
Studying Epigenetics of Cardiovascular Diseases on Chip Guide

Abstract
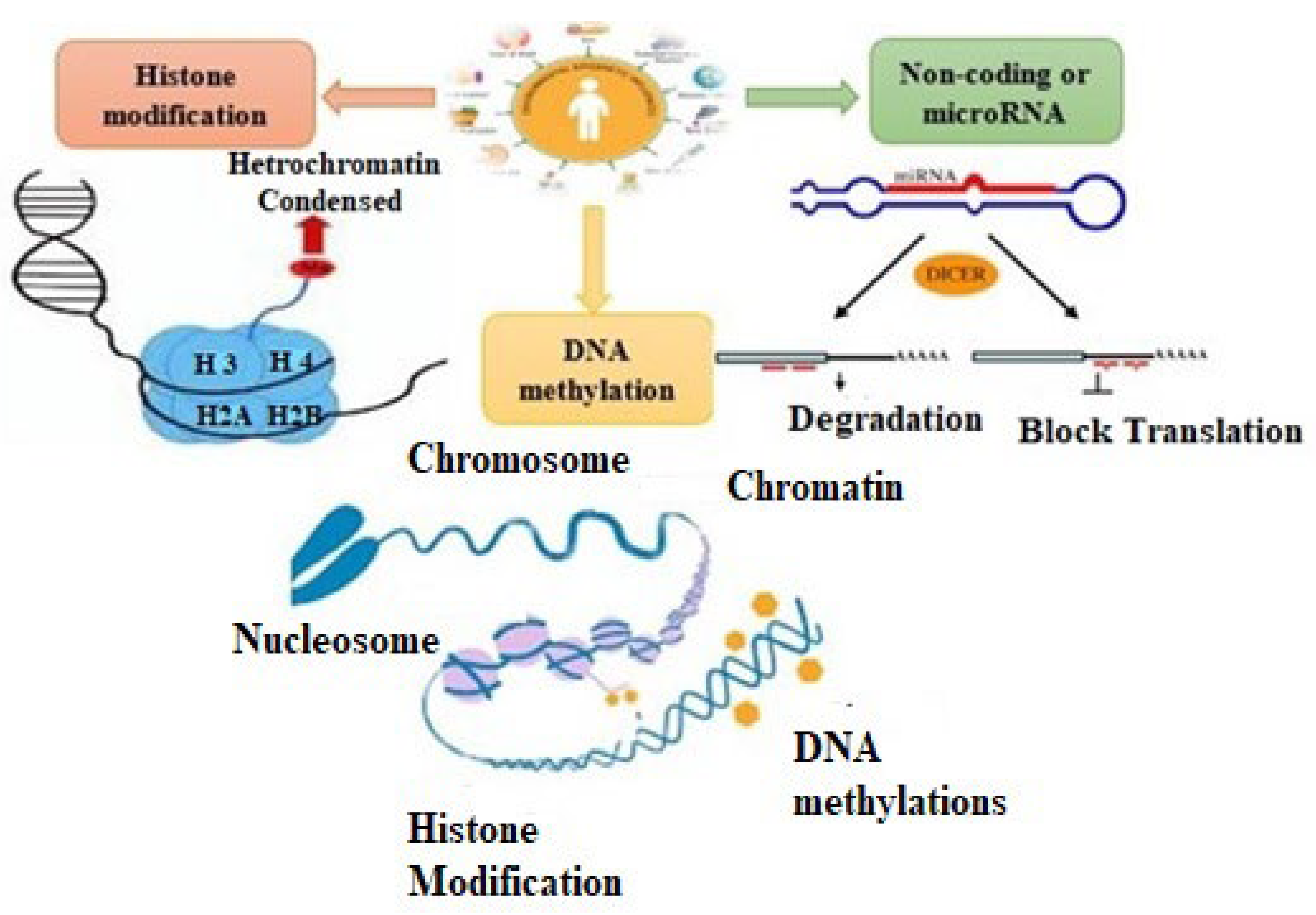
1. Introduction
1.1. DNA Methylation
1.2. Histone Modification
1.3. Noncoding or MicroRNAs
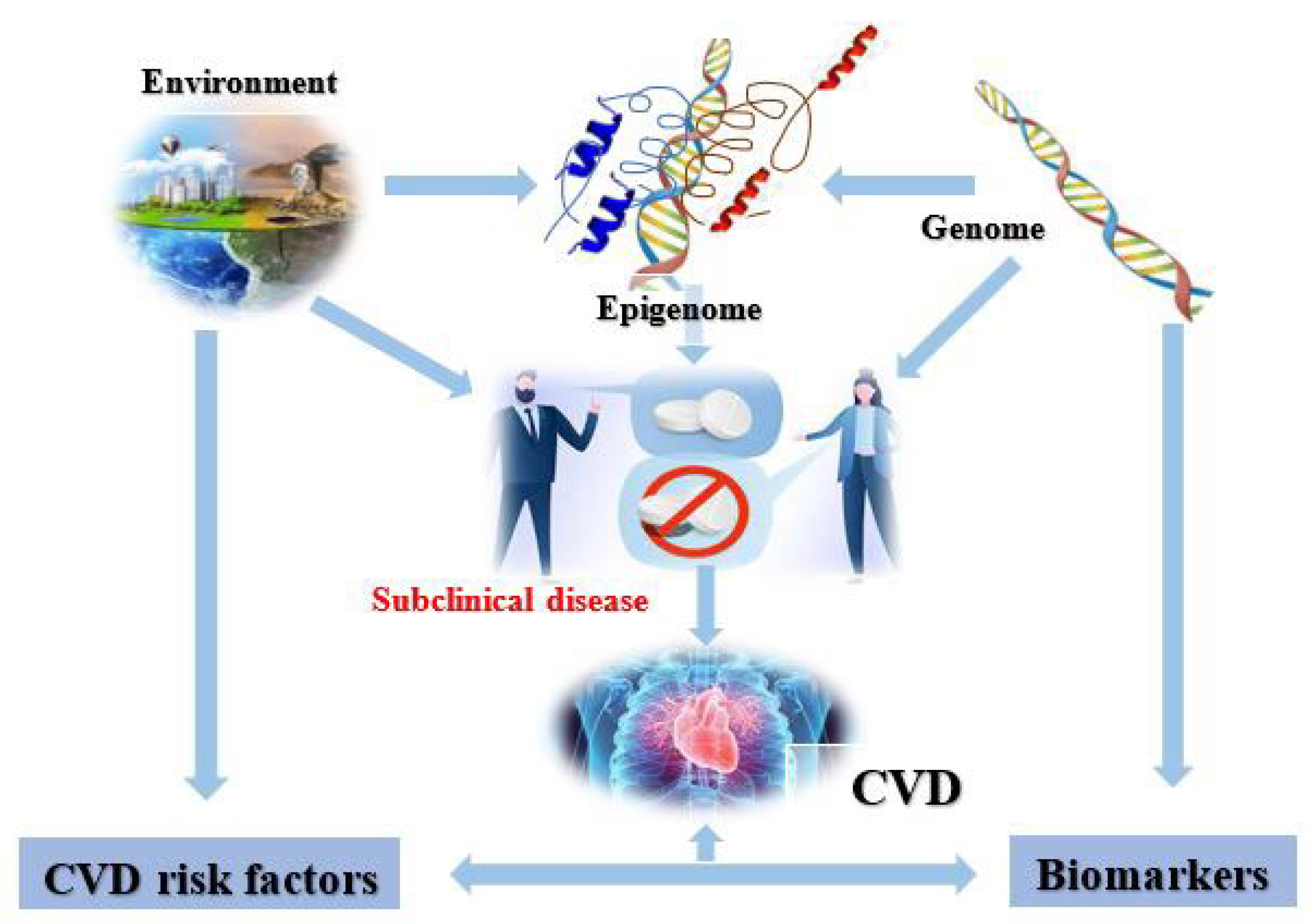
2. Cardiovascular Epigenetics
3. Epigenetic Modifications in CVD
3.1. Role of DNA Methylation in CVD
3.2. Role of Histone Modification in CVD
3.3. Role of microRNA in CVD

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Micrornas | Methodology | Site of Expression | Fold Expression | References |
|---|---|---|---|---|
| Heart failure | ||||
| miR-212, -129, -21 | Array | Human heart tissue | >1.5× upregulated | [51] |
| miR-133b, -92 | downregulated | |||
| miR-342, -214, -181b, -125 | Array | Left heart ventricle | Upregulation | [52] |
| miR-497, -139, -125b | qRT-PCR | PBMCs | >2× downregulation | [53] |
| miR-29b | >2× upregulation | |||
| miR-214 | Bead-based hybridization | Left heart ventricle | 2–2.7× downregulation | [54] |
| miR-24, -214, -125b, -195 | Northern blots | Left heart ventricle | 1.3–3× upregulation | [55] |
| Coronary artery disease | ||||
| miR-624, miR-340 | Array | Platelets | 1.5× upregulation | [50] |
| miR-147 | qRT-PCR | PBMCs | 4× downregulation | [56] |
| miR-370, -134 | 3.1–12× upregulation | |||
| miR-499, -126-133a | qRT-PCR | Plasma | 2–20× upregulation | [57] |
| miR-208a, -133 | Array | Plasma | Upregulation | [48] |
| miR-17-92 | Downregulation | |||
| Acute myocardial infarction | ||||
| miR-328 | qRT-PCR | Plasma | Upregulation | [58] |
| miR-306 | Array | PBMCs | Upregulation | [59] |
| miR-1291 | Downregulation | |||
| miR-423-5p | qRT-PCR | Plasma | 3–10× upregulation | [60] |
| miR-375, miR-122 | qRT-PCR | Plasma | Downregulation | [61] |
| miR-21 | qRT-PCR | Rat myocytes | Border cells: upregulation, infarcted cells: downregulation | [62] |
| miR-223 | qRT-PCR | Plasma | Downregulation | [63] |
4. CVD Risk Factors
4.1. Diabetes Mellitus
4.2. Hypoxia: A Pathophysiological Factor
4.3. Aging
4.4. Dyslipidemia
5. Cardiovascular Diseases and the Epigenome
5.1. Coronary Heart Disease (CHD)
5.2. Pulmonary Arterial Hypertension or PAH
6. Epigenetic Dysfunctional Responses in CVD
6.1. Mitochondrial Dysfunction
6.2. Fibrosis
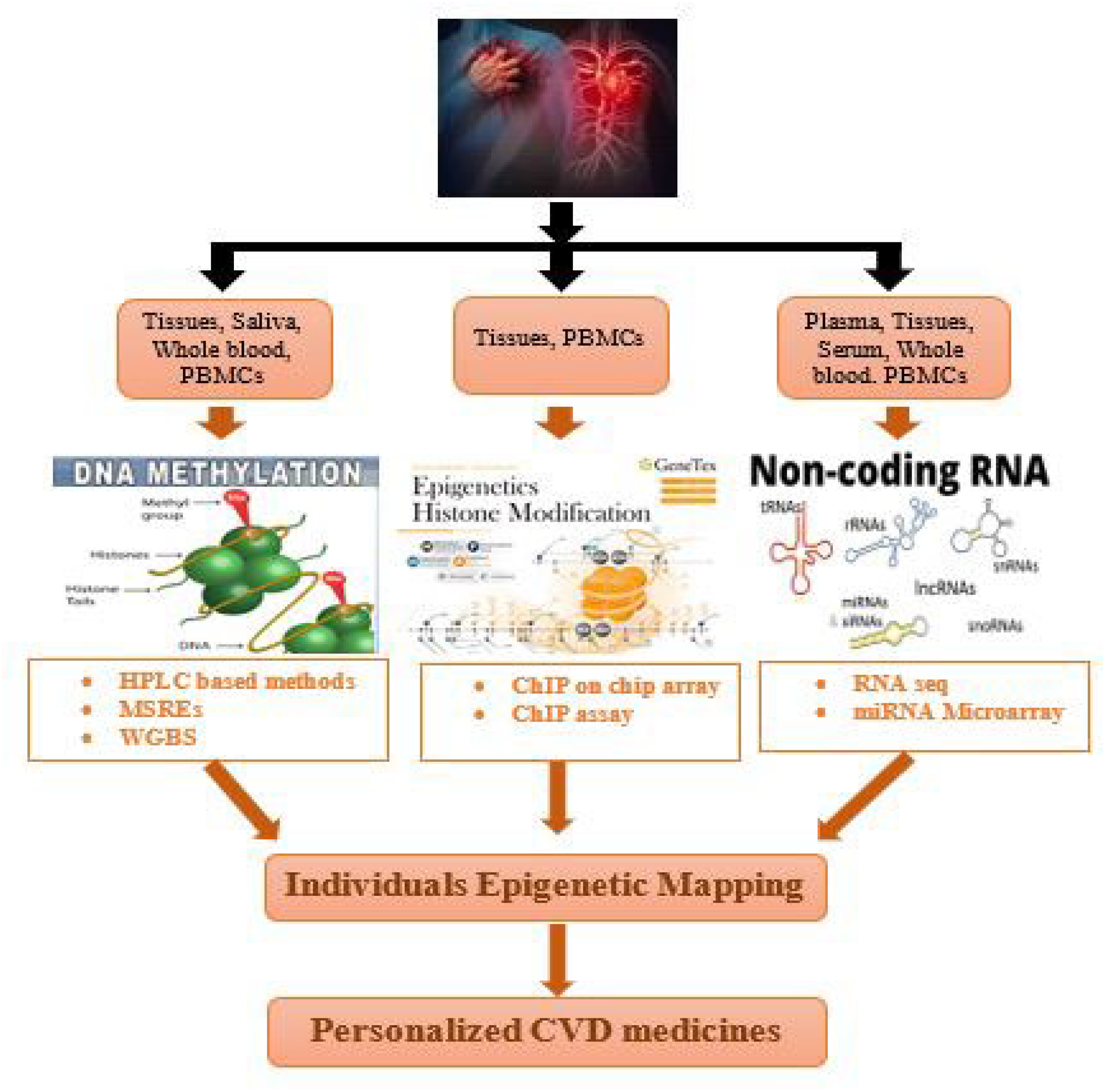
7. Techniques for Individual Epigenetic Mapping of Cardiovascular Disorders
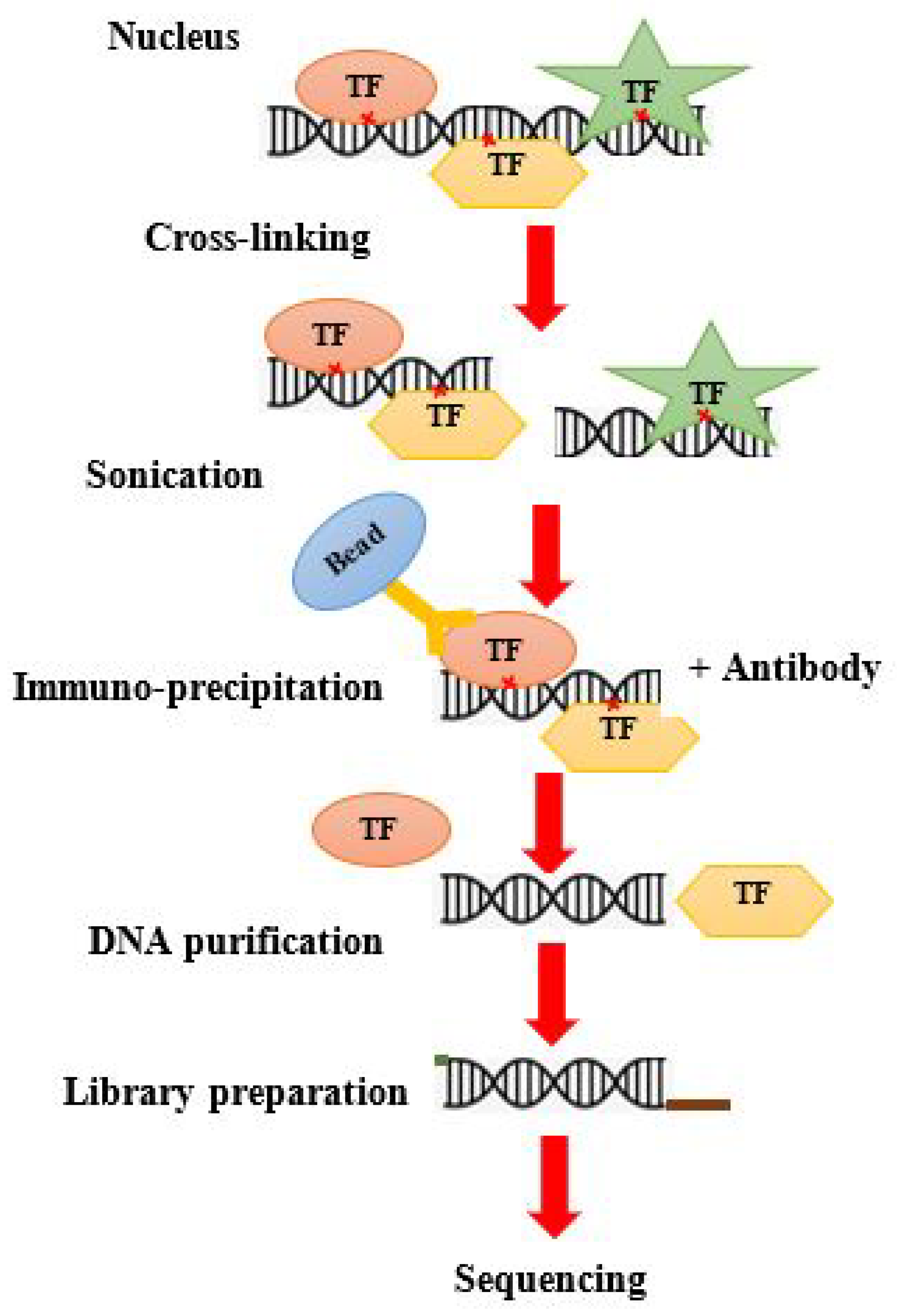
8. ChIP-on-Chip Guide
8.1. Traditional ChIP Seq
8.2. Single-Cell ChIP Seq
9. Role of ChIP Seq in Determining Epigenetic Signature Underlying Cardiac Hypertrophy
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, P.; Tyagi, S.C. Epigenetic mechanisms underlying cardiac degeneration and regeneration. Int. J. Cardiol. 2014, 173, 1–11. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, C.-F.; Tang, W.W. Epigenetics in Cardiac Hypertrophy and Heart Failure. JACC Basic Transl. Sci. 2019, 4, 976–993. [Google Scholar] [CrossRef] [PubMed]
- Muka, T.; Koromani, F.; Portilla, E.; O’Connor, A.; Bramer, W.M.; Troup, J.; Chowdhury, R.; Dehghan, A.; Franco, O.H. The role of epigenetic modifications in cardiovascular disease: A systematic review. Int. J. Cardiol. 2016, 212, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Baccarelli, A.; Ghosh, S. Environmental exposures, epigenetics and cardiovascular disease. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 323–329. [Google Scholar] [CrossRef]
- Miranda, T.B.; Jones, P.A. DNA methylation: The nuts and bolts of repression. J. Cell. Physiol. 2007, 213, 384–390. [Google Scholar] [CrossRef]
- Matouk, C.C.; Marsden, P.A. Epigenetic Regulation of Vascular Endothelial Gene Expression. Circ. Res. 2008, 102, 873–887. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the Histone Code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Cooper, M.E.; El-Osta, A.J.C. Epigenetics: Mechanisms and implications for diabetic complications. Circ. Res. 2010, 107, 1403–1413. [Google Scholar] [CrossRef]
- Keating, S.; El-Osta, A.J.E. Transcriptional regulation by the Set7 lysine methyltransferase. Epigenetics 2013, 8, 361–372. [Google Scholar] [CrossRef]
- Poller, W.; Dimmeler, S.; Heymans, S.; Zeller, T.; Haas, J.; Karakas, M.; Leistner, D.M.; Jakob, P.; Nakagawa, S.; Blankenberg, S.; et al. Non-coding RNAs in cardiovascular diseases: Diagnostic and therapeutic perspectives. Eur. Heart J. 2018, 39, 2704–2716. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Jaé, N.; Holdt, L.; Dimmeler, S. Long noncoding RNAs: From clinical genetics to therapeutic targets? J. Am. Coll. Cardiol. 2016, 67, 1214–1226. [Google Scholar] [CrossRef] [PubMed]
- Baccarelli, A.; Rienstra, M.; Benjamin, E.J. Cardiovascular epigenetics: Basic concepts and results from animal and human studies. Circ. Cardiovasc. Genet. 2010, 3, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Karaplis, A.C.; Ackerman, S.L.; Pogribny, I.P.; Melnyk, S.; Lussier-Cacan, S.; Chen, M.F.; Pai, A.; John, S.W.; Smith, R.S.; et al. Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreased methylation capacity, with neuropathology and aortic lipid deposition. Hum. Mol. Genet. 2001, 10, 433–444. [Google Scholar] [CrossRef]
- Lund, G.; Andersson, L.; Lauria, M.; Lindholm, M.; Fraga, M.F.; Villar-Garea, A.; Ballestar, E.; Esteller, M.; Zaina, S. DNA Methylation Polymorphisms Precede Any Histological Sign of Atherosclerosis in Mice Lacking Apolipoprotein E. J. Biol. Chem. 2004, 279, 29147–29154. [Google Scholar] [CrossRef] [PubMed]
- Makar, K.W.; Wilson, C.B. DNA methylation is a nonredundant repressor of the Th2 effector program. J. Immunol. 2004, 173, 4402–4406. [Google Scholar] [CrossRef]
- Kajstura, J.; Cheng, W.; Reiss, K.; Clark, W.A.; Sonnenblick, E.H.; Krajewski, S.; Reed, J.C.; Olivetti, G.; Anversa, P. Apoptotic and necrotic myocyte cell deaths are independent contributing variables of infarct size in rats. Lab. Investig. 1996, 74, 86–107. [Google Scholar]
- Mishra, P.K.; Chavali, V.; Metreveli, N.; Tyagi, S.C. Ablation of MMP9 induces survival and differentiation of cardiac stem cells into cardiomyocytes in the heart of diabetics: A role of extracellular matrix. Can. J. Physiol. Pharmacol. 2012, 90, 353–360. [Google Scholar] [CrossRef]
- Strahl, B.D.; Ohba, R.; Cook, R.G.; Allis, C.D. Methylation of histone H3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei in Tetrahymena. Proc. Natl. Acad. Sci. USA 1999, 96, 14967–14972. [Google Scholar] [CrossRef]
- Wang, G.K.; Zhu, J.Q.; Zhang, J.T.; Li, Q.; Li, Y.; He, J.; Jing, Q. Circulating microRNA: A novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur. Heart J. 2010, 31, 659–666. [Google Scholar] [CrossRef]
- Rayner, K.J.; Suárez, Y.; Dávalos, A.; Parathath, S.; Fitzgerald, M.L.; Tamehiro, N.; Fisher, E.A.; Moore, K.J.; Fernández-Hernando, C. MiR-33 Contributes to the Regulation of Cholesterol Homeostasis. Science 2010, 328, 1570–1573. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef]
- Van Weerd, J.H.; Koshiba-Takeuchi, K.; Kwon, C.; Takeuchi, J.K. Epigenetic factors and cardiac development. Cardiovasc. Res. 2011, 91, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Schober, A.; Weber, C.J. Mechanisms of microRNAs in atherosclerosis. Schober, A.; Weber, C.J. Mechanisms of microRNAs in atherosclerosis. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 583–616. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.K.; Kuypers, N.J.; Singh, S.R.; Leiberh, N.D.; Chavali, V.; Tyagi, S.C. Cardiac Stem Cell Niche, MMP9, and Culture and Differentiation of Embryonic Stem Cells. Methods Mol. Biol. 2013, 1035, 153–163. [Google Scholar]
- Tijsen, A.J.; Creemers, E.E.; Moerland, P.; De Windt, L.J.; van der Wal, A.; Kok, W.E.; Pinto, Y.M. MiR423-5p As a Circulating Biomarker for Heart Failure. Circ. Res. 2010, 106, 1035–1039. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.W.; Lee, S.-K.J. UTX, a histone H3-lysine 27 demethylase, acts as a critical switch to activate the cardiac developmental program. Dev. Cell 2012, 22, 25–37. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Kamal, M.; Lindblad-Toh, K.; Bekiranov, S.; Bailey, D.K.; Huebert, D.J.; McMahon, S.; Karlsson, E.K.; Kulbokas, E.J.; Gingeras, T.R.; et al. Genomic Maps and Comparative Analysis of Histone Modifications in Human and Mouse. Cell 2005, 120, 169–181. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Xiao, B.; Neppl, R.L.; Kallin, E.M.; Li, J.; Chen, T.; Wang, D.-Z.; Xiao, X.; Zhang, Y. DOT1L regulates dystrophin expression and is critical for cardiac function. Genes Dev. 2011, 25, 263–274. [Google Scholar] [CrossRef]
- Agger, K.; Cloos, P.A.C.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, S.; Weber, J.; Baxter, D.; Galas, D.J. Export of microRNAs and microRNA-protective protein by mammalian cells. Nucleic Acids Res. 2010, 38, 7248–7259. [Google Scholar] [CrossRef]
- Khyzha, N.; Alizada, A.; Wilson, M.D.; Fish, J.E. Epigenetics of Atherosclerosis: Emerging Mechanisms and Methods. Trends Mol. Med. 2017, 23, 332–347. [Google Scholar] [CrossRef]
- Smolarek, I.; Wyszko, E.; Barciszewska, A.M.; Nowak, S.; Gawronska, I.; Jablecka, A.; Barciszewska, M.Z. Global DNA methylation changes in blood of patients with essential hypertension. Med. Sci. Monit. 2010, 16, 155. [Google Scholar]
- Kim, M.; Long, T.I.; Arakawa, K.; Wang, R.; Yu, M.C.; Laird, P.W. DNA Methylation as a Biomarker for Cardiovascular Disease Risk. PLoS ONE 2010, 5, e9692. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 2010, 11, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.-P.; Oh, S.P.; Fuchs, M.; Zhou, N.-D.; Ch’Ng, L.-E.; Newsome, D.; Bronson, R.T.; Li, E.; Livingston, D.M.; Eckner, R. Gene Dosage–Dependent Embryonic Development and Proliferation Defects in Mice Lacking the Transcriptional Integrator p300. Cell 1998, 93, 361–372. [Google Scholar] [CrossRef]
- Shikama, N.; Lutz, W.; Kretzschmar, R.; Sauter, N.; Roth, J.; Marino, S.; Wittwer, J.; Scheidweiler, A.; Eckner, R. Essential function of p300 acetyltransferase activity in heart, lung and small intestine formation. EMBO J. 2003, 22, 5175–5185. [Google Scholar] [CrossRef] [PubMed]
- Yanazume, T.; Hasegawa, K.; Morimoto, T.; Kawamura, T.; Wada, H.; Matsumori, A.; Kawase, Y.; Hirai, M.; Kita, T. Cardiac p300 Is Involved in Myocyte Growth with Decompensated Heart Failure. Mol. Cell. Biol. 2003, 23, 3593–3606. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kawamura, T.; Morimoto, T.; Ono, K.; Wada, H.; Kawase, Y.; Matsumori, A.; Nishio, R.; Kita, T.; Hasegawa, K. Histone Acetyltransferase Activity of p300 Is Required for the Promotion of Left Ventricular Remodeling After Myocardial Infarction in Adult Mice In Vivo. Circulation 2006, 113, 679–690. [Google Scholar] [CrossRef]
- Fish, J.E.; Matouk, C.C.; Rachlis, A.; Lin, S.; Tai, S.C.; D’Abreo, C.; Marsden, P.A. The Expression of Endothelial Nitric-oxide Synthase Is Controlled by a Cell-specific Histone Code. J. Biol. Chem. 2005, 280, 24824–24838. [Google Scholar] [CrossRef]
- Ohtani, K.; Vlachojannis, G.J.; Koyanagi, M.; Boeckel, J.N.; Urbich, C.; Farcas, R.; Bonig, H.; Marquez, V.E.; Zeiher, A.M.; Dimmeler, S. Epigenetic regulation of endothelial lineage committed genes in pro-angiogenic hematopoietic and endothelial progenitor cells. Circ. Res. 2011, 109, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Zhou, V.W.; Goren, A.; Bernstein, B.E. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 2011, 12, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Vallaster, M.; Vallaster, C.D.; Wu, S.M. Epigenetic mechanisms in cardiac development and disease. Acta Biochim. Biophys. Sin. 2011, 44, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-P.; Yang, J.; Han, P.; Cheng, H.-L.; Shang, C.; Ashley, E.; Zhou, B.; Chang, C.P. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature 2010, 466, 62–67. [Google Scholar]
- Menghini, R.; Casagrande, V.; Cardellini, M.; Martelli, E.; Terrinoni, A.; Amati, F.; Nicotera, M.; Ippoliti, A.; Novelli, G.; Melino, G.; et al. MicroRNA 217 Modulates Endothelial Cell Senescence via Silent Information Regulator 1. Circulation 2009, 120, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandra, Y.; Devanna, P.; Limana, F.; Straino, S.; Di Carlo, A.; Brambilla, P.G.; Rubino, M.; Carena, M.C.; Spazzafumo, L.; De Simone, M.; et al. Circulating microRNAs are new and sensitive biomarkers of myocardial infarction. Eur. Heart J. 2010, 31, 2765–2773. [Google Scholar] [CrossRef]
- Harris, T.A.; Yamakuchi, M.; Ferlito, M.; Mendell, J.T.; Lowenstein, C.J. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc. Natl. Acad. Sci. USA 2008, 105, 1516–1521. [Google Scholar] [CrossRef]
- Zernecke, A.; Bidzhekov, K.; Noels, H.; Shagdarsuren, E.; Gan, L.; Denecke, B.; Hristov, M.; Köppel, T.; Jahantigh, M.N.; Lutgens, E.; et al. Delivery of MicroRNA-126 by Apoptotic Bodies Induces CXCL12-Dependent Vascular Protection. Sci. Signal. 2009, 2, ra81. [Google Scholar] [CrossRef]
- Hoekstra, M.; van der Lans, C.A.; Halvorsen, B.; Gullestad, L.; Kuiper, J.; Aukrust, P.; van Berkel, T.J.; Biessen, E.A. The peripheral blood mononuclear cell microRNA signature of coronary artery disease. Biochem. Biophys. Res. Commun. 2010, 394, 792–797. [Google Scholar] [CrossRef]
- Sondermeijer, B.M.; Bakker, A.; Halliani, A.; De Ronde, M.W.J.; Marquart, A.A.; Tijsen, A.J.; Mulders, T.A.; Kok, M.G.M.; Battjes, S.; Maiwald, S.; et al. Platelets in Patients with Premature Coronary Artery Disease Exhibit Upregulation of miRNA340* and miRNA624*. PLoS ONE 2011, 6, e25946. [Google Scholar]
- Keller, S.; Sanderson, M.P.; Stoeck, A.; Altevogt, P. Exosomes: From biogenesis and secretion to biological function. Immunol. Lett. 2006, 107, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Stoorvogel, W.J.B. Functional transfer of microRNA by exosomes. J. Am. Soc. Hematol. 2012, 119, 646–648. [Google Scholar] [CrossRef] [PubMed]
- El-Hefnawy, T.; Raja, S.; Kelly, L.; Bigbee, W.L.; Kirkwood, J.M.; Luketich, J.D.; Godfrey, T.E. Characterization of Amplifiable, Circulating RNA in Plasma and Its Potential as a Tool for Cancer Diagnostics. Clin. Chem. 2004, 50, 564–573. [Google Scholar] [CrossRef]
- Sucharov, C.; Bristow, M.R.; Port, J.D. miRNA expression in the failing human heart: Functional correlates. J. Mol. Cell. Cardiol. 2008, 45, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Kong, S.W.; Lu, J.; Bisping, E.; Zhang, H.; Allen, P.D.; Golub, T.R.; Pieske, B.; Pu, W.T. Altered microRNA expression in human heart disease. Physiol. Genom. 2007, 31, 367–373. [Google Scholar] [CrossRef]
- Dong, S.; Cheng, Y.; Yang, J.; Li, J.; Liu, X.; Wang, X.; Wang, D.; Krall, T.J.; Delphin, E.S.; Zhang, C. MicroRNA Expression Signature and the Role of MicroRNA-21 in the Early Phase of Acute Myocardial Infarction. J. Biol. Chem. 2009, 284, 29514–29525. [Google Scholar] [CrossRef]
- Meyer, G.P.; Wollert, K.C.; Lotz, J.; Pirr, J.; Rager, U.; Lippolt, P.; Hahn, A.; Fichtner, S.; Schaefer, A.; Arseniev, L.; et al. Intracoronary bone marrow cell transfer after myocardial infarction: 5-year follow-up from the randomized-controlled BOOST trial. Eur. Heart J. 2009, 30, 2978–2984. [Google Scholar] [CrossRef]
- Collino, F.; Deregibus, M.C.; Bruno, S.; Sterpone, L.; Aghemo, G.; Viltono, L.; Tetta, C.; Camussi, G. Microvesicles Derived from Adult Human Bone Marrow and Tissue Specific Mesenchymal Stem Cells Shuttle Selected Pattern of miRNAs. PLoS ONE 2010, 5, e11803. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Ham, O.; Cha, M.-J.; Song, B.-W.; Choi, E.; Kim, I.-K.; Chang, W.; Lim, S.; Lee, C.Y.; Park, J.-H.; et al. The promotion of cardiogenic differentiation of hMSCs by targeting epidermal growth factor receptor using microRNA-133a. Biomaterials 2013, 34, 92–99. [Google Scholar] [CrossRef]
- Meder, B.; Keller, A.; Vogel, B.; Haas, J.; Sedaghat-Hamedani, F.; Kayvanpour, E.; Just, S.; Borries, A.; Rudloff, J.; Leidinger, P.; et al. MicroRNA signatures in total peripheral blood as novel biomarkers for acute myocardial infarction. Basic Res. Cardiol. 2010, 106, 13–23. [Google Scholar] [CrossRef]
- Fichtlscherer, S.; De Rosa, S.; Fox, H.; Schwietz, T.; Fischer, A.; Liebetrau, C.; Weber, M.; Hamm, C.W.; Röxe, T.; Müller-Ardogan, M.; et al. Circulating MicroRNAs in Patients with Coronary Artery Disease. Circ. Res. 2010, 107, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Corsten, M.F.; Dennert, R.; Jochems, S.; Kuznetsova, T.; Devaux, Y.; Hofstra, L.; Wagner, D.R.; Staessen, J.A.; Heymans, S.; Schroen, B. Circulating MicroRNA-208b and MicroRNA-499 Reflect Myocardial Damage in Cardiovascular Disease. Circ. Cardiovasc. Genet. 2010, 3, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Ai, J.; Zhang, R.; Li, Y.; Pu, J.; Lu, Y.; Jiao, J.; Li, K.; Yu, B.; Li, Z.; Wang, R.; et al. Circulating microRNA-1 as a potential novel biomarker for acute myocardial infarction. Biochem. Biophys. Res. Commun. 2010, 391, 73–77. [Google Scholar] [CrossRef]
- Costantino, S.; Libby, P.; Kishore, R.; Tardif, J.-C.; El-Osta, A.; Paneni, F. Epigenetics and precision medicine in cardiovascular patients: From basic concepts to the clinical arena. Eur. Heart J. 2017, 39, 4150–4158. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Antonicelli, R.; Lorenzi, M.; D’Alessandra, Y.; Lazzarini, R.; Santini, G.; Spazzafumo, L.; Lisa, R.; La Sala, L.; Galeazzi, R.; et al. Diagnostic potential of circulating miR-499-5p in elderly patients with acute non ST-elevation myocardial infarction. Int. J. Cardiol. 2013, 167, 531–536. [Google Scholar] [CrossRef]
- Villeneuve, L.M.; Natarajan, R.J. The role of epigenetics in the pathology of diabetic complications. Am. J. Physiol.-Renal Physiol. 2010, 299, F14–F25. [Google Scholar] [CrossRef]
- Kuroda, A.; Rauch, T.A.; Todorov, I.; Ku, H.T.; Al-Abdullah, I.H.; Kandeel, F.; Mullen, Y.; Pfeifer, G.P.; Ferreri, K. Insulin gene expression is regulated by DNA methylation. PLoS ONE 2009, 4, e6953. [Google Scholar] [CrossRef]
- Kong, A.; DIAGRAM Consortium; Steinthorsdottir, V.; Masson, G.; Thorleifsson, G.; Sulem, P.; Besenbacher, S.; Jonasdottir, A.; Sigurdsson, A.; Kristinsson, K.T.; et al. Parental origin of sequence variants associated with complex diseases. Nature 2009, 462, 868–874. [Google Scholar] [CrossRef]
- Paneni, F.; Costantino, S.; Cosentino, F. Molecular pathways of arterial aging. Clin. Sci. 2014, 128, 69–79. [Google Scholar] [CrossRef]
- Costantino, S.; Paneni, F.; Battista, R.; Castello, L.; Capretti, G.; Chiandotto, S.; Tanese, L.; Russo, G.; Pitocco, D.; Lanza, G.A.; et al. Impact of Glycemic Variability on Chromatin Remodeling, Oxidative Stress, and Endothelial Dysfunction in Patients with Type 2 Diabetes and with Target HbA1c Levels. Diabetes 2017, 66, 2472–2482. [Google Scholar] [CrossRef]
- Sapienza, C.; Lee, J.; Powell, J.; Erinle, O.; Yafai, F.; Reichert, J.; Siraj, E.S.; Madaio, M. DNA methylation profiling identifies epigenetic differences between diabetes patients with ESRD and diabetes patients without nephropathy. Epigenetics 2011, 6, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Paneni, F.; Costantino, S.; Battista, R.; Castello, L.; Capretti, G.; Chiandotto, S.; Scavone, G.; Villano, A.; Pitocco, D.; Lanza, G.; et al. Adverse Epigenetic Signatures by Histone Methyltransferase Set7 Contribute to Vascular Dysfunction in Patients with Type 2 Diabetes Mellitus. Circ. Cardiovasc. Genet. 2015, 8, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Avogaro, A.; de Kreutzenberg, S.V.; Federici, M.; Fadini, G.P. The Endothelium Abridges Insulin Resistance to Premature Aging. J. Am. Heart Assoc. 2013, 2, e000262. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.B.; Denko, N.; Barton, M.C. Hypoxia induces a novel signature of chromatin modifications and global repression of transcription. Mutat. Res. Mol. Mech. Mutagen. 2008, 640, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Fish, J.E.; Yan, M.S.; Matouk, C.C.; Bernard, R.S.; Ho, J.J.D.; Gavryushova, A.; Srivastava, D.; Marsden, P.A. Hypoxic Repression of Endothelial Nitric-oxide Synthase Transcription Is Coupled with Eviction of Promoter Histones. J. Biol. Chem. 2010, 285, 810–826. [Google Scholar] [CrossRef]
- Zampetaki, A.; Zeng, L.; Margariti, A.; Xiao, Q.; Li, H.; Zhang, Z.; Pepe, A.E.; Wang, G.; Habi, O.; DeFalco, E.; et al. Histone Deacetylase 3 Is Critical in Endothelial Survival and Atherosclerosis Development in Response to Disturbed Flow. Circulation 2010, 121, 132–142. [Google Scholar] [CrossRef]
- Xiao, F.-H.; Wang, H.-T.; Kong, Q.-P. Dynamic DNA methylation during aging: A “prophet” of age-related outcomes. Front. Genet. 2019, 10, 107. [Google Scholar] [CrossRef]
- Blecua, P.; Martinez-Verbo, L.; Esteller, M. The DNA methylation landscape of hematological malignancies: An update. Mol. Oncol. 2020, 14, 1616–1639. [Google Scholar] [CrossRef]
- Arai, F.; Stumpf, P.S.; Ikushima, Y.M.; Hosokawa, K.; Roch, A.; Lutolf, M.P.; Suda, T.; MacArthur, B.D. Machine Learning of Hematopoietic Stem Cell Divisions from Paired Daughter Cell Expression Profiles Reveals Effects of Aging on Self-Renewal. Cell Syst. 2020, 11, 640–652.e5. [Google Scholar] [CrossRef]
- Menghini, R.; Casagrande, V.; Federici, M. MicroRNAs in Endothelial Senescence and Atherosclerosis. J. Cardiovasc. Transl. Res. 2013, 6, 924–930. [Google Scholar] [CrossRef]
- Sayols-Baixeras, S.; Irvin, M.R.; Elosua, R.; Arnett, D.K.; Aslibekyan, S.W. Epigenetics of Lipid Phenotypes. Curr. Cardiovasc. Risk Rep. 2016, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tobi, E.; Lumey, L.H.; Talens, R.P.; Kremer, D.; Putter, H.; Stein, A.; Slagboom, P.; Heijmans, B.T. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum. Mol. Genet. 2009, 18, 4046–4053. [Google Scholar] [CrossRef]
- Irvin, M.R.; Zhi, D.; Joehanes, R.; Mendelson, M.; Aslibekyan, S.; Claas, S.A.; Thibeault, K.S.; Patel, N.; Day, K.; Jones, L.W.; et al. Epigenome-Wide Association Study of Fasting Blood Lipids in the Genetics of Lipid-Lowering Drugs and Diet Network Study. Circulation 2014, 130, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Ono, K. Functions of microRNA-33a/b and microRNA therapeutics. J. Cardiol. 2016, 67, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Infante, T.; Del Viscovo, L.; De Rimini, M.L.; Padula, S.; Caso, P.; Napoli, C. Network Medicine: A Clinical Approach for Precision Medicine and Personalized Therapy in Coronary Heart Disease. J. Atheroscler. Thromb. 2020, 27, 279–302. [Google Scholar] [CrossRef]
- Schiano, C.; Benincasa, G.; Franzese, M.; Della Mura, N.; Pane, K.; Salvatore, M.; Napoli, C. Epigenetic-sensitive pathways in personalized therapy of major cardiovascular diseases. Pharmacol. Ther. 2020, 210, 107514. [Google Scholar] [CrossRef]
- Westerman, K.; Fernández-Sanlés, A.; Patil, P.; Sebastiani, P.; Jacques, P.; Starr, J.M.; JDeary, I.; Liu, Q.; Liu, S.; Elosua, R.; et al. Epigenomic assessment of cardiovascular disease risk and interactions with traditional risk metrics. J. Am. Heart Assoc. 2020, 9, e015299. [Google Scholar] [CrossRef]
- Schiano, C.; Benincasa, G.; Infante, T.; Franzese, M.; Castaldo, R.; Fiorito, C.; Mansueto, G.; Grimaldi, V.; Della Valle, G.; Fatone, G.; et al. Integrated analysis of DNA methylation profile of HLA-G gene and imaging in coronary heart disease: Pilot study. PLoS ONE 2020, 15, e0236951. [Google Scholar] [CrossRef]
- Westerman, K.; Sebastiani, P.; Jacques, P.; Liu, S.; DeMeo, D.; Ordovás, J.M. DNA methylation modules associate with incident cardiovascular disease and cumulative risk factor exposure. Clin. Epigenet. 2019, 11, 1–14. [Google Scholar] [CrossRef]
- Chelladurai, P.; Dabral, S.; Basineni, S.R.; Chen, C.-N.; Schmoranzer, M.; Bender, N.; Feld, C.; Nötzold, R.R.; Dobreva, G.; Wilhelm, J.; et al. Isoform-specific characterization of class I histone deacetylases and their therapeutic modulation in pulmonary hypertension. Sci. Rep. 2020, 10, 12864. [Google Scholar] [CrossRef]
- Maron, B.J.; Maron, M.S.; Maron, B.A.; Loscalzo, J. Moving beyond the sarcomere to explain heterogeneity in hypertrophic cardiomyopathy: JACC review topic of the week. J. Am. Coll. Cardiol. 2019, 73, 1978–1986. [Google Scholar] [CrossRef]
- Reyes-Palomares, A.; Gu, M.; Grubert, F.; Berest, I.; Sa, S.; Kasowski, M.; Arnold, C.; Shuai, M.; Srivas, R.; Miao, Y.; et al. Remodeling of active endothelial enhancers is associated with aberrant gene-regulatory networks in pulmonary arterial hypertension. Nat. Commun. 2020, 11, 1673. [Google Scholar] [CrossRef]
- Bround, M.J.; Wambolt, R.; Luciani, D.S.; Kulpa, J.E.; Rodrigues, B.; Brownsey, R.W.; Allard, M.F.; Johnson, J.D. Cardiomyocyte ATP Production, Metabolic Flexibility, and Survival Require Calcium Flux through Cardiac Ryanodine Receptors in Vivo. J. Biol. Chem. 2013, 288, 18975–18986. [Google Scholar] [CrossRef] [PubMed]
- Baccarelli, A.A.; Byun, H.-M. Platelet mitochondrial DNA methylation: A potential new marker of cardiovascular disease. Clin. Epigenet. 2015, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Humeres, C.; Frangogiannis, N.G. Fibroblasts in the infarcted, remodeling, and failing heart. JACC Basic Transl. Sci. 2019, 4, 449–467. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.J.; Collier, P.; Tea, I.; Neary, R.; Watson, J.A.; Robinson, C.; Phelan, D.; Ledwidge, M.; McDonald, K.; McCann, A.; et al. Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum. Mol. Genet. 2013, 23, 2176–2188. [Google Scholar] [CrossRef]
- Fox, C.S.; Hall, J.L.; Arnett, D.K.; Ashley, E.A.; Delles, C.; Engler, M.B.; Freeman, M.W.; Johnson, J.A.; Lanfear, D.E.; Liggett, S.B.; et al. Future translational applications from the contemporary genomics era: A scientific statement from the American Heart Association. Circulation 2015, 131, 1715–1736. [Google Scholar] [CrossRef] [PubMed]
- Altman, R.B.; Ashley, E.A. Using “big data” to dissect clinical heterogeneity. Circulation 2015, 131, 232–233. [Google Scholar] [CrossRef][Green Version]
- Antman, E.M.; Loscalzo, J. Precision medicine in cardiology. Nat. Rev. Cardiol. 2016, 13, 591–602. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, Y. Profiling chromatin regulatory landscape: Insights into the development of ChIP-seq and ATAC-seq. Mol. Biomed. 2020, 1, 1–13. [Google Scholar] [CrossRef]
- Ren, B.; Robert, F.; Wyrick, J.J.; Aparicio, O.; Jennings, E.G.; Simon, I.; Zeitlinger, J.; Schreiber, J.; Hannett, N.; Kanin, E.; et al. Genome-Wide Location and Function of DNA Binding Proteins. Science 2000, 290, 2306–2309. [Google Scholar] [CrossRef] [PubMed]
- Park, P.J. ChIP–seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.; Hirst, M.; Bainbridge, M.; Bilenky, M.; Zhao, Y.; Zeng, T.; Euskirchen, G.; Bernier, B.; Varhol, R.; Delaney, A.; et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 2007, 4, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-Wide Mapping of in Vivo Protein-DNA Interactions. Science 2007, 316, 1497–1502. [Google Scholar] [CrossRef]
- Truax, A.D.; Greer, S.F. ChIP and Re-ChIP Assays: Investigating Interactions between Regulatory Proteins, Histone Modifications, and the DNA Sequences to Which They Bind. In Transcriptional Regulation; Springer: Berlin/Heidelberg, Germany, 2012; pp. 175–188. [Google Scholar]
- Rotem, A.; Ram, O.; Shoresh, N.; Sperling, R.A.; Goren, A.; Weitz, D.A.; Bernstein, B.E. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol. 2015, 33, 1165–1172. [Google Scholar] [CrossRef]
- Grosselin, K.; Durand, A.; Marsolier, J.; Poitou, A.; Marangoni, E.; Nemati, F.; Dahmani, A.; Lameiras, S.; Reyal, F.; Frenoy, O.; et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat. Genet. 2019, 51, 1060–1066. [Google Scholar] [CrossRef]
- Izumo, S.; Nadal-Ginard, B.; Mahdavi, V. Protooncogene induction and reprogramming of cardiac gene expression produced by pressure overload. Proc. Natl. Acad. Sci. USA 1988, 85, 339–343. [Google Scholar] [CrossRef]
- Movassagh, M.; Choy, M.-K.; Knowles, D.A.; Cordeddu, L.; Haider, S.; Down, T.; Siggens, L.; Vujic, A.; Simeoni, I.; Penkett, C.; et al. Distinct Epigenomic Features in End-Stage Failing Human Hearts. Circulation 2011, 124, 2411–2422. [Google Scholar] [CrossRef]
- Haas, J.; Frese, K.S.; Park, Y.J.; Keller, A.; Vogel, B.; Lindroth, A.; Weichenhan, D.; Franke, J.; Fischer, S.; Bauer, A.; et al. Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol. Med. 2013, 5, 413–429. [Google Scholar] [CrossRef]
- Ovchinnikova, E.S.; Schmitter, D.; Vegter, E.L.; Ter Maaten, J.M.; Valente, M.A.; Liu, L.C.; Van Der Harst, P.; Pinto, Y.M.; De Boer, R.A.; Meyer, S.; et al. Signature of circulating microRNAs in patients with acute heart failure. Eur. J. Heart Fail. 2015, 18, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Ellis, K.L.; Cameron, V.A.; Troughton, R.W.; Frampton, C.M.; Ellmers, L.J.; Richards, A.M. Circulating microRNAs as candidate markers to distinguish heart failure in breathless patients. Eur. J. Heart Fail. 2013, 15, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Seronde, M.-F.; Vausort, M.; Gayat, E.; Goretti, E.; Ng, L.L.; Squire, I.B.; Vodovar, N.; Sadoune, M.; Samuel, J.-L.; Thum, T.; et al. Circulating microRNAs and Outcome in Patients with Acute Heart Failure. PLoS ONE 2015, 10, e0142237. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Zhang, Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011, 25, 1345–1358. [Google Scholar] [CrossRef]
- Ernst, J.; Kheradpour, P.; Mikkelsen, T.S.; Shoresh, N.; Ward, L.; Epstein, C.B.; Zhang, X.; Wang, L.; Issner, R.; Coyne, M.; et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011, 473, 43–49. [Google Scholar] [CrossRef]
- Dong, X.; Greven, M.C.; Kundaje, A.; Djebali, S.; Brown, J.B.; Cheng, C.; Gingeras, T.; Gerstein, M.B.; Guigó, R.; Birney, E.; et al. Modeling gene expression using chromatin features in various cellular contexts. Genome Biol. 2012, 13, 1–17. [Google Scholar] [CrossRef]
- Papait, R.; Cattaneo, P.; Kunderfranco, P.; Greco, C.; Carullo, P.; Guffanti, A.; Viganò, V.; Stirparo, G.G.; Latronico, M.V.G.; Hasenfuss, G.; et al. Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 20164–20169. [Google Scholar] [CrossRef]




| Epigenetic Modification | Binding Domains | Targets | Phenotypes | Altered Gene Expressions | References |
|---|---|---|---|---|---|
| DNA methylation | None | CpG islands | Change in gene expression of angiogenic factors and heart failure | Up- or downregulation | [17,18] |
| Histone modification | |||||
| Ribosylation | PARP1 | HDACs, PARP1, histones, and brg1 | Enhanced fetal β-MHC expression, complex with Brg1 and HDACs, heart failure, and cardiac hypertrophy | Upregulation | [19] |
| Phosphorylation | PKD, AMPK, JAK2, Rsk2, Aurora | HDACs, H2B, H4Y41, S28/S10, H3 | Transcriptional activation, cardiac hypertrophy regulation, cellular proliferation, and mitotic activity | Up- or downregulation | [20,21,22] |
| Deacetylation | HDAC Class II (9,5,4) | Tails of histones | Cardiac hypertrophy negative regulation and inhibition of MEF2 activity (myocyte enhancer factor 2) | [23,24] | |
| Acetylation | P300, CBP (CREBP-binding protein) | K19, K16, K12, K8, H4K5, H3K4 | Regulation of cardiac hypertrophy | Upregulation | [25,26] |
| Demethylation | UTX, JMJD2A | H3K27me3, H3K36me3, H3K4me3 | Embryo lethality, heart malformation, and stimulation of cardiac hypertrophy | Up- or downregulation | [27,28] |
| Methylation | DOTIL, PTIP | H3K79me, H3K27me3, H3K9me3 H3K4me3, H3K4me2 | Dilated cardiomyopathy, heart failure and angiogenesis, and activation of fetal cardiac gene | Up- or downregulation | [29,30,31] |
| Technique | Abbreviation | Description |
|---|---|---|
| Whole-genome bisulfite sequencing | WGBS | This is an NGS technique used to evaluate the status of DNA methylation of cytosine residues across the genome and to directly determine the ratio of methylated molecules. DNA samples are treated with sodium bisulfite that only converts unmethylated cytosine into uracil and leaves the methylated cytosine unchanged. |
| RNA sequencing | RNA seq | This is used for determination of the cellular transcriptome. It allows the evaluation of differences in gene expression, mutations or SNPs, gene fusion, post-transcriptional modification, and alternative gene sliced transcripts using different treatments or groups. Along with mRNA transcripts, this technique also determines ribosomal profiling and different RNA populations, including small RNAs such as tRNA and miRNA, as well as total RNA. |
| ChIP-on-chip | ChIP-on-chip | This is a technology that combines two techniques, i.e., DNA microarray (chip) and chromatin immune precipitation (ChIP). It determines the in vivo interaction between DNA and proteins. |
| Epigenome-wide association studies | EWAS | This is used to determine the connection between specific identifiable traits or phenotypes in large human cohorts and a genome-wide set of epigenetic biomarkers. It determines whether there is an actual correlation between epigenetic perturbation and given phenotype. |
| Genotype–Tissue Expression project | GTEx | This project is used to provide valuable understanding regarding the mechanisms of gene regulation using existing knowledge of human gene regulation and expression in multiple tissues, not only from healthy subjects but also from various human diseases. |
| Assay for transposase-accessible chromatin sequencing | ATAC-seq | This is an HTS technology that provides access to a genome-wide map of chromatin. It provides specific information regarding genome-wide positions of the following:
|
| Methods | Strategy | Mapping Rate | Cell State | Device for Cell Sorting |
|---|---|---|---|---|
| CUT and Tag | ChIP-free, Tn5-barcoding (1 round) | 97% | Native | Costly Takara ICELL8 |
| Co-BATCH | ChIP-free, Tn5-barcoding (for 2 rounds) | 94% | Fixed and native | FACS |
| sc-itChIP-seq | ChIP and Tn5-barcoding (1 round) | 94% | Fixed and native | FACS |
| scDrop-ChIP | Microfluidic system and ChIP for droplet formation | 70% | Native | Costly microfluidic device |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alghamdi, B.A.; Aljohani, I.M.; Alotaibi, B.G.; Ahmed, M.; Almazmomi, K.A.; Aloufi, S.; Alshamrani, J. Studying Epigenetics of Cardiovascular Diseases on Chip Guide. Cardiogenetics 2022, 12, 218-234. https://doi.org/10.3390/cardiogenetics12030021
Alghamdi BA, Aljohani IM, Alotaibi BG, Ahmed M, Almazmomi KA, Aloufi S, Alshamrani J. Studying Epigenetics of Cardiovascular Diseases on Chip Guide. Cardiogenetics. 2022; 12(3):218-234. https://doi.org/10.3390/cardiogenetics12030021
Chicago/Turabian StyleAlghamdi, Bandar Ali, Intisar Mahmoud Aljohani, Bandar Ghazi Alotaibi, Muhammad Ahmed, Kholod Abduallah Almazmomi, Salman Aloufi, and Jowhra Alshamrani. 2022. "Studying Epigenetics of Cardiovascular Diseases on Chip Guide" Cardiogenetics 12, no. 3: 218-234. https://doi.org/10.3390/cardiogenetics12030021
APA StyleAlghamdi, B. A., Aljohani, I. M., Alotaibi, B. G., Ahmed, M., Almazmomi, K. A., Aloufi, S., & Alshamrani, J. (2022). Studying Epigenetics of Cardiovascular Diseases on Chip Guide. Cardiogenetics, 12(3), 218-234. https://doi.org/10.3390/cardiogenetics12030021