-responsive epigenetic repressor HDAC4′s N-terminal fragment (HDAC4-NT) [
94]. Heart remodeling and failure were prevented by overexpressing HDAC4-NT. Exercise has also been shown to raise HDAC4-NT levels, and animals lacking a cardiomyocyte-specific copy of Hdac4 were less able to exercise. In both mice and humans, our work identified a regulatory axis wherein calcium handling—a well-established critical component of cardiac function—is influenced by the epigenetic modification of a metabolic pathway [
95,
96].
A recent study found that a long non-coding RNA (lncRNA) that controls the LDL receptor (LDLR) controls the absorption of LDL into human cells. This finding establishes a major relationship between LDLR, a critical target for therapy, and an epigenetic process that depends on lncRNA [
97]. Targeting this long non-coding RNA (lncRNA) using GalNAc-coupled siRNAs allowed for the direct targeting of liver cells and improved absorption of cholesterol. According to different research, stenotic vascular disease dependent on smooth muscle cells is improved by inhibiting the HDAC9 complex [
98].
Neointima development was decreased when the HDAC9 complex was targeted, either by ASOs that targeted MALAT1 or by inhibiting the methyltransferase EZH2. One research study used ASOs, while the other used GalNAc-coupled siRNAs; both made use of cutting-edge technologies that had previously been tested in clinical trials.
Even though just a small number of medications with epigenetic modes of action have been approved, little is known about the subject. Nonetheless, nucleic acid-based substances and peptides from different sources are included in the toolkit for epigenetic treatment [
87,
97,
99]. Peptides can influence both the maturation of miRNAs and the production of long noncoding RNAs (lncRNAs), which might indirectly change the epigenome.
5.5. Exploring Gene Therapy Options: Protein Augmentation vs. RNAi-Mediated Depletion
5.5.1. Principles of Gene Therapies
All therapeutic transport of nucleic acid sequences to damaged organs or tissues of patients is referred to as “gene therapy”; according to regulatory requirements, these treatments are known as Gene Therapy Medicinal Products (GTMPs). Early approaches to therapy focused on experimental gene supplementation strategies to restore downregulated or absent proteins important to pathophysiology in diseases that are acquired or monogenic. More recently, gene editing and silencing technologies have entered the therapeutic space. Currently, a phase 1/2a first-in-man clinical study is investigating gene supplementation for homozygous familial hypercholesterolemia (FH), a disorder characterized by a lack of the LDL receptor (LDLR) [
48].
LDL-C levels are used as a surrogate biomarker for LDLR expression in the experiment, which uses recombinant adeno-associated virus (rAAV)-based liver-directed human LDLR gene therapy as a GTMP.
There has been a thorough evaluation of GTMPs that target peripheral arterial disease (PAD), heart failure, and cardiomyopathies (CMPs) [
100,
101]. These treatments, which are mostly gene replenishment strategies based on viruses, have advanced to the early stages of clinical trials. While the FLOURISH trial (phase 3), which will involve a single intracoronary administration of adenovirus type 5 (Ad5)-based human adenylyl cyclase 6 (Ad5-hAC6) for patients with heart failure with reduced ejection fraction (HFrEF), is still in the clinical development stage, the CUPID trial series—which used rAAV1-SERCA2a-based treatment for HFrEF and dilated cardiomyopathy through the same invasive administration method—was prematurely stopped at phase 2b because of insufficient myocardial SERCA2a gene delivery.
5.5.2. Exploring Gene Therapy Options for Heart Failure and Cardiomyopathies
Heart-directed gene therapy procedures have been difficult to establish since the heart has proven to be a difficult target organ for rAAV vectors. Usually, percutaneous catheter-based delivery into veins or coronary arteries is used in these procedures. Percutaneous catheter-based delivery systems offer a balanced approach, providing precise and targeted delivery of therapeutic agents with fewer risks compared with surgical methods. They are minimally invasive, reducing systemic exposure and associated side effects compared with intravenous methods [
102]. Animal models are often used in scientific research; however, the limitations of animal models, particularly the reliance on young and healthy animals, significantly impact the translatability of nucleic acid therapies to human clinical practice. These models often fail to accurately represent the complexity of human diseases, the impact of aging, the varied immune responses seen in human patients, and the diverse patient populations that therapies aim to treat. However, these routes have successfully delivered sufficient myocardial genes in both small and large animal models, resulting in enhanced cardiac function in preclinical studies, and clinical trials like CUPID (Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease). The conclusions of the CUPID clinical study were that gene therapy targeting SERCA2a is safe and shows potential efficacy for treating heart failure, though long-term benefits were mixed. These findings have significant implications for the future of gene therapy in heart failure, highlighting the need for refined targeting, understanding patient variability, combining therapies, enhancing delivery systems, focusing on clinical endpoints, and the necessity of conducting further research [
103,
104,
105,
106,
107].
The development of guided rAAV evolution technologies presents a viable avenue to produce synthetic rAAVs specific to the heart that may be administered systemically by intravenous injection. This will enable accurate transduction of the heart with little off-target activity. This development is essential to opening the heart for therapeutic purposes. Therefore, until effective delivery mechanisms are developed, it could be wise to postpone the clinical development of other established heart failure targets, including S100A1 or the GRK2 inhibitor bARKct. By utilizing enhanced clinical delivery systems, these tools may facilitate the creation of next-generation cardiac GTMPs with adequate myocardial transduction efficiency for gene silencing, gene editing, and supplementation strategies to treat different types of acquired heart failure and hereditary cardiomyopathies [
108,
109,
110]. However, considering the results of Ad5-hAC6 clinical development so far, it is also imperative to investigate if, in contrast to current scientific consensus, Ad5 might function as a good carrier for cardiac indications such as heart failure.
Creating efficient delivery systems for gene therapy specific to the heart faces several obstacles. Overcoming the obstacles to creating efficient delivery systems involves addressing challenges in targeted delivery, immune response, sustained gene expression, vector safety, and cellular uptake. Advances in viral vector engineering, non-viral delivery methods, precision targeting techniques, immune modulation, gene construct optimization, and sustained release formulations have collectively contributed to improving the efficacy and safety of gene therapy for heart conditions. Despite significant progress, ongoing research and innovation are crucial to realizing the full potential of gene therapy in treating heart diseases [
103,
104,
105,
106,
107,
108,
109,
110].
5.5.3. Targeting Tomorrow: Novel Insights from Human Genome and Epigenome Research
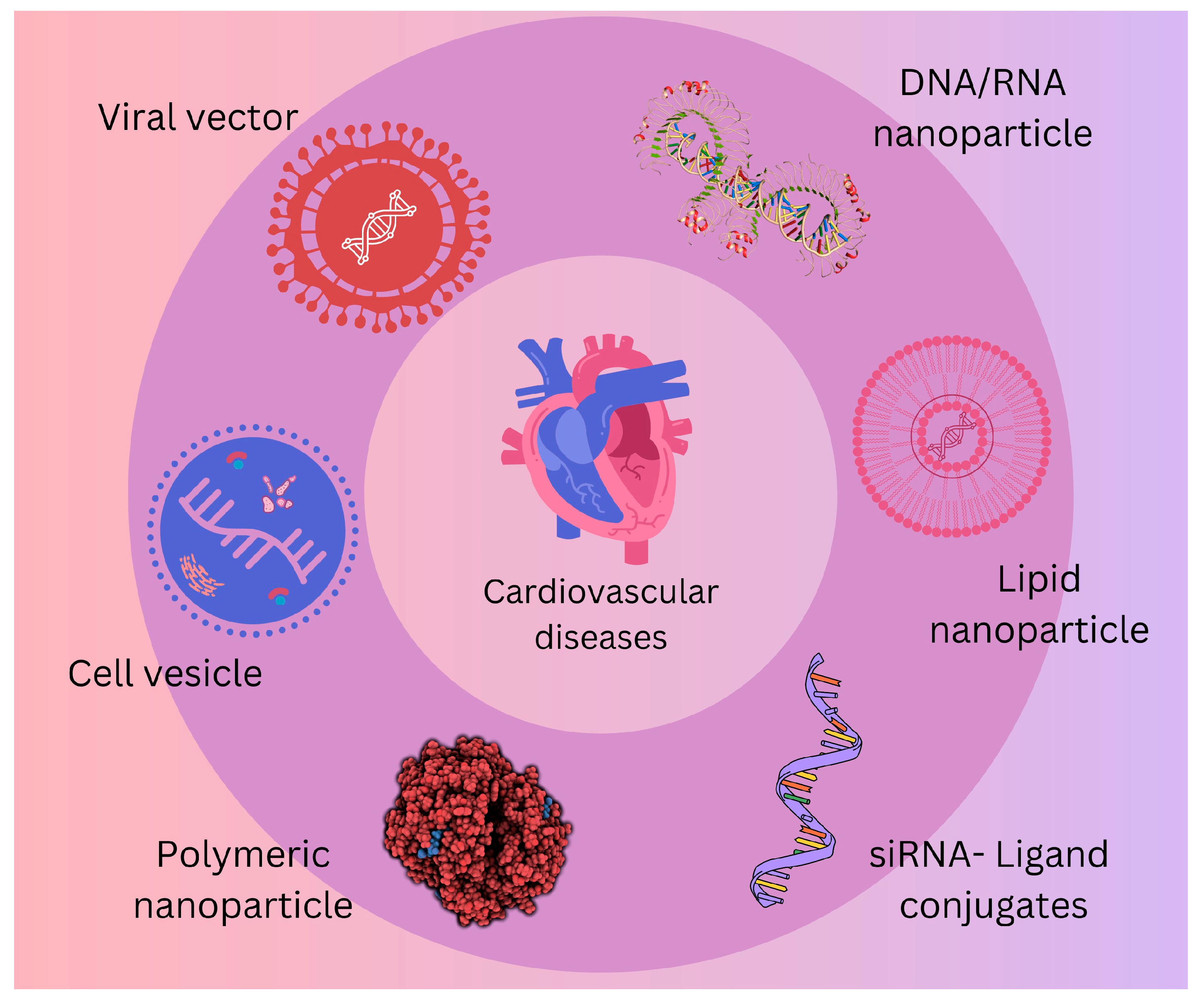
To create novel therapeutic ncRNA tools suited for therapeutic needs, an increasing variety of endogenous ncRNAs are being used [
111,
112]. Interestingly, non-coding (regulatory, architectural) transcripts, including microRNAs (miRs), lncRNAs, circRNAs, and others, are not only used as tools but also as a broad repertoire of possible therapeutic targets, as presented in
Figure 2 [
113,
114,
115,
116,
117]. This area of non-coding transcripts is largely unexplored.
Growing data suggest that many non-coding genomic regions are not single units that act like traditional protein-coding genes. Rather, they are complex networks of RNA processing machinery that control several biological functions, including cellular migration and proliferation [
118]. Furthermore, they could coordinate intricate biological processes occurring at the systemic level, such as innate and acquired immunological responses. Several CVDs have been linked to the dysregulation of these systems, and restoring proper control may have a significant impact on the course of the illness or recovery [
119]. According to recent research, there may be “master regulators” that function at the non-coding and epigenome levels of the genome, offering fresh and intriguing targets for treatment [
48].
5.6. Facing Challenges: Clinical Translation of Next-Generation Nucleic Acid Therapies
Obstacles to clinical translation of the compelling and broad choices outlined above range from clinical safety and regulatory problems to molecular drug design and delivery. Although animal experiments offer priceless proof-of-principle results, they frequently use young, otherwise healthy animals, which departs from clinical practice. Notwithstanding this drawback, several innovative clinical studies have effectively demonstrated the technical and therapeutic viability of nucleic acid treatment modalities, bolstering a strong base of substantial animal research. However, safety concerns are a major concern because every new medication and treatment target may reveal unique and unanticipated difficulties. These issues—which include immunological activation, cellular off-target effects, systemic mistargeting and accumulation, and delayed-onset safe effects—represent important factors to consider as one moves closer to clinical use [
48].
6. What Does the Future Bring?
Introducing genetic testing into the assessment process for CVDs offers a unique opportunity to better tailor a patient’s diagnosis and treatment plan. This can also be utilized in some circumstances to target family members for clinical and genetic assessment. However, there are still a lot of significant gaps that make it challenging to incorporate these useful methods into more conventional clinical practice.
The spectrum of individuals who can benefit from these new resources is increased by inclusion and diversity in genomic research, which also increases our understanding of the genetics underlying CVDs. The goal of regulatory measures has been to increase diversity and include more research participants in genetic investigations. Validation studies in historically underrepresented ancestry groups, however, can show diminished or non-significant results. Additionally, large inequalities in health outcomes persist among persons from underrepresented ancestry groups [
120]. Numerous studies have shown that individuals from other ancestry groups have a higher incidence of variants of unknown significance, which leads to a worse diagnostic yield from genetic testing as compared with those of European ancestry [
121].
Conducting implementation research and prospective studies is paramount in assessing the multifaceted impacts of genetic testing on the diagnosis and treatment of cardiovascular diseases. It is imperative to thoroughly evaluate not only the psychological effects and economic ramifications but also the overall effectiveness of incorporating genetic testing into clinical practice. To achieve this, it is essential to establish and maintain collaborative efforts among various stakeholders, including clinical geneticists, payers, primary care and specialty physicians, academic researchers, and health policy experts. By fostering these partnerships, we can conduct comprehensive and forward-thinking clinical implementation studies that provide valuable insights into the utility and feasibility of new genetic testing methods in real-world healthcare settings.
7. Conclusions
Considerable progress has been made in the past thirty years in establishing the connection between genetic risk factors and CVDs. Through extensive research, numerous genetic variants associated with cardiovascular conditions have been identified, shedding light on the underlying mechanisms and pathways involved. In several cases, genetic testing has not only enhanced patients’ risk assessments but has also guided treatment decisions, enabling healthcare providers to tailor interventions based on individual genetic profiles. Additionally, genetic testing has facilitated cascade screening for at-risk biological family members, allowing for the early detection and intervention of potential cardiovascular issues.
Recent advancements in CVD therapies have focused on novel drug treatments, gene therapy, regenerative medicine, and advanced device technologies. Gene therapy aims to improve calcium concentration in heart cells to enhance cardiac function. Trials like the CUPID study have shown mixed but promising results, indicating potential for future refinement. Emerging applications like CRISPR-Cas9 include editing genes associated with inherited cardiovascular diseases, though clinical use is still in the early stages. Immunomodulation represents a promising option, where certain cytokine inhibitors are shown to reduce recurrent cardiovascular events in patients with a history of myocardial infarction and elevated inflammatory markers, supporting the role of inflammation in CVDs. Finally, a biomarker-guided therapy utilizing genetic and proteomic markers to tailor treatments to individual patient profiles should help in improving outcomes and reducing adverse effects. These advancements represent significant progress in the treatment and management of CVDs, offering hope for improved patient outcomes through innovative and personalized approaches. However, despite these advancements, further implementation and rigorous clinical trial research are needed to generate robust evidence supporting the successful incorporation of genetic testing into routine medical practice. Continued efforts in this direction will be vital in optimizing patient care and improving outcomes in the field of cardiovascular medicine.
Author Contributions
Conceptualization, A.K. and L.B.; writing—original draft preparation, A.K.; writing—review and editing, L.B. and V.S.; supervision, L.B. and V.S.; project administration, L.B. and V.S.; funding acquisition, L.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
This work was written as part of the scientific project approved by the Croatian Science Foundation titled “Endothelial dysfunction, inflammation and oxidative stress in patients undergoing cardiac surgery—EDIOS” (IPS-2023-02-9650; grant approved: 5 October 2023), and the scientific project “uniri-iskusni-biomed-23-88-3035: Influence of elective primary coronary intervention and coronary artery bypass grafting on the beating heart on endothelial dysfunction, oxidative stress and inflammatory response in patients with ischemic heart disease”.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- World Health Organization. Cardiovascular Diseases (CVDs); WHO: Geneva, Switzerland, 2021; Available online: https://www.who.int/en/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 1 January 2020).
- Flora, G.D.; Nayak, M.K. A Brief Review of Cardiovascular Diseases, Associated Risk Factors and Current Treatment Regimes. Curr. Pharm. Des. 2019, 25, 4063–4084. [Google Scholar] [CrossRef] [PubMed]
- Voinescu, O.R.; Ionac, A.; Sosdean, R.; Ionac, I.; Ana, L.S.; Kundnani, N.R.; Morariu, S.; Puiu, M.; Chirita-Emandi, A. Genotype-Phenotype Insights of Inherited Cardiomyopathies-A Review. Medicina 2024, 60, 543. [Google Scholar] [CrossRef] [PubMed]
- McKenna, W.J.; Maron, B.J.; Thiene, G. Classification, Epidemiology, and Global Burden of Cardiomyopathies. Circ. Res. 2017, 121, 722–730. [Google Scholar] [CrossRef]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011, 8, 1308–1339. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M. Genetic evaluation of cardiomyopathy—A Heart Failure Society of America practice guideline. J. Card. Fail. 2018, 24, 281–302. [Google Scholar] [CrossRef]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: A report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2020, 142, e558–e631. [Google Scholar] [PubMed]
- Njoroge, J.N.; Mangena, J.C.; Aribeana, C.; Parikh, V.N. Emerging Genotype-Phenotype Associations in Dilated Cardiomyopathy. Curr. Cardiol. Rep. 2022, 24, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; van Tintelen, P.J.; McKenna, W.J.; Hauer, R.N.W.; Anastastakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brunckhorst, C.; Bucciarelli-Ducci, C.; et al. Arrhythmogenic right ventricular cardiomyopathy: Evaluation of the current diagnostic criteria and differential diagnosis. Eur. Heart J. 2020, 41, 1414–1429. [Google Scholar] [CrossRef]
- Gray, M.P.; Fatkin, D.; Ingles, J.; Robertson, E.N.; Figtree, G.A. Genetic testing in cardiovascular disease. Med. J. Aust. 2024, 220, 428–434. [Google Scholar] [CrossRef]
- McKenna, W.J.; Judge, D.P. Epidemiology of the inherited cardiomyopathies. Nat. Rev. Cardiol. 2021, 18, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Ingles, J.; Goldstein, J.; Thaxton, C.; Caleshu, C.; Corty, E.W.; Crowley, S.B.; Dougherty, K.; Harrison, S.M.; McGlaughon, J.; Milko, L.V.; et al. Evaluating the Clinical Validity of Hypertrophic Cardiomyopathy Genes. Circ. Genom. Precis. Med. 2019, 2, e002460. [Google Scholar] [CrossRef]
- Harper, A.R.; Goel, A.; Grace, C.; Thomson, K.L.; Petersen, S.E.; Xu, X.; Waring, A.; Ormondroyd, E.; Kramer, C.M.; Ho, C.Y.; et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat. Genet. 2021, 53, 135–142. [Google Scholar] [CrossRef]
- Weintraub, R.G.; Semsarian, C.; Macdonald, P. Dilated cardiomyopathy. Lancet 2017, 390, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Cowan, J.; Jordan, E.; Kinnamon, D.D. The Complex and Diverse Genetic Architecture of Dilated Cardiomyopathy. Circ. Res. 2021, 128, 1514–1532. [Google Scholar] [CrossRef]
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.J.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L.; et al. ClinGen. ClinGen--the Clinical Genome Resource. N. Engl. J. Med. 2015, 372, 2235–2242. [Google Scholar] [CrossRef] [PubMed]
- Mazzarotto, F.; Tayal, U.; Buchan, R.J.; Midwinter, W.; Wilk, A.; Whiffin, N.; Govind, R.; Mazaika, E.; de Marvao, A.; Dawes, T.J.W.; et al. Reevaluating the Genetic Contribution of Monogenic Dilated Cardiomyopathy. Circulation 2020, 141, 387–398. [Google Scholar] [CrossRef]
- Horvat, C.; Johnson, R.; Lam, L.; Munro, J.; Mazzarotto, F.; Roberts, A.M.; Herman, D.S.; Parfenov, M.; Haghighi, A.; McDonough, B.; et al. A gene-centric strategy for identifying disease-causing rare variants in dilated cardiomyopathy. Genet. Med. 2019, 21, 133–143. [Google Scholar] [CrossRef]
- Skriver, S.V.; Krett, B.; Poulsen, N.S.; Krag, T.; Walas, H.R.; Christensen, A.H.; Bundgaard, H.; Vissing, J.; Vissing, C.R. (Skeletal Muscle Involvement in Patients with Truncations of Titin and Familial Dilated Cardiomyopathy. JACC Heart Fail. 2024, 12, 740–753. [Google Scholar] [CrossRef]
- Fatkin, D.; Huttner, I.G.; Kovacic, J.C.; Seidman, J.G.; Seidman, C.E. Precision medicine in the management of dilated cardiomyopathy: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2019, 74, 2921–2938. [Google Scholar] [CrossRef]
- Fatkin, D.; Calkins, H.; Elliott, P.; James, C.A.; Peters, S.; Kovacic, J.C. Contemporary and Future Approaches to Precision Medicine in Inherited Cardiomyopathies: JACC Focus Seminar 3/5. J. Am. Coll. Cardiol. 2021, 77, 2551–2572. [Google Scholar] [CrossRef]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: Executive summary. Heart Rhythm. 2019, 16, e373–e407. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Kumar, S.; Elliott, P.; Kalman, J.M.; Fatkin, D. Arrhythmic Genotypes in Familial Dilated Cardiomyopathy: Implications for Genetic Testing and Clinical Management. Heart Lung Circ. 2019, 28, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Tadros, H.J.; Miyake, C.Y.; Kearney, D.L.; Kim, J.J.; Denfield, S.W. The Many Faces of Arrhythmogenic Cardiomyopathy: An Overview. Appl. Clin. Genet. 2023, 16, 181–203. [Google Scholar] [CrossRef]
- Graziano, F.; Zorzi, A.; Cipriani, A.; De Lazzari, M.; Bauce, B.; Rigato, I.; Brunetti, G.; Pilichou, K.; Basso, C.; Perazzolo Marra, M.; et al. The 2020 “Padua Criteria” for Diagnosis and Phenotype Characterization of Arrhythmogenic Cardiomyopathy in Clinical Practice. J. Clin. Med. 2022, 11, 279. [Google Scholar] [CrossRef] [PubMed]
- James, C.A.; Jongbloed, J.D.H.; Hershberger, R.E.; Morales, A.; Judge, D.P.; Syrris, P.; Pilichou, K.; Domingo, A.M.; Murray, B.; Cadrin-Tourigny, J.; et al. International Evidence Based Reappraisal of Genes Associated with Arrhythmogenic Right Ventricular Cardiomyopathy Using the Clinical Genome Resource Framework. Circ. Genom. Precis. Med. 2021, 14, e003273. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Stramba-Badiale, M.; Crotti, L.; Pedrazzini, M.; Besana, A.; Bosi, G.; Gabbarini, F.; Goulene, K.; Insolia, R.; Mannarino, S.; et al. Prevalence of the congenital long-QT syndrome. Circulation 2009, 120, 1761–1767. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Semsarian, C.; Márquez, M.F.; Sepehri Shamloo, A.; Ackerman, M.J.; Ashley, E.A.; Sternick, E.B.; Barajas-Martinez, H.; Behr, E.R.; Bezzina, C.R.; et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the State of Genetic Testing for Cardiac Diseases. Heart Rhythm. 2022, 19, e1–e60. [Google Scholar] [CrossRef]
- Wanhainen, A.; Van Herzeele, I.; Bastos Goncalves, F.; Bellmunt Montoya, S.; Berard, X.; Boyle, J.R.; D’Oria, M.; Prendes, C.F.; Karkos, C.D.; Kazimierczak, A.; et al. Editor’s Choice—European Society for Vascular Surgery (ESVS) 2024 Clinical Practice Guidelines on the Management of Abdominal Aorto-Iliac Artery Aneurysms. Eur. J. Vasc. Endovasc. Surg. Off. J. Eur. Soc. Vasc. Surg. 2024, 67, 192–331. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Ackerman, M.J.; George, A.L.; Wilde, A.A.M. Impact of genetics on the clinical management of channelopathies. J. Am. Coll. Cardiol. 2013, 62, 169–180. [Google Scholar] [CrossRef]
- Qureshi, F.; Ali, A.; John, P.; Jadhav, A.P.; Venkateshwari, A.; Rao, H.; Jayakrishnan, M.P.; Narasimhan, C.; Shenthar, J.; Thangaraj, K.; et al. Mutational analysis of SCN5A gene in long QT syndrome. Meta Gene 2015, 6, 6–35. [Google Scholar] [CrossRef] [PubMed]
- Zdravkovic, S.; Wienke, A.; Pedersen, N.L.; Marenberg, M.E.; Yashin, A.I.; De Faire, U. Heritability of death from coronary heart disease: A 36-year follow-up of 20 966 Swedish twins. J. Intern. Med. 2002, 252, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Won, H.H.; Natarajan, P.; Dobbyn, A.; Jordan, D.M.; Roussos, P.; Lage, K.; Raychaudhuri, S.; Stahl, E.; Do, R. Disproportionate Contributions of Select Genomic Compartments and Cell Types to Genetic Risk for Coronary Artery Disease. PLoS Genet. 2015, 11, e1005622. [Google Scholar] [CrossRef]
- Inouye, M.; Abraham, G.; Nelson, C.P.; Wood, A.M.; Sweeting, M.J.; Dudbridge, F.; Lai, F.Y.; Kaptoge, S.; Brozynska, M.; Wang, T.; et al. Genomic Risk Prediction of Coronary Artery Disease in 480,000 Adults: Implications for Primary Prevention. J. Am. Coll. Cardiol. 2018, 72, 1883–1893. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, G.; Wang, C.; Wang, X.; Qian, Z.M.; Cai, M.; Vaughn, M.G.; Bingheim, E.; Li, H.; Gao, Y.; et al. Associations of risk factor burden and genetic predisposition with the 10-year risk of atrial fibrillation: Observations from a large prospective study of 348,904 participants. BMC Med. 2023, 21, 88. [Google Scholar] [CrossRef]
- Marston, N.A.; Garfinkel, A.C.; Kamanu, F.K.; Melloni, G.M.; Roselli, C.; Jarolim, P.; Berg, D.D.; Bhatt, D.L.; Bonaca, M.P.; Cannon, C.P.; et al. A polygenic risk score predicts atrial fibrillation in cardiovascular disease. Eur. Heart J. 2023, 44, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Kloosterman, M.; Santema, B.T.; Roselli, C.; Nelson, C.P.; Koekemoer, A.; Romaine, S.P.R.; Van Gelder, I.C.; Lam, C.S.P.; Artola, V.A.; Lang, C.C.; et al. Genetic risk and atrial fibrillation in patients with heart failure. Eur. J. Heart Fail. 2020, 22, 519–527. [Google Scholar] [CrossRef]
- Okubo, Y.; Nakano, Y.; Ochi, H.; Onohara, Y.; Tokuyama, T.; Motoda, C.; Amioka, M.; Hironobe, N.; Okamura, S.; Ikeuchi, Y.; et al. Predicting atrial fibrillation using a combination of genetic risk score and clinical risk factors. Heart Rhythm. 2020, 17, 699–705. [Google Scholar] [CrossRef]
- Homburger, J.R.; Neben, C.L.; Mishne, G.; Zhou, A.Y.; Kathiresan, S.; Khera, A.V. Low coverage whole genome sequencing enables accurate assessment of common variants and calculation of genome-wide polygenic scores. Genome Med. 2019, 11, 74. [Google Scholar] [CrossRef]
- Ramms, B.; Patel, S.; Nora, C.; Pessentheiner, A.R.; Chang, M.W.; Green, C.R.; Golden, G.J.; Secrest, P.; Krauss, R.M.; Metallo, C.M.; et al. ApoC-III ASO promotes tissue LPL activity in the absence of apoE-mediated TRL clearance. J. Lipid Res. 2019, 60, 1379–1395. [Google Scholar] [CrossRef]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.C.; Baum, S.J.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. AKCEA-APO(a)-LRx Study Investigators. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef]
- Hegele, R.A.; Tsimikas, S. Lipid-Lowering Agents. Circ. Res. 2019, 124, 386–404. [Google Scholar] [CrossRef] [PubMed]
- Gales, L. Tegsedi (Inotersen): An Antisense Oligonucleotide Approved for the Treatment of Adult Patients with Hereditary Transthyretin Amyloidosis. Pharmaceuticals 2019, 12, 78. [Google Scholar] [CrossRef]
- Gertz, M.A.; Scheinberg, M.; Waddington-Cruz, M.; Heitner, S.B.; Karam, C.; Drachman, B.; Khella, S.; Whelan, C.; Obici, L. Inotersen for the treatment of adults with polyneuropathy caused by hereditary transthyretin-mediated amyloidosis. Expert. Rev. Clin. Pharmacol. 2019, 12, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Robinson, E.L.; Port, J.D. Utilization and Potential of RNA-Based Therapies in Cardiovascular Disease. JACC Basic. Transl. Sci. 2022, 7, 956–969. [Google Scholar] [CrossRef]
- Landmesser, U.; Poller, W.; Tsimikas, S.; Most, P.; Paneni, F.; Lüscher, T.F. From traditional pharmacological towards nucleic acid-based therapies for cardiovascular diseases. Eur. Heart J. 2020, 41, 3884–3899. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Witztum, J.L.; Bennett, F.; Baker, B.F. RNA-Targeted Therapeutics. Cell Metab. 2019, 29, 501. [Google Scholar] [CrossRef] [PubMed]
- Viney, N.J.; van Capelleveen, J.C.; Geary, R.S.; Xia, S.; Tami, J.A.; Yu, R.; Marcovina, S.M.; Hughes, S.G.; Graham, M.J.; Crooke, R.M.; et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): Two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016, 388, 2239–2253. [Google Scholar] [CrossRef]
- Levin, A.A. Treating disease at the RNA level with oligonucleotides. N. Engl. J. Med. 2019, 380, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Krichevsky, A.M.; Uhlmann, E.J. Oligonucleotide therapeutics as a new class of drugs for malignant brain tumors: Targeting mRNAs, regulatory RNAs, mutations, combinations, and beyond. Neurotherapeutics 2019, 16, 319–347. [Google Scholar] [CrossRef]
- Farkas, N.; Scaria, P.V.; Woodle, M.C.; Dagata, J.A. Physical-chemical measurement method development for self-assembled, core-shell nanoparticles. Sci. Rep. 2019, 9, 1655. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Jacobson, S.G.; Drack, A.V.; Ho, A.C.; Charng, J.; Garafalo, A.V.; Roman, A.J.; Sumaroka, A.; Han, I.C.; Hochstedler, M.D.; et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat. Med. 2019, 25, 225–228. [Google Scholar] [CrossRef]
- Witztum, J.L.; Gaudet, D.; Freedman, S.D.; Alexander, V.J.; Digenio, A.; Williams, K.R.; Yang, Q.; Hughes, S.G.; Geary, R.S.; Arca, M.; et al. Volanesorsen and Triglyceride Levels in Familial Chylomicronemia Syndrome. N. Engl. J. Med. 2019, 381, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.J.; Lee, R.G.; Brandt, T.A.; Tai, L.J.; Fu, W.; Peralta, R.; Yu, R.; Hurh, E.; Paz, E.; McEvoy, B.W.; et al. Cardiovascular and Metabolic Effects of ANGPTL3 Antisense Oligonucleotides. N. Engl. J. Med. 2017, 377, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, D.; Brisson, D.; Tremblay, K.; Alexander, V.J.; Singleton, W.; Hughes, S.G.; Geary, R.S.; Baker, B.F.; Graham, J.; Croo, R.M.; et al. Targeting APOC3 in the familial chylomicronemia syndrome. N. Engl. J. Med. 2014, 371, 2200–2206. [Google Scholar] [CrossRef] [PubMed]
- Alexander, V.J.; Xia, S.; Hurh, E.; Hughes, S.G.; O’Dea, L.; Geary, R.S.; Witztum, J.L.; Tsimikas, S. N-acetyl galactosamine-conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur. Heart J. 2019, 40, 2785–2796. [Google Scholar] [CrossRef]
- Stitziel, N.O.; Khera, A.V.; Wang, X.; Bierhals, A.J.; Vourakis, A.C.; Sperry, A.E.; Natarajan, P.; Klarin, D.; Emdin, C.A.; Zekavat, S.M.; et al. ANGPTL3 Deficiency and Protection Against Coronary Artery Disease. J. Am. Coll. Cardiol. 2017, 69, 2054–2063. [Google Scholar] [CrossRef]
- Minicocci, I.; Santini, S.; Cantisani, V.; Stitziel, N.; Kathiresan, S.; Arroyo, J.A.; Martí, G.; Pisciotta, L.; Noto, D.; Cefalù, A.B.; et al. Clinical characteristics and plasma lipids in subjects with familial combined hypolipidemia: A pooled analysis. J. Lipid Res. 2013, 54, 3481–3490. [Google Scholar] [CrossRef]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Singh, A.; Falk, R.H. ‘A new staging system for cardiac transthyretin amyloidosis’: Is it already on the verge of obsolescence? Eur. Heart J. 2018, 39, 2807–2809. [Google Scholar] [CrossRef]
- Siegismund, C.S.; Escher, F.; Lassner, D.; Kühl, U.; Gross, U.; Fruhwald, F.; Wenzel, P.; Münzel, T.; Frey, N.; Linke, R.P.; et al. Intramyocardial inflammation predicts adverse outcome in patients with cardiac AL amyloidosis. Eur. J. Heart Fail. 2018, 20, 751–757. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Qureshi, A.; Ghosh, S.; Ashish, K.; Heise, L.R.; Hajra, A.; Ghosh, R.K. Safety and Efficacy of Extremely Low LDL-Cholesterol Levels and Its Prospects in Hyperlipidemia Management. J. Lipids 2018, 23, 8598054. [Google Scholar] [CrossRef]
- Nishikido, T.; Ray, K.K. Inclisiran for the treatment of dyslipidemia. Expert Opin. Investig. Drugs 2018, 27, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Nicholls, S.J.; Langsted, A.; Ray, K.K.; Tybjærg-Hansen, A. Advances in lipid-lowering therapy through gene-silencing technologies. Nat. Rev. Cardiol. 2018, 15, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Solomon, S.D.; Adams, D.; Kristen, A.; Grogan, M.; González-Duarte, A.; Maurer, M.S.; Merlini, G.; Damy, T.; Slama, M.S.; Brannagan, T.H., 3rd; et al. Effects of Patisiran, an RNA Interference Therapeutic, on Cardiac Parameters in Patients with Hereditary Transthyretin-Mediated Amyloidosis. Circulation 2019, 13, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Poller, W.; Dimmeler, S.; Heymans, S.; Zeller, T.; Haas, J.; Karakas, M.; Leistner, D.M.; Jakob, P.; Nakagawa, S.; Blankenberg, S.; et al. Non-coding RNAs in cardiovascular diseases: Diagnostic and therapeutic perspectives. Eur. Heart J. 2018, 39, 2704–2716. [Google Scholar] [CrossRef]
- Gebert, L.F.R.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef]
- van Rooij, E.; Purcell, A.L.; Levin, A.A. Developing microRNA therapeutics. Circ. Res. 2012, 110, 496–507. [Google Scholar] [CrossRef]
- Barwari, T.; Joshi, A.; Mayr, M. MicroRNAs in cardiovascular disease. J. Am. Coll. Cardiol. 2016, 68, 2577–2584. [Google Scholar] [CrossRef]
- Rogg, E.M.; Abplanalp, W.T.; Bischof, C.; John, D.; Schulz, M.H.; Krishnan, J.; Fischer, A.; Poluzzi, C.; Schaefer, L.; Bonauer, A.; et al. Analysis of Cell Type-Specific Effects of MicroRNA-92a Provides Novel Insights into Target Regulation and Mechanism of Action. Circulation 2018, 138, 2545–2558. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantt, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef]
- Sassi, Y.; Avramopoulos, P.; Ramanujam, D.; Grüter, L.; Werfel, S.; Giosele, S.; Brunner, A.D.; Esfandyari, D.; Papadopoulou, A.S.; De Strooper, B.; et al. Cardiac myocyte miR-29 promotes pathological remodeling of the heart by activating Wnt signaling. Nat. Commun. 2017, 20, 1614. [Google Scholar] [CrossRef] [PubMed]
- Janssen, H.L.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef]
- Hanna, J.; Hossain, G.S.; Kocerha, J. The Potential for microRNA Therapeutics and Clinical Research. Front. Genet. 2019, 10, 478. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther. Nucleic Acids 2017, 8, 132–143. [Google Scholar] [CrossRef]
- Hoffman, M.M.; Ernst, J.; Wilder, S.P.; Kundaje, A.; Harris, R.S.; Libbrecht, M.; Giardine, B.; Ellenbogen, P.M.; Bilmes, J.A.; Birney, E.; et al. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 2013, 41, 827–841. [Google Scholar] [CrossRef]
- Siggens, L.; Ekwall, K. Epigenetics, chromatin and genome organization: Recent advances from the ENCODE project. J. Intern. Med. 2014, 276, 201–214. [Google Scholar] [CrossRef]
- The ENCODE Project Consortium. Perspectives on ENCODE. Nature 2020, 583, 693–698. [Google Scholar] [CrossRef]
- Costantino, S.; Mohammed, S.A.; Ambrosini, S.; Paneni, F. Epigenetic processing in cardiometabolic disease. Atherosclerosis 2019, 281, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Heuslein, J.L.; Gorick, C.M.; Price, R.J. Epigenetic regulators of the revascularization response to chronic arterial occlusion. Cardiovasc. Res. 2019, 115, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Nicorescu, I.; Dallinga, G.M.; de Winther, M.P.J.; Stroes, E.S.G.; Bahjat, M. Potential epigenetic therapeutics for atherosclerosis treatment. Atherosclerosis 2019, 281, 189–197. [Google Scholar] [CrossRef]
- Meder, B.; Haas, J.; Sedaghat-Hamedani, F.; Kayvanpour, E.; Frese, K.; Lai, A.; Nietsch, R.; Scheiner, C.; Mester, S.; Bordalo, D.M.; et al. Epigenome-Wide Association Study Identifies Cardiac Gene Patterning and a Novel Class of Biomarkers for Heart Failure. Circulation 2017, 136, 1528–1544. [Google Scholar] [CrossRef] [PubMed]
- Cochran, A.G.; Conery, A.R.; Sims, R.J., 3rd. Bromodomains: A new target class for drug development. Nat. Rev. Drug Discov. 2019, 18, 609–628. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Roberts, J.M.; Seo, H.S.; Souza, A.; Paulk, J.; Scott, T.G.; DeAngelo, S.L.; Dhe-Paganon, S.; Bradner, J.E. Design and characterization of bivalent BET inhibitors. Nat. Chem. Biol. 2012, 12, 1089–1096. [Google Scholar] [CrossRef]
- Tsujikawa, L.M.; Fu, L.; Das, S.; Halliday, C.; Rakai, B.D.; Stotz, S.C.; Sarsons, C.D.; Gilham, D.; Daze, E.; Wasiak, S.; et al. Apabetalone (RVX-208) reduces vascular inflammation in vitro and in CVD patients by a BET-dependent epigenetic mechanism. Clin. Epigenetics 2019, 11, 102. [Google Scholar] [CrossRef]
- Shishikura, D.; Kataoka, Y.; Honda, S.; Takata, K.; Kim, S.W.; Andrews, J.; Psaltis, P.J.; Sweeney, M.; Kulikowski, E.; Johansson, J.; et al. The Effect of Bromodomain and Extra-Terminal Inhibitor Apabetalone on Attenuated Coronary Atherosclerotic Plaque: Insights from the ASSURE Trial. Am. J. Cardiovasc. Drugs 2019, 19, 49–57. [Google Scholar] [CrossRef]
- Chen, K.; Li, Y.; Xu, L.; Qian, Y.; Liu, N.; Zhou, C.; Liu, J.; Zhou, L.; Xu, Z.; Jia, R.; et al. Comprehensive insight into endothelial progenitor cell-derived extracellular vesicles as a promising candidate for disease treatment. Stem Cell Res. Ther. 2022, 13, 238. [Google Scholar] [CrossRef]
- Zhu, C.; Piao, Z.; Jin, L. HDAC5 inhibition attenuates ventricular remodeling and cardiac dysfunction. Orphanet J. Rare Dis. 2023, 18, 266. [Google Scholar] [CrossRef]
- Costantino, S.; Paneni, F.; Mitchell, K.; Mohammed, S.A.; Hussain, S.; Gkolfos, C.; Berrino, L.; Volpe, M.; Schwarzwald, C.; Lüscher, T.F.; et al. Hyperglycaemia-induced epigenetic changes drive persistent cardiac dysfunction via the adaptor p66Shc. Int. J. Cardiol. 2018, 268, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Kronlage, M.; Dewenter, M.; Grosso, J.; Fleming, T.; Oehl, U.; Lehmann, L.H.; Falcão-Pires, I.; Leite-Moreira, A.F.; Volk, N.; Gröne, H.J.; et al. O-GlcNAcylation of Histone Deacetylase 4 Protects the Diabetic Heart from Failure. Circulation 2019, 140, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Mayourian, J.; Ceholski, D.K.; Gonzalez, D.M.; Cashman, T.J.; Sahoo, S.; Hajjar, R.J.; Costa, K.D. Physiologic, Pathologic, and Therapeutic Paracrine Modulation of Cardiac Excitation-Contraction Coupling. Circ. Res. 2018, 22, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Weber, T.; Hajjar, R.J. Human Cardiac Gene Therapy. Circ. Res. 2018, 123, 601–613. [Google Scholar] [CrossRef]
- Ray, R.M.; Hansen, A.H.; Slott, S.; Taskova, M.; Astakhova, K.; Morris, K.V. Control of LDL Uptake in Human Cells by Targeting the LDLR Regulatory Long Non-coding RNA BM450697. Mol. Ther. Nucleic Acids 2019, 17, 264–276. [Google Scholar] [CrossRef]
- Lino Cardenas, C.L.; Kessinger, C.W.; Chou, E.; Ghoshhajra, B.; Yeri, A.S.; Das, S.; Weintraub, N.L.; Malhotra, R.; Jaffer, F.A.; Lindsay, M.E. HDAC9 complex inhibition improves smooth muscle-dependent stenotic vascular disease. JCI Insight 2019, 4, e124706. [Google Scholar] [CrossRef]
- Janssens, Y.; Wynendaele, E.; Vanden Berghe, W.; De Spiegeleer, B. Peptides as epigenetic modulators: Therapeutic implications. Clin. Epigenetics 2019, 11, 101. [Google Scholar] [CrossRef]
- Hytönen, E.; Laurema, A.; Kankkonen, H.; Miyanohara, A.; Kärjä, V.; Hujo, M.; Laham-Karam, N.; Ylä-Herttuala, S. Bile-duct proliferation as an unexpected side-effect after AAV2-LDLR gene transfer to rabbit liver. Sci. Rep. 2019, 9, 6934. [Google Scholar] [CrossRef]
- Rodriguez-Calvo, R.; Masana, L. Review of the scientific evolution of gene therapy for the treatment of homozygous familial hypercholesterolaemia: Past, present and future perspectives. J. Med. Genet. 2019, 56, 711–717. [Google Scholar] [CrossRef]
- Ylä-Herttuala, S.; Baker, A.H. Cardiovascular Gene Therapy: Past, Present, and Future. Mol. Ther. 2017, 25, 1095–1106. [Google Scholar] [CrossRef]
- Bezzerides, V.J.; Caballero, A.; Wang, S.; Ai, Y.; Hylind, R.J.; Lu, F.; Heims-Waldron, D.A.; Chambers, K.D.; Zhang, D.; Abrams, D.J.; et al. Gene Therapy for Catecholaminergic Polymorphic Ventricular Tachycardia by Inhibition of Ca2+/Calmodulin-Dependent Kinase II. Circulation 2019, 140, 405–419. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Scott, L., Jr.; Campbell, H.M.; Pan, X.; Alsina, K.M.; Reynolds, J.; Philippen, L.E.; Hulsurkar, M.; Lagor, W.R.; Li, N.; et al. Atrial-Specific Gene Delivery Using an Adeno-Associated Viral Vector. Circ. Res. 2019, 124, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Domenger, C.; Grimm, D. Next-generation AAV vectors-do not judge a virus (only) by its cover. Hum. Mol. Genet. 2019, 8, R3–R14. [Google Scholar] [CrossRef]
- Gan, L.M.; Lagerström-Fermér, M.; Carlsson, L.G.; Arfvidsson, C.; Egnell, A.C.; Rudvik, A.; Kjaer, M.; Collén, A.; Thompson, J.D.; Joyal, J.; et al. Intradermal delivery of modified mRNA encoding VEGF-A in patients with type 2 diabetes. Nat. Commun. 2019, 10, 871. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, A.J.; Romeo, F.J.; Mazurek, R.; Ishikawa, K. Barriers in Heart Failure Gene Therapy and Approaches to Overcome Them. Heart Lung Circ. 2023, 32, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, K.; Riyad, J.M.; Garnett, T.; Kohlbrenner, E.; Mookerjee, A.; Elmastour, F.; Benard, L.; Chen, J.; Vanden Driessche, T.; Chuah, M.K.; et al. A Calsequestrin Cis-Regulatory Motif Coupled to a Cardiac Troponin T Promoter Improves Cardiac Adeno-Associated Virus Serotype 9 Transduction Specificity. Hum. Gene Ther. 2018, 29, 927–937. [Google Scholar] [CrossRef]
- Raake, P.W.J.; Barthelmes, J.; Krautz, B.; Buss, S.; Huditz, R.; Schlegel, P.; Weber, C.; Stangassinger, M.; Haberkorn, U.; Katus, H.A.; et al. Comprehensive cardiac phenotyping in large animals: Comparison of pressure-volume analysis and cardiac magnetic resonance imaging in pig post-myocardial infarction systolic heart failure. Int. J. Cardiovasc. Imaging 2019, 35, 1691–1699. [Google Scholar] [CrossRef]
- Bauer, R.; Enns, H.; Jungmann, A.; Leuchs, B.; Volz, C.; Schinkel, S.; Koch, W.J.; Raake, P.W.; Most, P.; Katus, H.A.; et al. Various effects of AAV9-mediated βARKct gene therapy on the heart in dystrophin-deficient (mdx) mice and δ-sarcoglycan-deficient (Sgcd-/-) mice. Neuromuscul. Disord. 2019, 29, 231–241. [Google Scholar] [CrossRef]
- Lv, B.; He, S.; Li, P.; Jiang, S.; Li, D.; Lin, J.; Feinberg, M.W. MicroRNA-181 in cardiovascular disease: Emerging biomarkers and therapeutic targets. FASEB J. 2024, 38, e23635. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, L.; Wang, C.; Xie, X.; Han, Y. Research Progress of Long Non-Coding RNA in Tumor Drug Resistance: A New Paradigm. Drug Des. Devel Ther. 2024, 18, 1385–1398. [Google Scholar] [CrossRef]
- Patel, A.P.; Natarajan, P. Completing the genetic spectrum influencing coronary artery disease: From germline to somatic variation. Cardiovasc. Res. 2019, 115, 830–843. [Google Scholar] [CrossRef]
- Hoeksema, M.A.; Glass, C.K. Nature and nurture of tissue-specific macrophage phenotypes. Atherosclerosis 2019, 281, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wen, Y.; He, S.; Yang, X.; Zhang, H.; Zhu, H. Pipelines for cross-species and genome-wide prediction of long noncoding RNA binding. Nat. Protoc. 2019, 14, 795–818. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Fang, P.; Li, W.J.; Zhang, J.; Wang, G.P.; Jiang, D.F.; Chen, F.P. LncRNA NEAT1 sponges miR-214 to regulate M2 macrophage polarization by regulation of B7-H3 in multiple myeloma. Mol. Immunol. 2020, 117, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Qiu, H.; He, J.; Liu, L.; Xue, W.; Fox, A.; Tickner, J.; Xu, J. The emerging roles of hnRNPK. J. Cell Physiol. 2020, 235, 1995–2008. [Google Scholar] [CrossRef]
- Zhang, P.; Wu, W.; Chen, Q.; Chen, M. Non-Coding RNAs and their Integrated Networks. J. Integr. Bioinform. 2019, 16, 20190027. [Google Scholar] [CrossRef]
- Winkels, H.; Gerdes, N.; Kahles, F.; Dutta, P.; Giannarelli, C.; Wolf, D. Editorial: Immune and autoimmune mechanisms in cardiovascular disease. Front. Cardiovasc. Med. 2023, 9, 1125959. [Google Scholar] [CrossRef]
- Coronary Artery Disease (C4D) Genetics Consortium. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat. Genet. 2011, 43, 339–344. [Google Scholar] [CrossRef]
- Koyama, S.; Ito, K.; Terao, C.; Akiyama, M.; Horikoshi, M.; Momozawa, Y.; Matsunaga, H.; Ieki, H.; Ozaki, K.; Onouchi, Y.; et al. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat. Genet. 2020, 52, 1169–1177. [Google Scholar] [CrossRef]
- Huang, H.; Verma, J.; Mok, V.; Bharadwaj, H.R.; Alrawashdeh, M.M.; Aratikatla, A.; Sudan, S.; Talukder, S.; Habaka, M.; Tse, G.; et al. Exploring Health Care Disparities in Genetic Testing and Research for Hereditary Cardiomyopathy: Current State and Future Perspectives. Glob. Med. Genet. 2024, 11, 36–47. [Google Scholar] [CrossRef]
| Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).


{kind=link}
{kind=link}