Risk of Cardiac Arrhythmias in Patients with Late-Onset Pompe Disease—Results from a Long Follow-Up in a Group of 12 Patients and Review of Literature

 and
and 
Abstract
1. Introduction
1.1. Clinical Features
1.2. Diagnosis and Treatment
2. Materials and Methods
2.1. Patients
2.2. Methods
2.3. Review of the Literature, Search and Selection
3. Results
4. Discussion
Review of Literature
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hannah, W.B.; Derks, T.G.J.; Drumm, M.L.; Grünert, S.C.; Kishnani, P.S.; Vissing, J. Glycogen storage diseases. Nat. Rev. Dis. Prim. 2023, 9, 46. [Google Scholar] [CrossRef]
- Hirschhorn, R.; Reuser, A. Glycogen storage disease type II: Acid alpha-glucosidase (acid maltase) deficiency. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C., Beaudet, A., Sly, W., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; Volume 3, pp. 3389–3420. [Google Scholar]
- Reuser, A.J.J.; Kroos, M.A.; Hermans, M.M.P.; Bijvoet, A.G.A.; Verbeet, M.P.; Van Diggelen, O.P.; Kleijer, W.J.; Van Der Ploeg, A.T. Glycogenosis type II (acid maltase deficiency). Muscle Nerve 1995, 18, S61–S69. [Google Scholar] [CrossRef]
- Di Rocco, M.D.; Buzzi, D.; Tarò, M. Glycogen storage disease type II: Clinical overview. Acta Myol. 2007, XXVI, 42–44. [Google Scholar]
- Kroos, M.; Pomponio, R.J.; van Vliet, L.; Palmer, R.E.; Phipps, M.; Van der Helm, R.; Halley, D.; Reuser, A.; The GAA Database Consortium. Update of the Pompe disease mutation database with 107 sequence variants and a format for severity rating. Hum. Mutat. 2008, 29, E13–E26. [Google Scholar] [CrossRef] [PubMed]
- Reuser, A.J.J.; Ploeg, A.T.; Chien, Y.; Llerena, J.; Abbott, M.; Clemens, P.R.; Kimonis, V.E.; Leslie, N.; Maruti, S.S.; Sanson, B.; et al. GAA variants and phenotypes among 1,079 patients with Pompe disease: Data from the Pompe Registry. Hum. Mutat. 2019, 40, 2146–2164. [Google Scholar] [CrossRef] [PubMed]
- Niño, M.Y.; In’t Groen, S.L.; Bergsma, A.J.; van der Beek, N.A.; Kroos, M.; Hoogeveen-Westerveld, M.; Van Der Ploeg, A.T.; Pijnappel, W.P. Extension of the Pompe mutation database by linking disease-associated variants to clinical severity. Hum. Mutat. 2019, 40, 1954–1967. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, Y.; Fuji, N.; Yamazaki, N.; Hirakiyama, A.; Kamioka, T.; Seo, J.-H.; Mashima, R.; Kosuga, M.; Okuyama, T. A molecular analysis of the GAA gene and clinical spectrum in 38 patients with Pompe disease in Japan. Mol. Genet. Metab. Rep. 2017, 14, 3–9. [Google Scholar] [CrossRef] [PubMed]
- De Filippi, P.; Errichiello, E.; Toscano, A.; Mongini, T.; Moggio, M.; Ravaglia, S.; Filosto, M.; Servidei, S.; Musumeci, O.; Giannini, F.; et al. Distribution of Exonic Variants in Glycogen Synthesis and Catabolism Genes in Late Onset Pompe Disease (LOPD). Curr. Issues Mol. Biol. 2023, 45, 2847–2860. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, S. Trafficking of lysosomal enzymes in normal and disease states. J. Clin. Investig. 1986, 77, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wisselaar, H.A.; Kroos, M.A.; Hermans, M.M.; van Beeumen, J.; Reuser, A.J. Structural and functional changes of lysosomal acid al-pha-glucosidase during intracellular transport and maturation. J. Biol. Chem. 1993, 268, 2223–2231. [Google Scholar] [CrossRef]
- Dasouki, M.; Jawdat, O.; Almadhoun, O.; Pasnoor, M.; McVey, A.L.; Abuzinadah, A.; Herbelin, L.; Barohn, R.J.; Dimachkie, M.M. Pompe Disease: Literature review and case series. Neurol. Clin. 2014, 32, 751–776. [Google Scholar] [CrossRef] [PubMed]
- Malicdan, M.C.V.; Nishino, I. Autophagy in Lysosomal Myopathies. Brain Pathol. 2011, 22, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Raben, N.; Wong, A.; Ralston, E.; Myerowitz, R. Autophagy and mitochondria in Pompe disease: Nothing is so new as what has long been forgotten. Am. J. Med. Genet. Part C Semin. Med. Genet. 2012, 160C, 13–21. [Google Scholar] [CrossRef] [PubMed]
- van den Hout, H.M.; Hop, W.; van Diggelen, O.P.; Smeitink, J.A.M.; Smit, G.P.A.; Poll-The, B.-T.T.; Bakker, H.D.; Loonen, M.C.B.; De Klerk, J.B.C.; Reuser, A.J.J.; et al. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics 2003, 112, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Hwu, W.-L.; Mandel, H.; Nicolino, M.; Yong, F.; Corzo, D.; Infantile-Onset Pompe Disease Natural History Study Group. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J. Pediatr. 2006, 148, 671–676.e2. [Google Scholar] [CrossRef] [PubMed]
- Winkel, L.P.F.; Hagemans, M.L.C.; Doorn, P.A.; Loonen, M.C.B.; Hop, W.J.C.; Reuser, A.J.J.; Ploeg, A.T. The natural course of non–classic Pompe’s disease; a review of 225 published cases. J. Neurol. 2005, 252, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Toscano, A.; Rodolico, C.; Musumeci, O. Multisystem late onset Pompe disease (LOPD): An update on clinical aspects. Ann. Transl. Med. 2019, 7, 284. [Google Scholar] [CrossRef]
- Hagemans, M.L.C.; Winkel, L.P.F.; Van Doorn, P.A.; Hop, W.J.C.; Loonen, M.C.B.; Reuser, A.J.J.; Van der Ploeg, A.T. Clinical manifestation and natural course of late-onset Pompe’s disease in 54 Dutch patients. Brain 2005, 128, 671–677. [Google Scholar] [CrossRef]
- Filosto, M.; Todeschini, A.; Cotelli, M.S.; Vielmi, V.; Rinaldi, F.; Rota, S.; Scarpelli, M.; Padovani, A. Non-muscle involvement in late-onset Glycogenosis II. Acta Myol. 2013, XXXII, 91–94. [Google Scholar]
- Labella, B.; Piccinelli, S.C.; Risi, B.; Caria, F.; Damioli, S.; Bertella, E.; Poli, L.; Padovani, A.; Filosto, M. A Comprehensive Update on Late-Onset Pompe Disease. Biomolecules 2023, 13, 1279. [Google Scholar] [CrossRef]
- Preisler, N.; Lukacs, Z.; Vinge, L.; Madsen, K.L.; Husu, E.; Hansen, R.S.; Duno, M.; Andersen, H.; Laub, M.; Vissing, J. Late-onset Pompe disease is prevalent in unclassified limb-girdle muscular dystrophies. Mol. Genet. Metab. 2013, 110, 287–289. [Google Scholar] [CrossRef]
- Lukacs, Z.; Cobos, P.N.; Wenninger, S.; Willis, T.A.; Guglieri, M.; Roberts, M.; Quinlivan, R.; Hilton-Jones, D.; Evangelista, T.; Zierz, S.; et al. Prevalence of Pompe disease in 3,076 patients with hyperCKemia and limb-girdle muscular weakness. Neurology 2016, 87, 295–298. [Google Scholar] [CrossRef]
- Silvestri, N.J.; Wolfe, G.I. Asymptomatic/pauci-symptomatic creatine kinase elevations (hyperckemia). Muscle Nerve 2013, 47, 805–815. [Google Scholar] [CrossRef]
- Morandi, L.; Angelini, C.; Prelle, A.; Pini, A.; Grassi, B.; Bernardi, G.; Politano, L.; Bruno, C.; De Grandis, D.; Cudia, P.; et al. High plasma creatine kinase: Review of the literature and proposal for a diagnostic algorithm. Neurol. Sci. 2006, 27, 303–311. [Google Scholar] [CrossRef]
- Kyriakides, T.; Angelini, C.; Schaefer, J.; Sacconi, S.; Siciliano, G.; Vilchez, J.J.; Hilton-Jones, D. EFNS guidelines on the diagnostic approach to pauci- or asymptomatic hyperCKemia. Eur. J. Neurol. 2010, 17, 767–773. [Google Scholar] [CrossRef]
- Berger, K.I.; Chan, Y.; Rom, W.N.; Oppenheimer, B.W.; Goldring, R.M. Progression from respiratory dysfunction to failure in late-onset Pompe disease. Neuromuscul. Disord. 2016, 26, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Mellies, U.; Stehling, F.; Dohna-Schwake, C.; Ragette, R.; Teschler, H.; Voit, T. Respiratory failure in Pompe disease: Treatment with noninvasive ventilation. Neurology 2005, 64, 1465–1467. [Google Scholar] [CrossRef] [PubMed]
- Boentert, M.; Prigent, H.; Várdi, K.; Jones, H.N.; Mellies, U.; Simonds, A.K.; Wenninger, S.; Cortés, E.B.; Confalonieri, M. Practical Recommendations for Diagnosis and Management of Respiratory Muscle Weakness in Late-Onset Pompe Disease. Int. J. Mol. Sci. 2016, 17, 1735. [Google Scholar] [CrossRef]
- DeRuisseau, L.R.; Fuller, D.D.; Qiu, K.; DeRuisseau, K.C.; Donnelly, W.H.; Mah, C.; Reier, P.J.; Byrne, B.J. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc. Natl. Acad. Sci. USA 2009, 106, 9419–9424. [Google Scholar] [CrossRef] [PubMed]
- Soliman, O.I.I.; Van Der Beek, N.A.M.E.; Van Doorn, P.A.; Vletter, W.B.; Nemes, A.; Van Dalen, B.M.; Cate, F.J.T.; Van Der Ploeg, A.T.; Geleijnse, M.L. Cardiac involvement in adults with Pompe disease. J. Intern. Med. 2008, 264, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Herbert, M.; Cope, H.; Li, J.S.; Kishnani, P.S. Severe Cardiac Involvement Is Rare in Patients with Late-Onset Pompe Disease and the Common c.-32-13T>G Variant: Implications for Newborn Screening. J. Pediatr. 2018, 198, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-H.; Qiu, W.-J.; Lee, J.; Chien, Y.-H.; Hwu, W.-L.; Zschocke, J.; Gibson, K.M. Hypertrophic Cardiomyopathy in Pompe Disease Is Not Limited to the Classic Infantile-Onset Phenotype. JIMD Rep. 2014, 17, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Sacconi, S.; Wahbi, K.; Theodore, G.; Garcia, J.; Salviati, L.; Bouhour, F.; Vial, C.; Duboc, D.; Laforêt, P.; Desnuelle, C. Atrio-ventricular block requiring pacemaker in patients with late onset Pompe disease. Neuromuscul. Disord. 2014, 24, 648–650. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Bailey, L.A.; Estrada, J.; Rehder, C.W.; Li, J.S.; Rogers, J.G.; Bali, D.S.; Buckley, A.F.; Kishnani, P.S. Severe Cardiomyopathy as the Isolated Presenting Feature in an Adult with Late-Onset Pompe Disease: A Case Report. JIMD Rep. 2016, 31, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Walczak-Galezewska, M.; Skrypnik, D.; Szulinska, M.; Musialik, K.M.; Skrypnik, K.; Bogdanski, P. Late-onset Pompe disease in a 54 year-old sportsman with an episode of syncope: A case report. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3665–3667. [Google Scholar]
- Hossain, M.A.; Miyajima, T.; Akiyama, K.; Eto, Y. A Case of Adult-onset Pompe Disease with Cerebral Stroke and Left Ventricular Hypertrophy. J. Stroke Cerebrovasc. Dis. 2018, 27, 3046–3052. [Google Scholar] [CrossRef]
- Wens, S.C.A.; Kuperus, E.; Mattace-Raso, F.U.S.; Kruijshaar, M.E.; Brusse, E.; van Montfort, K.C.A.G.M.; de Boer, M.S.; Sijbrands, E.J.G.; van der Ploeg, A.T.; van Doorn, P.A. Increased aortic stiffness and blood pressure in non-classic Pompe disease. J. Inherit. Metab. Dis. 2014, 37, 391–397. [Google Scholar] [CrossRef]
- van der Beek, N.; Soliman, O.; van Capelle, C.; Geleijnse, M.; Vletter, W.; Kroos, M.; Reuser, A.; Frohn-Mulder, I.; van Doorn, P.; van der Ploeg, A. Cardiac evaluation in children and adults with Pompe disease sharing the common c.−32-13T>G genotype rarely reveals abnormalities. J. Neurol. Sci. 2008, 275, 46–50. [Google Scholar] [CrossRef]
- Bembi, B.; Cerini, E.; Danesino, C.; Donati, M.A.; Gasperini, S.; Morandi, L.; Musumeci, O.; Parenti, G.; Ravaglia, S.; Seidita, F.; et al. Diagnosis of glycogenosis type II. Neurology 2008, 71, S4–S11. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Steiner, R.D.; Bali, D.; Berger, K.; Byrne, B.J.; Case, L.E.; Crowley, J.F.; Downs, S.; Howell, R.R.; Kravitz, R.M.; et al. Pompe disease diagnosis and management guideline. Genet. Med. 2006, 8, 267–288. [Google Scholar] [CrossRef]
- Musumeci, O.; Toscano, A. Diagnostic tools in late onset Pompe disease (LOPD). Ann. Transl. Med. 2019, 7, 286. [Google Scholar] [CrossRef] [PubMed]
- Ozdamar, S.E.; Koc, A.F.; Tekce, H.D.; Kotan, D.; Ekmekci, A.H.; Sengun, I.S.; Yuceyar, A.N.; Uluc, K. Expert opinion on the diagnostic odyssey and management of late-onset Pompe disease: A neurologist’s perspective. Front. Neurol. 2023, 14, 1095134. [Google Scholar] [CrossRef] [PubMed]
- Er, T.-K.; Chen, C.-C.; Chien, Y.-H.; Liang, W.-C.; Kan, T.-M.; Jong, Y.-J. Development of a feasible assay for the detection of GAA mutations in patients with Pompe disease. Clin. Chim. Acta 2014, 429, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Savarese, M.; Torella, A.; Musumeci, O.; Angelini, C.; Astrea, G.; Bello, L.; Bruno, C.; Comi, G.P.; Di Fruscio, G.; Piluso, G.; et al. Targeted gene panel screening is an effective tool to identify undiagnosed late onset Pompe disease. Neuromuscul. Disord. 2018, 28, 586–591. [Google Scholar] [CrossRef] [PubMed]
- van der Meijden, J.C.; Kruijshaar, M.E.; Harlaar, L.; Rizopoulos, D.; van der Beek, N.A.M.E.; van der Ploeg, A.T. Long-term follow-up of 17 patients with childhood Pompe disease treated with enzyme replacement therapy. J. Inherit. Metab. Dis. 2018, 41, 1205–1214. [Google Scholar] [CrossRef]
- Kuperus, E.; Kruijshaar, M.E.; Wens, S.C.; de Vries, J.M.; Favejee, M.M.; van der Meijden, J.C.; Rizopoulos, D.; Brusse, E.; van Doorn, P.A.; van der Ploeg, A.T.; et al. Long-term benefit of enzyme replacement therapy in Pompe disease: A 5-year prospective study. Neurology 2017, 89, 2365–2373. [Google Scholar] [CrossRef] [PubMed]
- Parini, R.; De Lorenzo, P.; Dardis, A.; Burlina, A.; Cassio, A.; Cavarzere, P.; Concolino, D.; Della Casa, R.; Deodato, F.; Donati, M.A.; et al. Long term clinical history of an Italian cohort of infantile onset Pompe disease treated with enzyme replacement therapy. Orphanet J. Rare Dis. 2018, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- van der Ploeg, A.T.; Clemens, P.R.; Corzo, D.; Escolar, D.M.; Florence, J.; Groeneveld, G.J.; Herson, S.; Kishnani, P.S.; Laforet, P.; Lake, S.L.; et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N. Engl. J. Med. 2010, 362, 1396–1406. [Google Scholar] [CrossRef]
- Strothotte, S.; Strigl-Pill, N.; Grunert, B.; Kornblum, C.; Eger, K.; Wessig, C.; Deschauer, M.; Breunig, F.; Glocker, F.X.; Vielhaber, S.; et al. Enzyme replacement therapy with alglucosidase alfa in 44 patients with late-onset glycogen storage disease type 2: 12-month results of an observational clinical trial. J. Neurol. 2009, 257, 91–97. [Google Scholar] [CrossRef]
- van Capelle, C.I.; Poelman, E.; Frohn-Mulder, I.M.; Koopman, L.P.; Hout, J.M.v.D.; Régal, L.; Cools, B.; Helbing, W.A.; van der Ploeg, A.T. Cardiac outcome in classic infantile Pompe disease after 13 years of treatment with recombinant human acid alpha-glucosidase. Int. J. Cardiol. 2018, 269, 104–110. [Google Scholar] [CrossRef]
- Lecis, M.; Rossi, K.; Guerzoni, M.E.; Mariotti, I.; Iughetti, L. Enzyme Replacement Therapy (ERT) on Heart Function Changes the Outcome in Patients with Infantile-Onset Pompe Disease: A Familial History. Case Rep. Pediatr. 2023, 2023, 8470341. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-F.; Yang, C.C.; Liao, H.-C.; Huang, L.-Y.; Chiang, C.-C.; Ho, H.-C.; Lai, C.-J.; Chu, T.-H.; Yang, T.-F.; Hsu, T.-R.; et al. Very Early Treatment for Infantile-Onset Pompe Disease Contributes to Better Outcomes. J. Pediatr. 2016, 169, 174–180.e1. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, M.A.; Almalik, M.H.; Mirghani, H.M. Early administration of enzyme replacement therapy for Pompe disease: Short-term follow-up results. J. Inherit. Metab. Dis. 2008, 31 (Suppl. S2), 431–436. [Google Scholar] [CrossRef] [PubMed]
- Winkel, L.P.F.; Hout, J.M.P.V.D.; Kamphoven, J.H.J.; Disseldorp, J.A.M.; Remmerswaal, M.; Arts, W.F.M.; Loonen, M.C.B.; Vulto, A.G.; Van Doorn, P.A.; De Jong, G.; et al. Enzyme replacement therapy in late-onset Pompe’s disease: A three-year follow-up. Ann. Neurol. 2004, 55, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Molnár, M.J.; Oláh, B.B.; Visy, K.V.; Grosz, Z.; Sebők, Á.; Dézsi, L.; Almássy, Z.; Kerényi, L.; Jobbágy, Z.; Jávor, L.; et al. A késői kezdetű Pompe-kórban szenvedők enzimpótló kezelésének hosszú távú követése [The long-term follow-up of enzyme replacement treatment in late onset Pompe disease]. Ideggyogy Sz 2020, 73, 151–159. (In Hungarian) [Google Scholar] [CrossRef] [PubMed]
- Winkler, M.; von Landenberg, C.; Kuchenbecker, K.; Reimann, J.; Kornblum, C. Long-term effects of enzyme replacement therapy in an elderly cohort of late-onset Pompe disease. Neuromuscul. Disord. 2022, 32, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Semplicini, C.; De Antonio, M.; Taouagh, N.; Béhin, A.; Bouhour, F.; Echaniz-Laguna, A.; Magot, A.; Nadaj-Pakleza, A.; Orlikowski, D.; Sacconi, S.; et al. Long-term benefit of enzyme replacement therapy with alglucosidase alfa in adults with Pompe disease: Prospective analysis from the French Pompe Registry. J. Inherit. Metab. Dis. 2020, 43, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal Storage Diseases: From Pathophysiology to Therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef]
- van Gelder, C.M.; Hoogeveen-Westerveld, M.; Kroos, M.A.; Plug, I.; van der Ploeg, A.T.; Reuser, A.J.J. Enzyme therapy and immune response in relation to CRIM status: The Dutch experience in classic infantile Pompe disease. J. Inherit. Metab. Dis. 2014, 38, 305–314. [Google Scholar] [CrossRef]
- Douillard-Guilloux, G.; Raben, N.; Takikita, S.; Ferry, A.; Vignaud, A.; Guillet-Deniau, I.; Favier, M.; Thurberg, B.L.; Roach, P.J.; Caillaud, C.; et al. Restoration of muscle functionality by genetic suppression of glycogen synthesis in a murine model of Pompe disease. Hum. Mol. Genet. 2009, 19, 684–696. [Google Scholar] [CrossRef]
- Xiao, J.; Westbroek, W.; Motabar, O.; Lea, W.A.; Hu, X.; Velayati, A.; Zheng, W.; Southall, N.; Gustafson, A.M.; Goldin, E.; et al. Discovery of a Novel Noniminosugar Acid α Glucosidase Chaperone Series. J. Med. Chem. 2012, 55, 7546–7559. [Google Scholar] [CrossRef]
- Clayton, N.P.; Nelson, C.A.; Weeden, T.; Taylor, K.M.; Moreland, R.J.; Scheule, R.K.; Phillips, L.; Leger, A.J.; Cheng, S.H.; Wentworth, B.M. Antisense Oligonucleotide-mediated Suppression of Muscle Glycogen Synthase 1 Synthesis as an Approach for Substrate Reduction Therapy of Pompe Disease. Mol. Ther. Nucleic Acids 2014, 3, e206. [Google Scholar] [CrossRef]
- Borie-Guichot, M.; Tran, M.L.; Génisson, Y.; Ballereau, S.; Dehoux, C. Pharmacological Chaperone Therapy for Pompe Disease. Molecules 2021, 26, 7223. [Google Scholar] [CrossRef] [PubMed]
- Kroos, M.A.; Pomponio, R.J.; Hagemans, M.L.; Keulemans, J.; Phipps, M.; DeRiso, M.; Palmer, R.E.; Ausems, M.G.; Van der Beek, N.A.; Van Diggelen, O.P.; et al. Broad spectrum of Pompe disease in patients with the same c.-32-13T->G haplotype. Neurology 2007, 68, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Boentert, M.; Florian, A.; Dräger, B.; Young, P.; Yilmaz, A. Pattern and prognostic value of cardiac involvement in patients with late-onset pompe disease: A comprehensive cardiovascular magnetic resonance approach. J. Cardiovasc. Magn. Reson. 2016, 18, 91. [Google Scholar] [CrossRef] [PubMed]
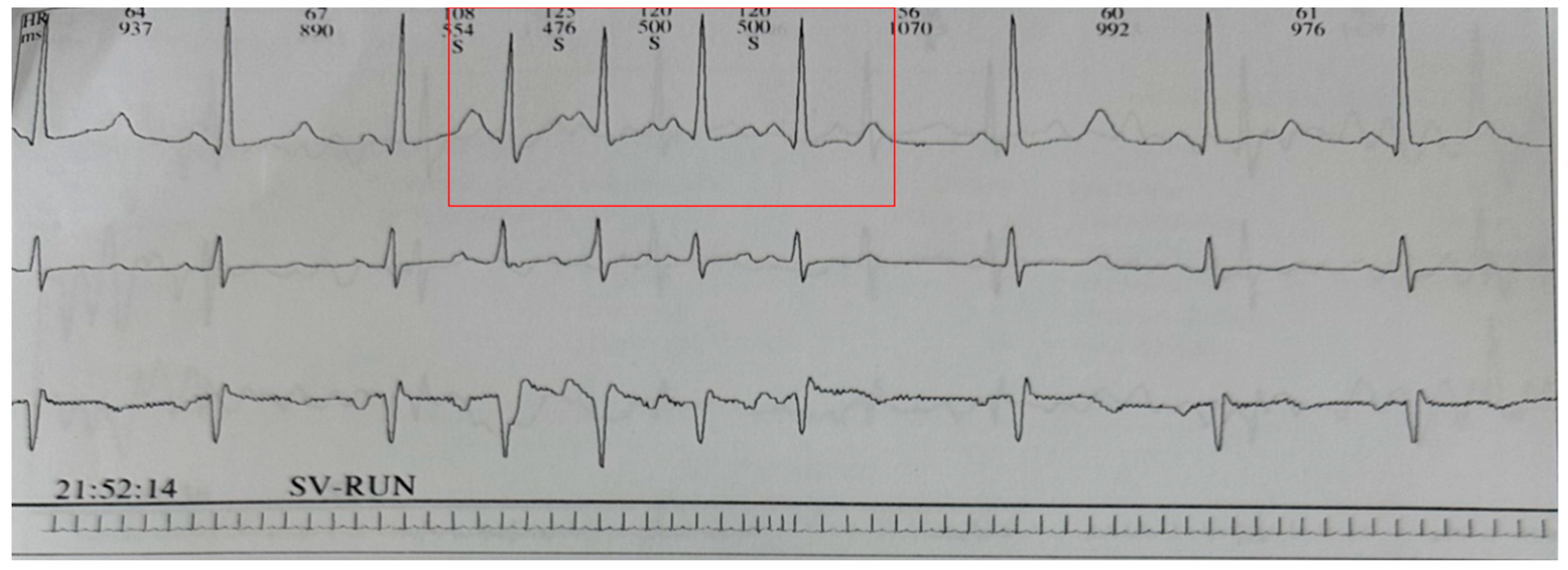

{kind=link}
| Patient ID | Current Age (Years) | Family History | GAA Gene Mutations | Age at Onset (Years) | Presentation | Age at First Control (In Years) | Age at Last Control (In Years) | FU (In Years) | Forced Vital Capacity (FVC) | |
|---|---|---|---|---|---|---|---|---|---|---|
| mL | % | |||||||||
| NA1 | 41 | no | CmHz:ex 2(IVS1-32-13T>G); ex 4:c.784G>A (p.Glu262Lys) | Increased CK values | 17 | 27 | 10 | 4120 | 84 | |
| NA2 | 66 | no | CmHz:ex 2(IVS1-32-13T>G); ex 7: c.1124G>T (p.Arg375Leu) | 54 | Muscle weakness | 63 | 66 | 3 | 2620 | 30 |
| NA3 | 58 | no | CmHz:ex 2(IVS1-32-13T>G); ex 7: c.1124G>T (p.Arg375Leu) | 30 | Post pregnancy muscle weakness | 38 | 58 | 20 | 1760 | 45 |
| NA4 | 42 | yes | CmHz:ex 2(IVS1-32-13T>G); ex 7: c.1124G>T (p.Arg375Leu) | Increased CK values | 34 | 41 | 7 | 3000 | 90 | |
| NA5 | 51 | yes | CmHz:ex 2(IVS1-32-13T>G); ex 7: c.1124G>T (p.Arg375Leu) | 18 | Muscle weakness | 31 | 51 | 20 | 3420 | 68 |
| NA6 | 57 | yes | CmHz:ex 2(IVS1-32-13T>G); ex 7: c.1124G>T (p.Arg375Leu) | 40 | Muscle weakness | 49 | 55 | 6 | 3020 | 100 |
| NA7 | 64 | no | CmHz:ex 2(IVS1-32-13T>G); ex 6: c.989G>A (p.W330Stop) | 34 | Muscle weakness | 38 | 64 | 26 | 3880 | 93 |
| NA8 | 73 | yes | CmHz:ex 2(IVS1-32-13T>G); ex 16: c.2237G>A (p.W746Stop) | 41 | Muscle weakness | 53 | 73 | 20 | 2600 | 63 |
| NA9 | 69 | yes | CmHz:ex 2(IVS1-32-13T>G); ex 16: c.2237G>A (p.W746Stop) | 54 | Muscle weakness | 54 | 59 | 5 | 1440 | 36 |
| NA10 | 56 | yes | CmHz:ex 2(IVS1-32-13T>G); ex 16: c.2237G>A (p.W746Stop) | 30 | Muscle weakness | 57 | 76 | 19 | 2680 | 68 |
| N11 | 29 | no | CmHz:ex 2(IVS1-32-13T>G); ex 12 c.1670T>G (p.Ile557Ser) | Increased CK values | 14 | 21 | 7 | 2220 | 76 | |
| N12 | 53 | no | CmHz: ex 4: c.784G>A (p.Glu262Lys); ex 5i c.959-6TC (p?) | 16 | Myalgia Exercise intolerance | 43 | 53 | 10 | 4630 | 92 |
| Mean ± SD | 55 ± 12.8 | 35 ± 13.6 | 40.9 ± 15.4 | 53.4 ± 16.9 | 12.7 ± 7.7 | 2796.4 ± 944.7 | 74.8 ± 20.9 | |||
| Cardiac Parameters at Baseline | Cardiac Parameters at Last Control | Treatment | ||||||||||||||||
| Patient ID | ECG-ECG Holter | Echocardiogram | ECG-ECG Holter | Echocardiogram | Pharmacological Treatment | Supporting Treatment | ||||||||||||
| HR | SVB | VB | SVT | Runs | AF | VTD (mm) | EF (%) | HR | SVB | VB | SVT | Runs | AF | VTD (mm) | EF (%) | |||
| NA1 | 50 | 0 | 0 | 0 | 0 | no | 51.1 | 69.0 | 75 | 0 | 0 | 0 | 0 | no | 51.4 | 64.7 | Magnesium pidolate | |
| NA2 | 82 | 0 | 0 | 0 | 0 | no | 46.0 | 62.0 | 72 | 0 | 0 | 0 | 0 | no | 50.0 | 68.0 | ||
| NA3 | 62 | 1870 | 0 | 0 | 1 | no | 45.4 | 66.2 | 55 | 0 | 0 | 0 | 0 | yes | 48.0 | 55.0 | Beta-blockers; dabigatran; sartans; flecainide;furosemide | PMK implant at 47 years |
| NA4 | 79 | 0 | 0 | 0 | 0 | no | 57.1 | 66.9 | 68 | 0 | 0 | 0 | 0 | no | 53.0 | 60.0 | Ace-inhibitors | |
| NA5 | 68 | 0 | 0 | 0 | 0 | no | 49.2 | 63.4 | 80 | 0 | 0 | 0 | 0 | no | 47.0 | 60.0 | Magnesium pidolate; ascorbic acid | |
| NA6 | 75 | 0 | 0 | 0 | 0 | no | 49.2 | 66.1 | 80 | 0 | 0 | 0 | 0 | no | 50.0 | 65.0 | ||
| NA7 | 60 | 0 | 0 | 0 | 0 | no | 45.5 | 66.9 | 80 | 0 | 0 | 0 | 0 | no | 50.4 | 55.0 | Magnesium pidolate; ascorbic acid | |
| NA8 | 71 | 0 | 0 | 0 | 0 | no | 57.0 | 56.0 | 75 | 0 | 0 | 0 | 0 | no | 54.0 | 62.0 | Ace-inhibitors | |
| NA9 | 75 | 0 | 0 | 0 | 0 | no | 53.7 | 54.5 | 83 | 0 | 0 | 0 | 0 | no | 52.3 | 57.2 | Ace-inhibitors | |
| NA10 | 88 | 0 | 0 | 0 | 0 | no | 48.8 | 70.0 | 82 | 0 | 0 | 0 | 0 | no | 50.0 | 56.0 | Ace-inhibitors; metformin | |
| NA11 | 75 | 0 | 0 | 0 | 0 | no | 50.1 | 70.0 | 90 | 0 | 0 | 0 | 0 | no | 45.6 | 71.6 | Ace-inhibitors | |
| Na12 | 88 | 0 | 0 | 0 | 0 | no | 42.4 | 67.9 | 71 | 0 | 0 | 0 | 0 | no | ||||
| Mean ± SD | 72.8 ± 11.4 | 49.6 ± 4.6 | 64.9 ± 5.1 | 75.9 ± 8.9 | 50.2 ± 2.5 | 61.3 ± 5.5 | ||||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palladino, A.; Passamano, L.; Scutifero, M.; Morra, S.; Picillo, E.; Papa, A.A.; Nigro, G.; Politano, L. Risk of Cardiac Arrhythmias in Patients with Late-Onset Pompe Disease—Results from a Long Follow-Up in a Group of 12 Patients and Review of Literature. Cardiogenetics 2024, 14, 38-50. https://doi.org/10.3390/cardiogenetics14010003
Palladino A, Passamano L, Scutifero M, Morra S, Picillo E, Papa AA, Nigro G, Politano L. Risk of Cardiac Arrhythmias in Patients with Late-Onset Pompe Disease—Results from a Long Follow-Up in a Group of 12 Patients and Review of Literature. Cardiogenetics. 2024; 14(1):38-50. https://doi.org/10.3390/cardiogenetics14010003
Chicago/Turabian StylePalladino, Alberto, Luigia Passamano, Marianna Scutifero, Salvatore Morra, Esther Picillo, Andrea Antonio Papa, Gerardo Nigro, and Luisa Politano. 2024. "Risk of Cardiac Arrhythmias in Patients with Late-Onset Pompe Disease—Results from a Long Follow-Up in a Group of 12 Patients and Review of Literature" Cardiogenetics 14, no. 1: 38-50. https://doi.org/10.3390/cardiogenetics14010003
APA StylePalladino, A., Passamano, L., Scutifero, M., Morra, S., Picillo, E., Papa, A. A., Nigro, G., & Politano, L. (2024). Risk of Cardiac Arrhythmias in Patients with Late-Onset Pompe Disease—Results from a Long Follow-Up in a Group of 12 Patients and Review of Literature. Cardiogenetics, 14(1), 38-50. https://doi.org/10.3390/cardiogenetics14010003