
Clinical Phenotypes of Cardiovascular and Heart Failure Diseases Can Be Reversed? The Holistic Principle of Systems Biology in Multifaceted Heart Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
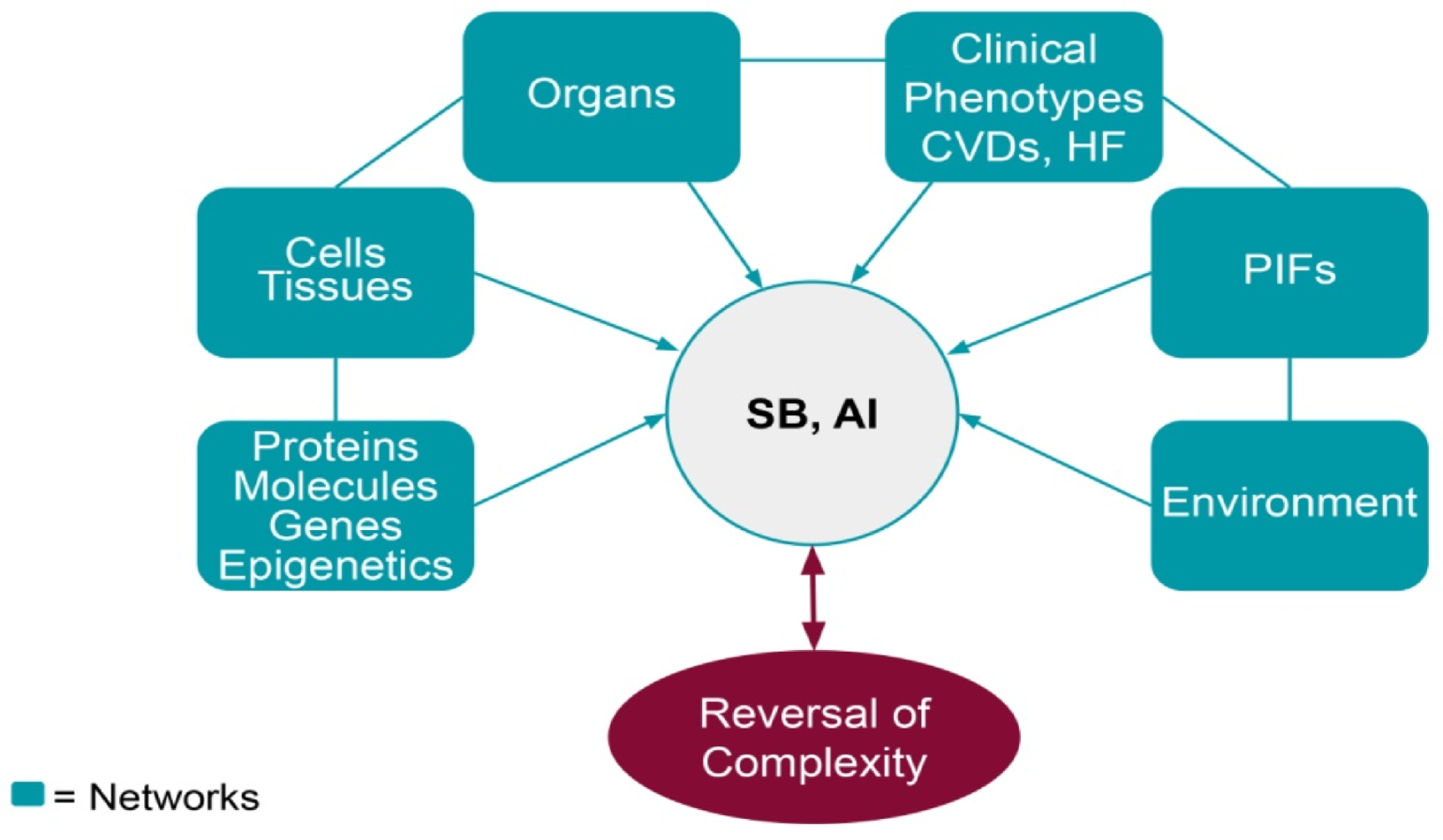
Highlights
- The paper is focused on the probability of reversal of the causes and effects of cardiovascular diseases (CVDs) and heart failure (HF).
- We proposed a methodology based on the application of the holistic principles of systems biology (SB) with the help of artificial intelligence (AI).
- This proposal requires a large amount of relative data; it employs a multi-omics approach and network analysis in order to understand the concept of clinical complexity.
- We believe this approach will help in the appreciation of the complexity of chronic heart diseases and explain the interconnection between their biological and clinical networks.
- We also believe that this methodology will enhance the prospects for research and clinical applications aimed at the reversal of clinical phenotypes.
Abstract
1. Introduction
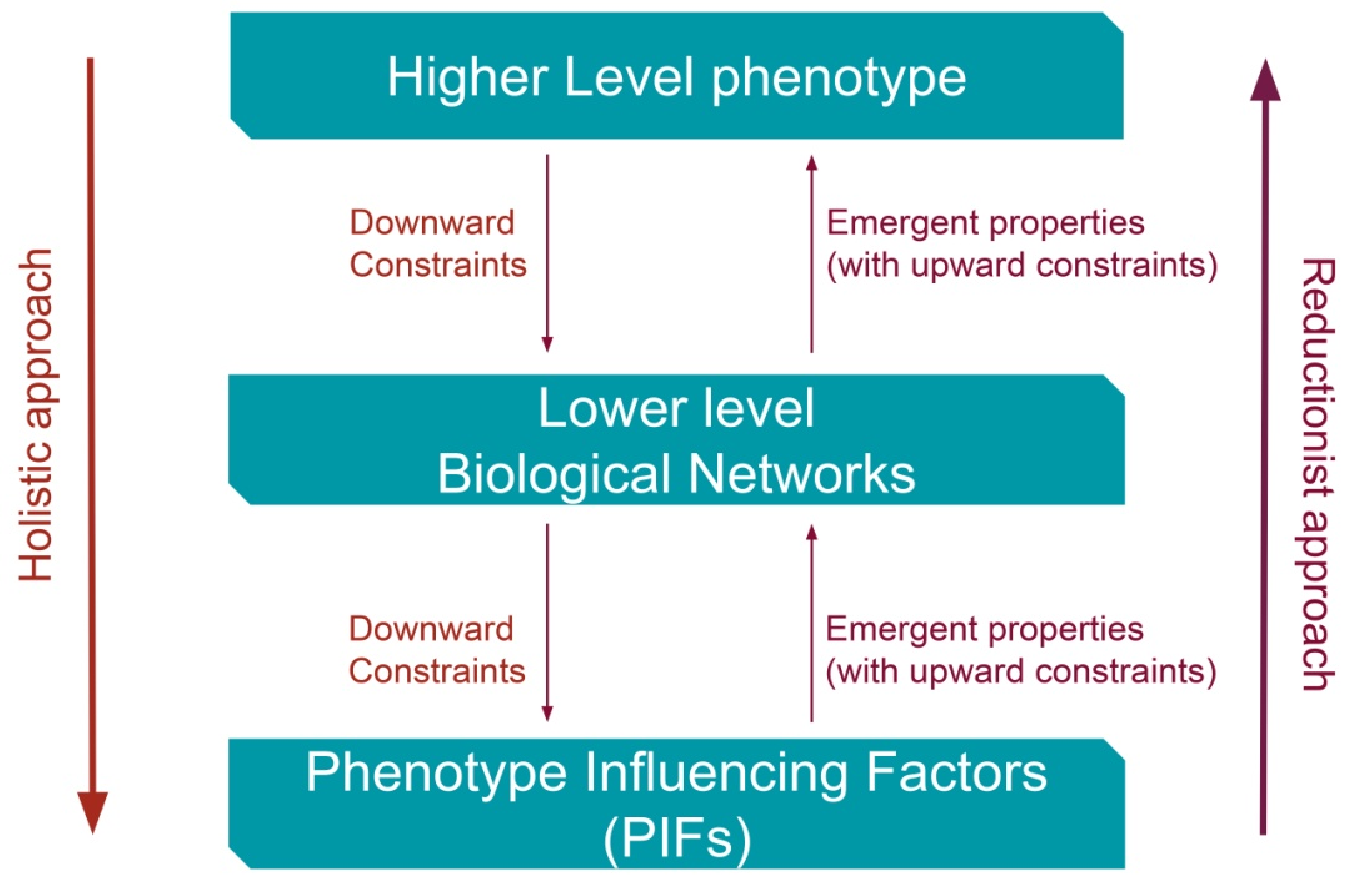
2. Constraints from Higher Levels of Disease (Phenotypes) towards Lower Disease Levels Can Reverse Causes and Effects of Illness?
3. Systems Biology
3.1. The Holistic Principle of Systems Biology
Artificial Intelligence
3.2. Systems Biology Directions
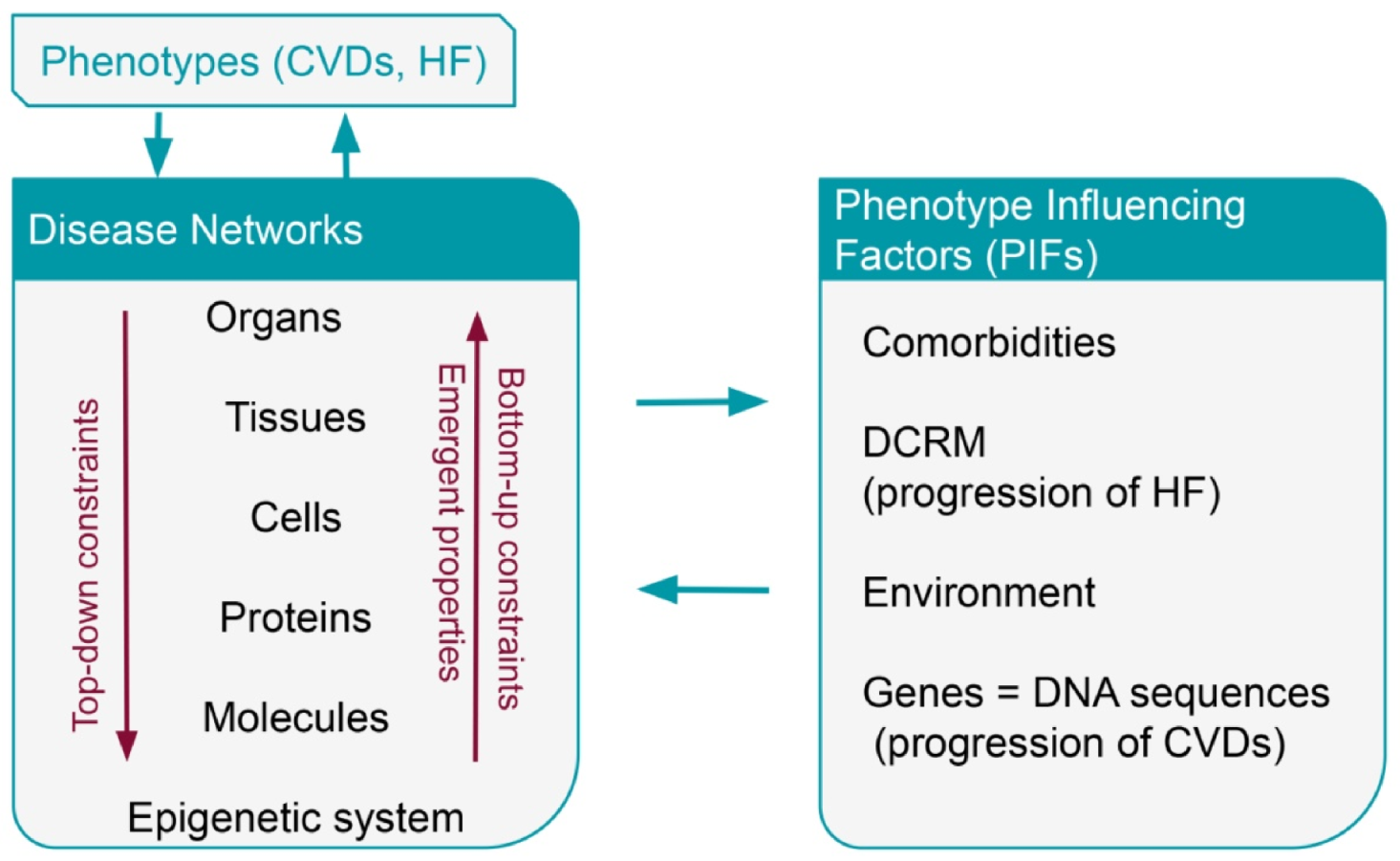
4. Phenotype Influencing Factors—PIFs
4.1. Comorbidities
4.1.1. Comorbidities in Coronary Artery Disease
4.1.2. Comorbidities in Sudden Cardiac Death
4.1.3. Comorbidities in Heart Failure
4.2. Environment
5. Progression, Prevention, and Risk Prediction
5.1. Progression of HF-Compensatory and Regulatory Mechanisms
5.2. Prevention and Prediction of Heart Failure
5.3. Progression of CAD
5.4. Prevention and Prediction of Cardiovascular Diseases
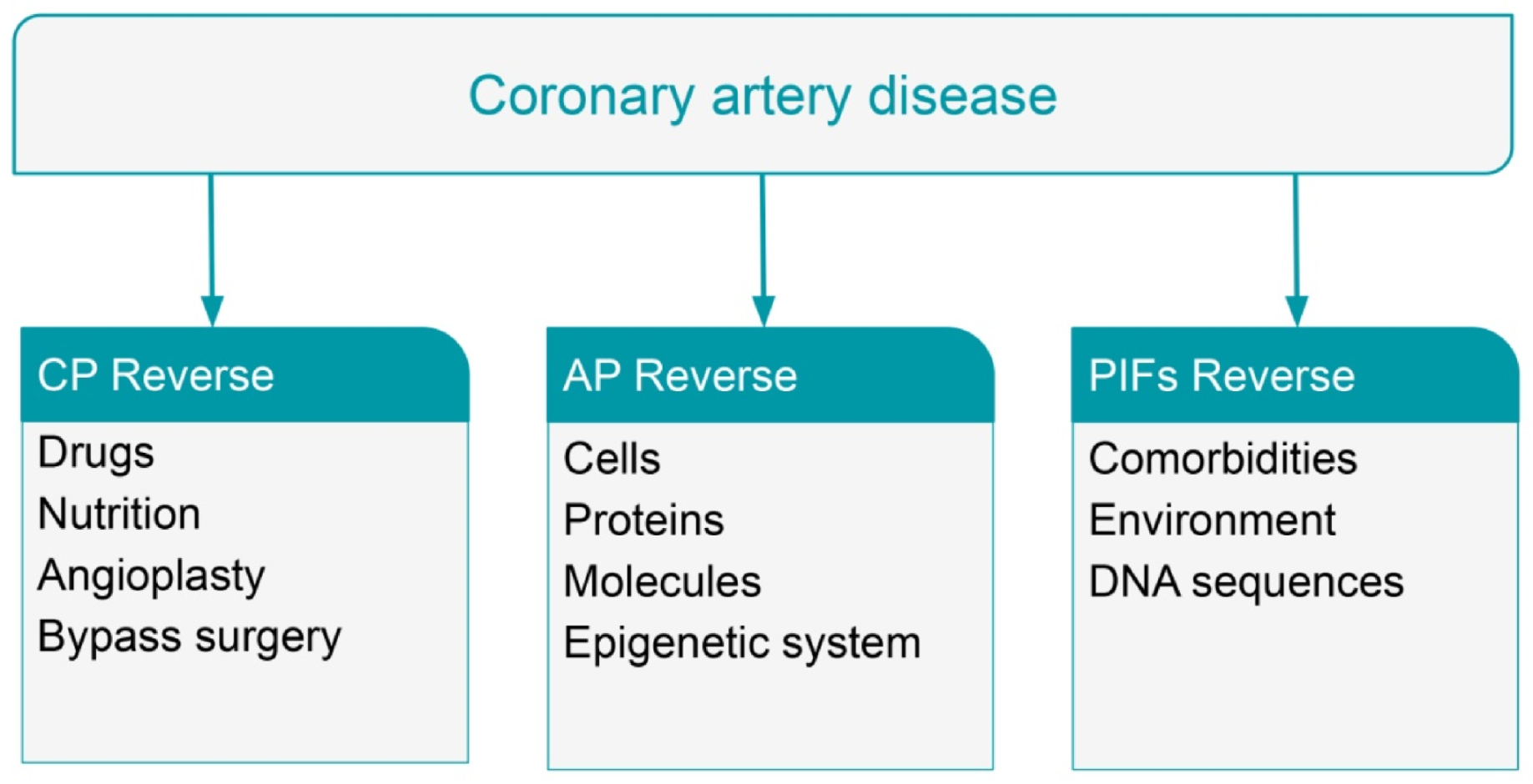
6. Reversal of Coronary Artery Disease with Statins and Coronary Aggressive Therapy
7. Reversal of Coronary Artery Disease with Nutritional Intervention
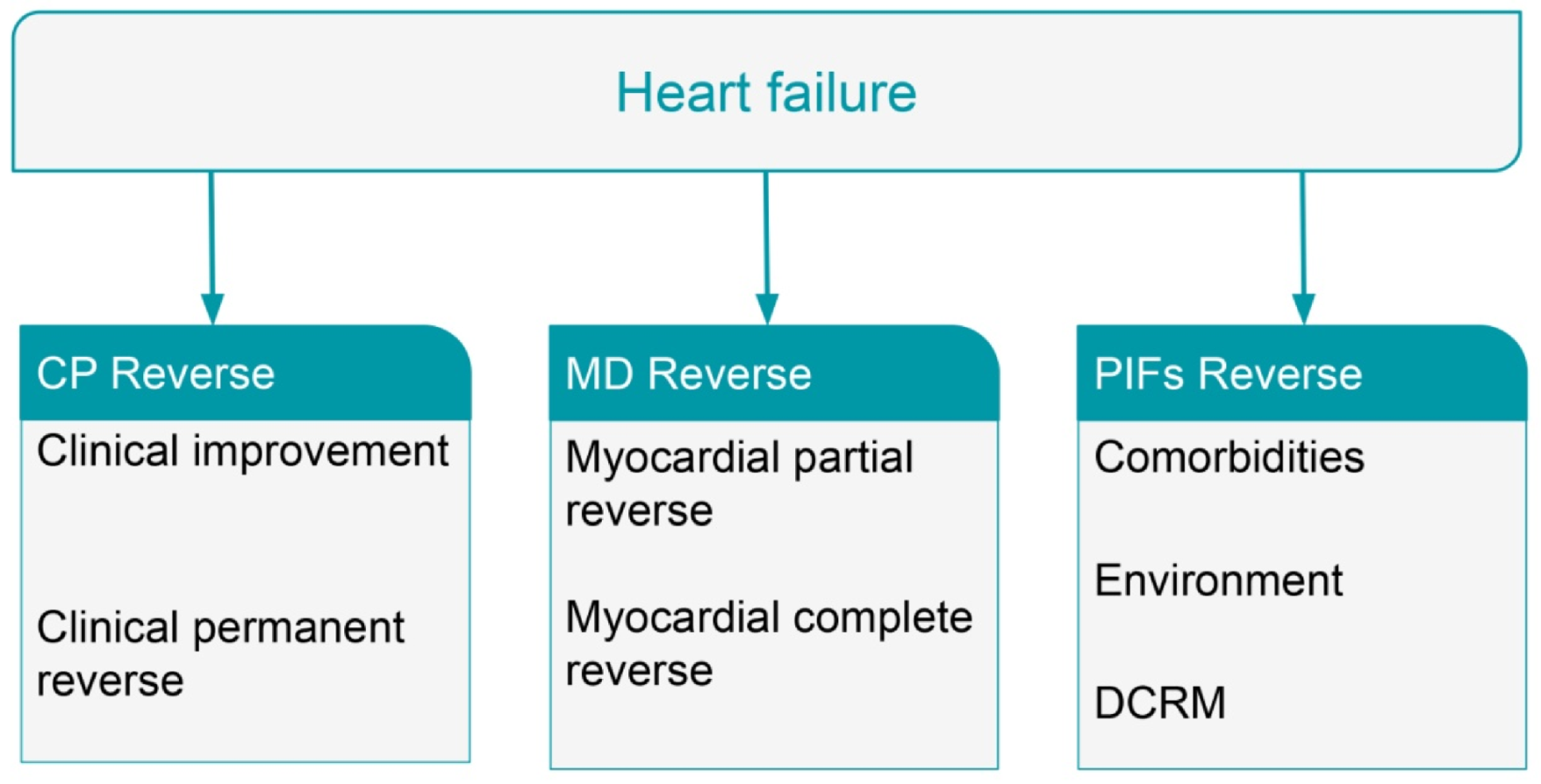
8. Reversal of Heart Failure
9. Methods for Network Analysis
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Fahed, A.C.; Jang, I.K. Plaque erosion and acute coronary syndromes: Phenotype, molecular characteristics and future directions. Nat. Rev. Cardiol. 2021, 18, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Johansson, I.; Lynoe, N. Medicine and Philosophy. A Twenty-First Century Introduction; Boston De Gruyter: Berlin, Germany, 2008. [Google Scholar] [CrossRef]
- Lourida, K.G.; Louridas, G.E. Constraints in clinical cardiology and personalized medicine: Interrelated concepts in clinical cardiology. Cardiogenetics 2021, 11, 7. [Google Scholar] [CrossRef]
- Chen, C.; Cui, J.; Zhang, W.; Shen, P. Robustness analysis identifies the plausible model of the bcl-2 apoptotic switch. FEBS Lett. 2007, 581, 5143–5150. [Google Scholar] [CrossRef]
- Steinacher, A.; Soyer, O. Evolutionary principles underlying structure and response dynamics of cellular networks. In Evolutionary Systems Biology; Soyer, O., Ed.; Springer: London, UK, 2012; pp. 225–247. [Google Scholar]
- Noble, D. Physiology is rocking the foundations of evolutionary biology. Exp. Physiol. 2013, 98, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Noble, D. Evolution beyond neo-Darwinism: A new conceptual framework. J. Exp. Biol. 2015, 218, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Noble, D. Genes and causation. Philos. Trans. R. Soc. 2008, 366, 3001–3015. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, M.; Wang, E. Dynamic modeling and analysis of cancer cellular network motifs. Integr. Biol. 2011, 3, 724–732. [Google Scholar] [CrossRef]
- Ellis, G.F.R.; Noble, D.; O’Connor, T. Top-down causation: An integrating theme within and across the sciences. Interface Focus 2012, 2, 1–3. [Google Scholar] [CrossRef]
- Kitano, H. Systems biology: A brief overview. Science 2002, 295, 1662–1664. [Google Scholar] [CrossRef]
- Louridas, G.E.; Lourida, K.G. Heart failure: A complex clinical process interpreted by systems biology approach and network medicine. Anadolou Kardiyol. Derg. 2014, 14, 178–185. [Google Scholar] [CrossRef]
- Braun, E.; Marom, S. Universality, Complexity and the Praxis of Biology: Two Case Studies. Stud. Hist. Philos. Biol. Biomed. Sci. 2015, 53, 68–72. [Google Scholar] [CrossRef]
- Green, S. Can biological complexity be reverse engineered? Stud. Hist. Philos. Biol. Biomed. Sci. 2015, 53, 73–83. [Google Scholar] [CrossRef]
- Kwon, D. Proteins in their natural habitats. Nature 2021, 598, 558–560. [Google Scholar] [CrossRef]
- Matheny, M.; Israni, S.T.; Ahmed, M.; Whicher, D. (Eds.) Artificial Intelligence in Health Care: The Hope, the Hype, the Promise, the Peril; National Academy of Medicine; NAM Special Publication: Washington, DC, USA, 2019. [Google Scholar]
- Ahmed, Z.; Mohamed, K.; Zeeshan, S.; Dong, X. Artificial intelligence with multi-functional machine learning platform development for better healthcare and precision medicine. Database 2020, 2020, baaa010. [Google Scholar] [CrossRef]
- Louridas, G.E.; Lourida, K.G. A conceptual paradigm of heart failure and systems biology approach. Intern. J. Cardiol. 2012, 159, 5–13. [Google Scholar] [CrossRef]
- Louridas, G.E.; Lourida, K.G. The complex cardiac atherosclerotic disorder: The elusive role of genetics and the new consensus of systems biology approach. J. Adv. Ther. Med. Innov. Sci. 2017, 2, 10–17. [Google Scholar] [CrossRef][Green Version]
- Noble, D. A theory of biological relativity: No privileged level of causation. Interface Focus 2012, 2, 55–64. [Google Scholar] [CrossRef]
- Louridas, G.E.; Lourida, K.G. Systems biology and constraint-based downward causation in medical clinical practice: A perspective concept for cardiology. In Advances in Medicine and Biology; Berhardt, K.V., Ed.; Nova Science Publishers Inc.: New York, NY, USA, 2020; ISBN 978-1-53617-491-5. [Google Scholar]
- Kohl, P.; Crampin, E.J.; Quinn, T.A.; Noble, D. Systems Biology: An Approach. Clin. Pharmacol. Ther. 2010, 88, 25–33. [Google Scholar] [CrossRef]
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes: The Task Force for the diagnosis and management of chronic coronary syndromes of the European Society of Cardiology (ESC). Eur. Heart J. 2020, 41, 407–477. [Google Scholar] [CrossRef]
- Yager, N.; Schulman-Marcus, J.; Torosoff, M. Coronary anatomy and comorbidities impact on elective PCI outcomes in left main and multivessel coronary artery disease. Catheter. Cardiovasc. Interv. 2021, 98, 436–444. [Google Scholar] [CrossRef]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Field, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Circulation 2018, 138, e272–e391. [Google Scholar] [CrossRef] [PubMed]
- Morin, D.P.; Homoud, M.K.; Mark Estes, N.A., 3rd. Prediction and prevention of sudden cardiac death. Card. Electrophysiol. Clin. 2017, 9, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Sapp, J. The risk and prevention of sudden death in patients with heart failure with reduced ejection fraction. Curr. Opin. Cardiol. 2020, 35, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.E.; Cowie, M.R.; Chow, A.W.C. Prediction and prevention of sudden cardiac death in heart failure. Heart 2005, 91, 674–680. [Google Scholar] [CrossRef]
- Moss, A.J.; Zareba, W.; Hall, W.J.; Klein, H.; Wilber, D.J.; Cannom, D.S.; Daubert, J.P.; Higgins, S.L.; Brown, M.W.; Andrews, M.L. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N. Engl. J. Med. 2002, 346, 877–883. [Google Scholar] [CrossRef]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef]
- Bardy, G.H.; Lee, K.L.; Mark, D.B.; Poole, J.E.; Packer, D.L.; Boineau, R.; Domanski, M.; Troutman, C.; Anderson, J.; Johnson, G.; et al. Amiodarone or an implantable defibrillator for congestive heart failure. N. Engl. J. Med. 2005, 352, 225–237. [Google Scholar] [CrossRef]
- Hayashi, M.; Shimizu, W.; Albert, C.M. The spectrum of epidemiology underlying sudden cardiac death. Circ. Res. 2015, 116, 1887–1906. [Google Scholar] [CrossRef]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2013, 62, e147–e239. [Google Scholar] [CrossRef]
- Wong, C.; Chen, S.; Iyngkaran, P. Cardiac imaging in heart failure with comorbidities. Curr. Cardiol. Rev. 2017, 13, 63–75. [Google Scholar] [CrossRef]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey Jr, D.E.; Colvin, M.M.; Drazner, M.H.; Filippatos, G.S.; Fonarow, G.C.; Givertz, M.M.; et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure. J. Am. Coll. Cardiol. 2017, 70, 776–803. [Google Scholar] [CrossRef]
- Zanoli, L.; Lentini, P.; Briet, M.; Castellino, P.; House, A.A.; London, G.M.; Malatino, L.; McCullough, P.A.; Mikhailidis, D.P.; Boutouyrie, P. Arterial stiffness in the heart disease of CKD. J. Am. Soc. Nephrol. 2019, 30, 918–928. [Google Scholar] [CrossRef]
- Xanthopoulos, A.; Starling, R.C.; Kitai, T.; Triposkiadis, F. Heart failure and liver disease: Cardiohepatic interactions. JACC Heart Fail. 2019, 7, 87–97. [Google Scholar] [CrossRef]
- Boully, C.; Hanon, O. Heart failure and comorbidities. Geriatr. Psychol. Neuropsychiatr. Vieil 2015, 13, 13–22. [Google Scholar] [CrossRef]
- Del Buono, M.G.; Iannaccone, G.; Scacciavillani, R.; Carbone, S.; Camilli, M.; Niccoli, G.; Borlaug, B.A.; Lavie, C.J.; Arena, R.; Crea, F.; et al. Heart failure with preserved ejection fraction diagnosis and treatment: An updated review of the evidence. Prog. Cardiovasc. Dis. 2020, 63, 570–584. [Google Scholar] [CrossRef]
- Louridas, G.E.; Lourida, K.G. Heart failure in patients with preserved ejection fraction: Questions concerning clinical progression. J. Cardiovasc. Dev. Dis. 2016, 3, 27. [Google Scholar] [CrossRef]
- The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N. Engl. J. Med. 1991, 325, 293–302. [Google Scholar] [CrossRef]
- Bohm, M.; Pogue, J.; Kindermann, I.; Pöss, J.; Koon, T.; Yusuf, S. Effect of comorbidities on outcomes and angiotensin converting enzyme inhibitor effects in patients with predominantly left ventricular dysfunction and heart failure. Eur. J. Heart Fail. 2014, 16, 325–333. [Google Scholar] [CrossRef]
- Grimm, W.; Koehler, U. Cardiac arrhythmias and sleep-disordered breathing in patients with heart failure. Int. J. Mol. Sci. 2014, 15, 18693–18705. [Google Scholar] [CrossRef]
- Vitovec, J.; Spinar, J.; Spinarova, L. Arrhythmias and conductance disturbances and heart failure. Vnitr. Lek. 2018, 64, 874–877. [Google Scholar] [CrossRef]
- Faber, T.S.; Zehender, M. Antiarrhythmic therapy in patients with heart failure. Ther. Umsch. 2000, 57, 324–332. [Google Scholar] [CrossRef]
- Louridas, G.; Lourida, K. Atrial fibrillation as a cause of incident heart failure: A discrete clinical phenotype. J. Atr. Fibrillation 2018, 10, 1807. [Google Scholar] [CrossRef]
- Cosselman, K.; Navas-Acien, A.; Kaufman, J. Environmental factors in cardiovascular disease. Nat. Rev. Cardiol. 2015, 12, 627–642. [Google Scholar] [CrossRef]
- Bhatnagar, A. Environmental Determinants of Cardiovascular Disease. Circ. Res. 2017, 121, 162–180. [Google Scholar] [CrossRef]
- Khera, A.V.; Emdin, C.A.; Drake, I.; Natarajan, P.; Bick, A.G.; Cook, N.R.; Chasman, D.I.; Baber, U.; Mehran, R.; Rader, D.J.; et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N. Engl. J. Med. 2016, 375, 2349–2358. [Google Scholar] [CrossRef]
- Tang, W.H.; Kitai, T.; Hazen, S.L. Gut Microbiota in Cardiovascular Health and Disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef]
- Kitai, T.; Tang, W.H.W. Gut microbiota in cardiovascular disease and heart failure. Clin. Sci. 2018, 132, 85–91. [Google Scholar] [CrossRef]
- Stewart, S.; Keates, A.; Redfern, A.; McMurray, J.J.V. Seasonal variations in cardiovascular disease. Nat. Rev. Cardiol. 2017, 14, 654–664. [Google Scholar] [CrossRef]
- Daiber, A.; Lelieveld, J.; Steven, S.; Oelze, M.; Kroller-Scxhon, S.; Sorensen, M.; Munzel, T. The "exposome" concept—how environmental risk factors influence cardiovascular health. Acta Biochim. Pol. 2019, 66, 269–283. [Google Scholar] [CrossRef]
- Louridas, G.E.; Louridas, A.G. Impact of chaos in the progression of heart failure. Int. J. Appl. Sci. Technol. 2012, 2, 24–30. [Google Scholar] [CrossRef]
- Louridas, G.E.; Lourida, K.G. Progressive nature of heart failure and systems biology. Int. Cardiovasc. Forum J. 2015, 3, 5–9. [Google Scholar] [CrossRef][Green Version]
- Fedak, P.W.; Verma, S.; Weisel, R.D.; Li, R.K. Cardiac remodeling and failure: From molecules to man (part 1). Cardiovasc. Path. 2005, 14, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Makkar, K.M.; Sanoski, C.A.; Spinler, S.A. Role of angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, and aldosterone antagonists in the prevention of atrial and ventricular arrhythmias. Pharmacotherapy 2009, 29, 31–48. [Google Scholar] [CrossRef] [PubMed]
- Konstam, M.A.; Rousseau, M.F.; Kronenberg, M.W.; Udelson, J.E.; Melin, J.; Stewart, D.; Dolan, N.; Edens, T.R.; Ahn, S.; Kinan, D.; et al. Effects of the angiotensin converting enzyme inhibitor enalapril on the long-term progression of left ventricular dysfunction in patients with heart failure. SOLVD Investigators. Circulation 1992, 86, 431–438. [Google Scholar] [CrossRef]
- Doughty, R.N.; Whalley, G.A.; Gamble, G.; MacMahon, S.; Sharpe, N. Left ventricular remodeling with carvedilol in patients with congestive heart failure due to ischemic heart disease. Australia-New Zealand Heart Failure Research Collaborative Group. J. Am. Coll. Cardiol. 1997, 29, 1060–1066. [Google Scholar] [CrossRef]
- Louridas, G.E.; Lourida, K.G. Conceptual foundations of systems biology explaining complex cardiac diseases. Healthcare 2017, 5, 10. [Google Scholar] [CrossRef]
- Wan, S.H.; Vogel, M.W.; Chen, H.H. Pre-Clinical Diastolic Dysfunction. J. Am. Coll. Cardiol. 2014, 63, 407–416. [Google Scholar] [CrossRef]
- Hamdani, N.; Bishu, K.G.; von Frieling-Salewsky, M.; Redfield, M.M.; Linke, W.A. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc. Res. 2013, 97, 464–471. [Google Scholar] [CrossRef]
- Jeong, E.M.; Monasky, M.M.; Gu, L.; Taglieri, D.M.; Patel, B.G.; Liu, H.; Wang, Q.; Greener, I.; Dudley, S.C., Jr.; Solaro, R.J. Tetrahydrobiopterin improves diastolic dysfunction by reversing changes in myofilament properties. J. Mol. Cell. Cardiol. 2013, 56, 44–54. [Google Scholar] [CrossRef]
- O’Meara, E.; Thibodeau-Jarry, N.; Ducharme, A.; Rouleau, J.L. The epidemic of heart failure: A lucid approach to stemming the rising tide. Can. J. Cardiol. 2014, 30, S442–S454. [Google Scholar] [CrossRef]
- Frigerio, M.; Oliva, F.; Turazza, F.M.; Bonow, R.O. Prevention and management of chronic heart failure in management of asymptomatic patients. Am. J. Cardiol. 2003, 91, 4F–9F. [Google Scholar] [CrossRef]
- Sahle, B.W.; Owen, A.J.; Chin, K.L.; Reid, C.M. Risk prediction models for incident heart failure: A systematic review of methodology and model performance. J. Card. Fail. 2017, 23, 680–687. [Google Scholar] [CrossRef]
- Sinha, A.; Gupta, D.K.; Yancy, C.W.; Shah, S.J.; Rasmussen-Torvik, L.J.; McNally, E.M.; Greenland, P.; Lloyd-Jones, D.M.; Sadiya, S.; Khan, S.S. Risk-based approach for the prediction and prevention of heart failure. Circ. Heart Fail. 2021, 14, e007761. [Google Scholar] [CrossRef]
- Ho, J.E.; Enserro, D.; Brouwers, F.P.; Kizer, J.R.; Shah, S.J.; Psaty, B.M.; Bartz, T.M.; Santhanakrishnan, R.; Lee, D.S.; Chan, C.; et al. Predicting heart failure with preserved and reduced ejection fraction: The international collaboration on heart failure subtypes. Circ. Heart Fail. 2016, 9, e003116. [Google Scholar] [CrossRef]
- Shinmura, K. Cardiac senescence, heart failure, and frailty: A triangle in elderly people. Keio J. Med. 2016, 65, 25–32. [Google Scholar] [CrossRef]
- Clerico, A.; Recchia, F.A.; Passino, C.; Emdin, M. Cardiac endocrine function is an essential component of the homeostatic regulation network: Physiological and clinical implications. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H17–H29. [Google Scholar] [CrossRef]
- Lanfear, D.E. Genetic variation in the natriuretic peptide system and heart failure. Heart Fail. Rev. 2010, 15, 219–228. [Google Scholar] [CrossRef]
- Hawkridge, A.M.; Heublein, D.M.; Bergen, H.R., III.; Cataliotti, A.; Burnett, J.C., Jr. Quantitative mass spectral evidence for the absence of circulating brain natriuretic peptide (BNP-32) in severe human heart failure. Proc. Natl. Acad. Sci. USA 2005, 102, 17442–17447. [Google Scholar] [CrossRef]
- Rabani, V.; Davani, S. Translational Approaches In Cardiovascular Diseases by “Omics”. Curr. Issues Mol. Biol. 2018, 28, 1–14. [Google Scholar] [CrossRef]
- Mebazaa, A.; Vanpoucke, G.; Thomas, G.; Verleysen, K.; Cohen-Solal, A.; Vanderheyden, M.; Bartunek, J.; Mueller, C.; Launay, J.M.; Van Landuyt, N.; et al. Unbiased plasma proteomics for novel diagnostic biomarkers in cardiovascular disease: Identification of quiescin Q6 as a candidate biomarker of acutely decompensated heart failure. Eur. Heart J. 2012, 33, 2317–2324. [Google Scholar] [CrossRef]
- Doehner, W. Diagnostic biomarkers in cardiovascular disease: The proteomics approach. Eur. Heart J. 2012, 33, 2249–2251. [Google Scholar] [CrossRef][Green Version]
- Stary, H.C. Natural history and histological classification of atherosclerotic lesions: An update. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1177–1178. [Google Scholar] [CrossRef]
- Nikpay, M.; Goel, A.; Won, H.H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Saleheen, D.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C.; et al. CARDIoGRAMplusC4D Consortium. A comprehensive 1000 genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015, 47, 1121–1130. [Google Scholar] [CrossRef]
- Palomaki, G.E.; Melillo, S.; Bradley, L.A. Association between 9p21 genomic markers and heart disease: A meta-analysis. JAMA 2010, 303, 648–656. [Google Scholar] [CrossRef]
- Krarup, N.T.; Borglykke, A.; Allin, K.H.; Sandholt, C.H.; Justesen, J.M.; Andersson, E.A.; Grarup, N.; Jørgensen, T.; Pedersen, O.; Hansen, T. A genetic risk score of 45 coronary artery disease risk variants associates with increased risk of myocardial infarction in 6041 Danish individuals. Atherosclerosis 2015, 240, 305–310. [Google Scholar] [CrossRef]
- O’Donnell, C.J.; Nabel, E.G. Genomics of Cardiovascular Disease. N. Engl. J. Med. 2011, 365, 2098–2109. [Google Scholar] [CrossRef]
- Bjorkegren, J.L.M.; Kovacic, J.C.; Dudley, J.T.; Schadt, E.E. Genome-wide significant loci: How important are they? Systems genetics to understand heritability of coronary artery disease and other common complex disorders. J. Am. Coll.Cardiol. 2015, 65, 830–845. [Google Scholar] [CrossRef]
- Assimes, T.L.; Roberts, R. Genetics: Implications for prevention and management of coronary artery disease. J. Am. Coll. Cardiol. 2016, 68, 2797–2818. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Fallin, M.D. Epigenetics at the crossroads of genes and the environment. JAMA 2015, 314, 1129–1130. [Google Scholar] [CrossRef]
- Joshi, A.; Rienks, M.; Theofilatos, K.; Mayr, M. Systems biology in cardiovascular disease: A multiomics approach. Nat. Rev. Cardiol. 2021, 18, 313–330. [Google Scholar] [CrossRef]
- Napoli, C.; Zullo, A.; Picascia, A.; Infante, T.; Francesco Paolo Mancini, F.P. Recent advances in proteomic technologies applied to cardiovascular disease. J. Cell. Biochem. 2013, 114, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Balestrieri, M.L.; Giovane, A.; Mancini, F.P. Proteomics and cardiovascular disease: An update. Curr. Med. Chem. 2008, 15, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Zuk, N.; Dubis, J.; Witkiewicz, W. The role of proteomics in prognosis and treatment of cardiovascular diseases. Przegl Lek. 2013, 70, 143–148. [Google Scholar] [PubMed]
- Mokou, M.; Lygirou, V.; Vlahou, A.; Mischak, H. Proteomics in cardiovascular disease: Recent progress and clinical implication and implementation. Expert Rev. Proteom. 2017, 14, 117–136. [Google Scholar] [CrossRef] [PubMed]
- Tunon, J.; Barbas, C.; Blanco-Colio, L.; Burillo, E.; Lorenzo, O.; Ventura, J.L.M.; Más, S.; Rupérez, F.J.; Egido, J. Proteomics and metabolomics in biomarker discovery for cardiovascular diseases: Progress and potential. Expert Rev. Proteom. 2016, 13, 857–871. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, M.L.; Mayr, M.; Gomes, A.V.; Delles, C.; Arrell, D.K.; Murphy, A.M.; Lange, R.A.; Costello, C.E.; Jin, Y.F.; Laskowitz, D.T.; et al. Transformative Impact of Proteomics on Cardiovascular Health and Disease. Circulation 2015, 132, 852–872. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.P.Y.; Ping, P.; Murphy, E. Proteomics research in cardiovascular medicine and biomarker discovery. J. Am. Coll. Cardiol. 2016, 68, 2819–2830. [Google Scholar] [CrossRef]
- Howes, J.M.; Keen, J.N.; Findlay, J.B.; Carter, A.M. The application of proteomics technology to thrombosis research: The identification of potential therapeutic targets in cardiovascular disease. Diabetes Vasc. Dis. Res. 2008, 5, 205–212. [Google Scholar] [CrossRef]
- Distelmaier, K.; Adlbrecht, C.; Jakowitsch, J.; Wagner, O.; Gerner, C.; Lang, I.M.; Kubicek, M. Proteomic profiling of acute coronary thrombosis reveals a local decrease in pigment epithelium-derived factor in acute myocardial infarction. Clin. Sci. 2012, 123, 111–119. [Google Scholar] [CrossRef]
- Tuñón, J.; Martín-Ventura, J.L.; Blanco-Colio, L.M.; Lorenzo, O.; López, J.A.; Egido, J. Proteomic strategies in the search of new biomarkers in atherothrombosis. J. Am. Coll. Cardiol. 2010, 55, 2009–2016. [Google Scholar] [CrossRef]
- Haase, T.; Börnigen, D.; Müller, C.; Tanja Zeller, T. Systems Medicine as an Emerging Tool for Cardiovascular Genetics. Front. Cardiovasc. Med. 2016, 3, 27. [Google Scholar] [CrossRef]
- Bellis, C.; Kulkarni, H.; Mamtani, M.; Kent, J.W., Jr.; Wong, G.; Weir, J.M.; Barlow, C.K.; Diego, V.; Almeida, M.; Dyer, T.D.; et al. Human plasma lipidome is pleiotropically associated with cardiovascular risk factors and death. Circulation. Cardiovasc. Genet. 2014, 7, 854–863. [Google Scholar] [CrossRef]
- Ussher, J.R.; Elmariah, S.; Gerszten, R.E.; Dyck, J.R.B. The emerging role of metabolomics in the diagnosis and prognosis of cardiovascular disease. J. Am. Coll. Cardiol. 2016, 68, 2850–2870. [Google Scholar] [CrossRef]
- Ganna, A.; Salihovic, S.; Sundstrom, J.; Broeckling, C.D.; Hedman, A.K.; Magnusson, P.K.E.; Pedersen, N.L.; Larsson, A.; Siegbahn, A.; Zilmer, M.; et al. Large-scale metabolomic profiling identifies novel biomarkers for incident coronary heart disease. PLoS Genet. 2014, 10, e1004801. [Google Scholar] [CrossRef]
- Rader, D.J.; Daugherty, A. Translating molecular discoveries into new therapies for atherosclerosis. Nature 2008, 451, 904–913. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The LDL Receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef]
- Wilson, D.J.; Gahan, M.; Haddad, L. A World Wide Web site for low-density lipoprotein receptor gene mutations in familial hypercholesterolemia: Sequence-based, tabular, and direct submission data handling. Am. J. Cardiol. 1998, 81, 1509–1511. [Google Scholar] [CrossRef]
- Frueh, J.; Maimari, N.; Lui, Y.; Kis, Z.; Mehta, V.; Pormehr, N.; Grant, C.; Chalkias, E.; Falck-Hansen, M.; Bovens, S.; et al. Systems and synthetic biology of the vessel wall. FEBS Lett. 2012, 586, 2164–2170. [Google Scholar] [CrossRef]
- Giannoglou, G.D.; Soulis, J.V.; Farmakis, T.M.; Farmakis, D.M.; Louridas, G.E. Haemodynamic factors and the important role of local low static pressure in coronary wall thickening. Int. J. Cardiol. 2002, 86, 27–40. [Google Scholar] [CrossRef]
- Szostak, J.; Martin, F.; Talikka, M.; Peitsch, M.C.; Hoeng, J. Semi-Automated curation allows causal network model building for the quantification of age-dependent plaque progression in ApoE-/- mouse. Gene Regul. Syst. Biol. 2016, 10, 95–103. [Google Scholar] [CrossRef]
- Cho, D.Y.; Kim, Y.A.; Przytycka, T.M. Chapter 5: Network biology approach to complex diseases. PLoS Comput. Biol. 2012, 8, e1002820. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Frantz, S.; Swirski, F.K.; Mulder, W.J.M.; Randolph, G.; Ertl, G.; Ntziachristos, V.; Piek, J.P.; Stroes, E.S.; Schwaiger, M.; et al. Imaging systemic inflammatory networks in ischemic heart disease. J. Am. Cardiol. 2015, 65, 1583–1591. [Google Scholar] [CrossRef]
- Wollenweber, T.; Roentgen, P.; Schäfer, A.; Schatka, I.; Zwadlo, C.; Brunkhorst, T.; Berding, G.; Bauersachs, J.; Bengel, F.M. Characterizing the inflammatory tissue response to acute myocardial infarction by clinical multi-modality noninvasive imaging. Circ. Cardiovasc. Imaging 2014, 7, 811–818. [Google Scholar] [CrossRef]
- Paynter, N.P.; Ridker, P.M.; Chasman, D.I. Are genetic tests for atherosclerosis ready for routine clinical use? Circ. Res. 2016, 118, 607–619. [Google Scholar] [CrossRef]
- Umans-Eckenhausen, M.A.; Sijbrands, E.J.; Kastelein, J.J.; Defesche, J.C. Low-density lipoprotein receptor gene mutations and cardiovascular risk in a large genetic cascade screening population. Circulation 2002, 106, 3031–3036. [Google Scholar] [CrossRef]
- Usifo, E.; Leigh, S.E.; Whittall, R.A.; Lench, N.; Alison Taylor, A.; Yeats, C.; Orengo, C.A.; Martin, A.C.R.; Celli, J.; Humphries, S.E. Low-density lipoprotein receptor gene familial hypercholesterolemia variant database: Update and pathological assessment. Ann. Hum. Genet. 2012, 76, 387–401. [Google Scholar] [CrossRef]
- Ridker, P.M. LDL cholesterol: Controversies and future therapeutic directions. Lancet 2014, 384, 607–617. [Google Scholar] [CrossRef]
- Perk, J.; De Backer, G.; Gohlke, H.; Graham, I.; Reiner, Z.; Verschuren, M.; Albus, C.; Benlian, P.; Boysen, G.; Cifkova, R.; et al. European Guidelines on cardiovascular disease prevention in clinical practice (version 2012). The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts). Eur. Heart J. 2012, 33, 1635–1701. [Google Scholar] [CrossRef]
- Ridker, P.M.; Buring, J.E.; Rifai, N.; Cook, N.R. Development and validation of improved algorithms for the assessment of global cardiovascular risk in women: The Reynolds Risk Score. JAMA 2007, 297, 611–619. [Google Scholar] [CrossRef]
- Hippisley-Cox, J.; Coupland, C.; Vinogradova, Y.; Robson, J.; Minhas, R.; Sheikh, A.; Peter Brindle, P. Predicting cardiovascular risk in England and Wales: Prospective derivation and validation of QRISK2. BMJ 2008, 336, 1475–1482. [Google Scholar] [CrossRef]
- Goff, D.C., Jr.; Lloyd-Jones, D.M.; Bennett, G.; Coady, S.; D’Agostino, R.B.; Gibbons, R.; Greenland, P.; Lackland, D.T.; Levy, D.; O’Donnell, C.J.; et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2014, 63, 2935–2959. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, S.; Choi, H.C.; Weisshaar, J.C. The spatial biology of transcription and translation in rapidly growing Escherichia coli. Front. Microbiol. 2015, 6, 636. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, A.; Mathe, E.A.; Li, L.; Liu, B.; Ma, Q. Elucidation of biological networks across complex diseases using single-cell omics. Trends Genet. 2020, 36, 951–966. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Stein, J.E.; Rimm, D.L.; Wang, D.W.; Bell, J.M.; Johnson, D.B.; Sosman, J.A.; Schalper, K.A.; Anders, R.A.; Wang, H.; et al. Comparison of Biomarker Modalities for Predicting Response to PD-1/PD-L1 Checkpoint Blockade: A Systematic Review and Meta-analysis. JAMA Oncol. 2019, 5, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Chhibber, A.; Kroetz, D.L.; Tantisira, K.G.; McGeachie, M.; Cheng, C.; Plenge, R.; Stahl, E.; Sadee, W.; Ritchie, M.D.; Sarah A Pendergrass, S.A. Genomic architecture of pharmacological efficacy and adverse events. Pharmacogenomics 2014, 15, 2025–2048. [Google Scholar] [CrossRef] [PubMed]
- Biesecker, L.G. Opportunities and challenges for the integration of massively parallel genomic sequencing into clinical practice: Lessons from the ClinSeq project. Genet. Med. 2012, 14, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.T.; Boerwinkle, S.E.; O’Connell, J.R.; Bailey, K.R.; Gong, Y.; Chapman, A.B.; McDonough, C.W.; Beitelshees, A.L.; Schwartz, G.L.; Gums, J.G.; et al. Genomic association analysis of common variants influencing antihypertensive response to hydrochlorothiazide. Hypertension 2013, 62, 391–397. [Google Scholar] [CrossRef]
- Ross, S.; Eikelboom, J.; Anand, S.S.; Eriksson, N.; Gerstein, H.C.; Mehta, S.; Connolly, S.J.; Rose, L.; Ridker, P.M.; Wallentin, L.; et al. Association of cyclooxygenase-2 genetic variant with cardiovascular disease. Eur. Heart J. 2014, 35, 2242–2248. [Google Scholar] [CrossRef]
- Polisecki, E.; Muallem, H.; Maeda, N.; Peter, I.; Robertson, M.; McMahon, A.D.; Ford, I.; Packard, C.; Shepherd, J.; Jukema, J.W.; et al. Prospective Study of Pravastatin in the Elderly at Risk (PROSPER) Investigators. Genetic variation at the LDL receptor and HMG-CoA reductase gene loci, lipid levels, statin response, and cardiovascular disease incidence in PROSPER. Atherosclerosis 2008, 200, 109–114. [Google Scholar] [CrossRef]
- Postmus, I.; Trompet, S.; Deshmukh, H.A.; Barnes, M.R.; Li, X.; Warren, H.R.; Chasman, D.I.; Zhou, K.; Arsenault, B.J.; Donnelly, L.A.; et al. Welcome Trust Case Control Consortium. Pharmacogenetic meta-analysis of genome-wide association studies of LDL cholesterol response to statins. Nat. Commun. 2014, 5, 5068. [Google Scholar] [CrossRef]
- Stewart, A. SLCO1B1 polymorphisms and statin-induced myopathy. PLoS Curr. 2013, 5. [Google Scholar] [CrossRef]
- Pereira, N.L.; Bavry, A.A.; Eagle, K.A. Tailored Antiplatelet Initiation to Lessen Outcomes due to Decreased Clopidogrel Response after Percutaneous Coronary Intervention-TAILOR PCI. In Proceedings of the ACC.21 Meeting, Virtual, 17 May 2021. [Google Scholar]
- Johnson, J.A.; Gong, L.; Whirl-Carrillo, M.; Gage, B.F.; Scott, S.A.; Stein, C.M.; Anderson, J.L.; Kimmel, S.E.; Lee, M.T.M.; Pirmohamed, M.; et al. Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin. Pharmacol. Ther. 2011, 90, 625–629. [Google Scholar] [CrossRef]
- Pirillo, A.; Casula, M.; Olmastroni, E.; Norata, G.D.; Catapano, A.L. Global epidemiology of dyslipidaemias. Nat. Rev. Cardiol. 2021, 18, 689–700. [Google Scholar] [CrossRef]
- EMG-Health Podcast: Unmet Need among Patients with Atherosclerotic Cardiovascular Disease Receiving Statin Therapy. Primary Prevention and Treatment of Atherosclerotic Cardiovascular Disease. Available online: https://www.emg-health.com/omnipresent/emg-health-podcast-unmet-needs-among-patients-with-atherosclerotic-cardiovascular-disease-receiving-statin/ (accessed on 1 December 2021).
- Adhyaru, B.B.; Jacobson, T.A. Safety and efficacy of statin therapy. Nat. Rev. Cardiol. 2018, 15, 757–769. [Google Scholar] [CrossRef]
- Gui, Y.J.; Liao, C.X.; Liu, Q.; Guo, Y.; Yang, T.; Chen, J.Y.; Wang, Y.T.; Hu, J.H.; Xu, D.Y. Efficacy and safety of statins and exercise combination therapy compared to statin monotherapy in patients with dyslipidaemia: A systematic review and meta-analysis. Eur. J. Prev. Cardiol. 2017, 24, 907–916. [Google Scholar] [CrossRef]
- Baigent, C.; Keech, A.; Kearney, P.M.; Blackwell, L.; Buck, G.; Pollicino, C.; Kirby, A.; Sourjina, T.; Peto, R.; Collins, R.; et al. Efficacy and safety of cholesterol-lowering treatment: Prospective meta-analysis of data from 90,056 participants in 14 randomized trials of statins. Lancet 2005, 366, 1267–1278. [Google Scholar] [CrossRef]
- Pownall, H.J.; Rosales, C.; Gillard, B.K.; Gotto, A.M., Jr. High-density lipoproteins, reverse cholesterol transport and atherogenesis. Nat. Rev. Cardiol. 2021, 18, 712–723. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk: The task force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Boden, W.E.; O’Rourke, R.A.; Teo, K.K.; Hartigan, P.M.; Maron, D.J.; Kostuk, W.J.; Knudtson, M.; Dada, M.; Paul Casperson, P.; Harris, C.L.; et al. Optimal medical therapy with or without PCI for stable coronary disease. N. Engl. J. Med. 2007, 356, 1503–1516. [Google Scholar] [CrossRef]
- Trogdon, J.G.; Finkelstein, E.A.; Nwaise, I.A.; Tangka, F.K.; Orenstein, D. The economic burden of chronic cardiovascular disease for major insurers. Health Promot. Pract. 2007, 8, 234–242. [Google Scholar] [CrossRef]
- Amitzur, G.; Einav, S. Three Cardiovascular Physiological Parameters, Endothelial Function, Arterial Stiffness and Autonomic System Function, Represent Unique Complementary Properties. Tel Aviv: Academia, Tel Aviv University. 2007. Available online: https://telaviv.academia.edu/GioraAmitzur (accessed on 1 March 2021).
- Yamaoka-Tojo, M. Endothelial function for cardiovascular disease prevention and management. Int. J. Clin. Cardiol. 2017, 4, 103. [Google Scholar] [CrossRef]
- Oikonomou, E.; Siasos, G.; Tsigkou, V.; Bletsa, E.; Panoilia, M.E.; Oikonomou, I.N.; Simanidis, I.; Spinou, M.; Papastavrou, A.; Kokosias, G.; et al. Coronary artery disease and endothelial dysfunction: Novel diagnostic and therapeutic approaches. Curr. Med. Chem. 2020, 27, 1052–1080. [Google Scholar] [CrossRef]
- de Koning, E.J.; Rabelink, T.J. Endothelial function in the post-prandial state. Atheroscler. Suppl. 2002, 3, 11–16. [Google Scholar] [CrossRef]
- Vogel, R.A.; Corretti, M.C.; Plotnick, G.D. Effect of a single high-fat meal on endothelial function in healthy subjects. Am. J. Cardiol. 1997, 79, 350–354. [Google Scholar] [CrossRef]
- Giannattasio, C.; Zoppo, A.; Gentile, G.; Failla, M.; Capra, A.; Maggi, F.M.; Catapano, A.; Mancia, G. Acute effect of high-fat meal on endothelial function in moderately dyslipidemic subjects. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 406–410. [Google Scholar] [CrossRef]
- Rathnayake, K.M.; Weech, M.; Jackson, K.G.; Lovegrove, J.A. Impact of meal fatty acid composition on postprandial lipaemia, vascular function and blood pressure in postmenopausal women. Nutr. Res. Rev. 2018, 31, 193–203. [Google Scholar] [CrossRef]
- Suwaidi, J.A.; Hamasaki, S.; Higano, S.T.; Nishimura, R.A.; Holmes, D.R., Jr.; A Lerman, A. Long-term follow-up of patients with mild coronary artery disease and endothelial dysfunction. Circulation 2000, 101, 948–954. [Google Scholar] [CrossRef]
- Esselstyn Jr, C.B.; Gendy, G.; Doyle, J.; Golubic, M.; Roizen, M.F. A way to reverse CAD? J. Fam. Pract. 2014, 63, 356–364. [Google Scholar]
- Dehghan, M.; Mente, A.; Teo, K.K.; Gao, P.; Sleight, P.; Dagenais, G.; Avezum, A.; Probstfield, J.L.; Dans, T.; Yusuf, S. Ongoing Telmisartan Alone and in Combination With Ramipril Global End Point Trial (ON TARGET)/Telmisartan Randomized ASSESSMENT Study in ACEI Intolerant Subjects With Cardiovascular Disease (TRANSCEND) Trial Investigators. Relationship between healthy diet and risk of cardiovascular disease among patients on drug therapies for secondary prevention: A prospective cohort study of 31,546 high-risk individuals from 40 countries. Circulation 2012, 126, 2705–2712. [Google Scholar] [CrossRef]
- Crowe, F.L.; Appleby, P.N.; Travis, R.C.; Key, T.J. Risk of hospitalization or death from ischemic heart disease among British vegetarians and nonvegetarians: Results from the EPIC-Oxford cohort study. Am. J. Clin. Nutr. 2013, 97, 597–603. [Google Scholar] [CrossRef]
- Huang, B.T.; Peng, Y.; Liu, W.; Zhang, C.; Chai, H.; Huang, F.Y.; Zuo, Z.L.; Liao, Y.B.; Xia, T.L.; Chen, M. Nutritional state predicts all-cause death independent of comorbidities in geriatric patients with coronary artery disease. J. Nutr. Health Aging 2016, 20, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Patnode, C.D.; Evans, C.V.; Senger, C.A.; Redmond, N.; Lin, G.S. Behavioral counseling to promote a healthful diet and physical activity for cardiovascular disease prevention in adults without know cardiovascular disease risk factors: Updated evidence report and systematic review for the US preventive services task force. JAMA 2017, 318, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Yiaslas, T.A.; Sood, A.; Ono, G.; Rogers-Soeder, T.S.; Kitazono, R.E.; Embree, J.; Spann, C.; Caputo, C.A.; Taylor, J.; Schaefer, S. The Design and Implementation of a Heart Disease Reversal Program in the Veterans Health Administration: Before and During the COVID-19 Pandemic. Fed. Pract. 2020, 37, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Tilinin, I.S. Design and Implementation of Reverse Risk Assessment Software. Master’s Theses, San Jose State University, San Jose, CA, USA, 2008. [Google Scholar]
- Ponikowski, P.; Voors, A.A.; Anker, S.D. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef] [PubMed]
- Leach, J.; Heallen, T.; Zhang, M.; Rahmani, M.; Morikawa, Y.; Hill, M.C.; Segura, A.; Willerson, J.T.; Martin, J.F. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 2017, 550, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, J.E.; Fang, J.C.; Margulies, K.B.; Mann, D.L. Heart failure with recovered left ventricular ejection fraction: JACC Scientific Expert Panel. J. Am. Coll. Cardiol. 2020, 76, 719–734. [Google Scholar] [CrossRef]
- Bermejo, R.A.; Babarro, E.G.; Canoa, J.N.L.; Román, A.V.; Otero, I.G.; Ayude, M.O.; Vazquez, P.P.; Rodríguez, I.G.; Castro, O.D.; Juanatey, J.R.G. Heart failure with recovered ejection fraction: Clinical characteristics, determinants and prognosis. CARDIOCHUS-CHOP registry. Cardiol. J. 2018, 25, 353–362. [Google Scholar] [CrossRef]
- Tanabe, K.; Sakamoto, T. Heart failure with recovered ejection fraction. J. Echocardiogr. 2019, 17, 5–9. [Google Scholar] [CrossRef]
- Gutowska, K.; Kogut, D.; Kardynska, M.; Formanowicz, P.; Smieja, J.; Puszynski, K. Petri nets and ODEs as complementary methods for comprehensive analysis on an example of the ATM-p53-NF-[Formula: See text]B signaling pathways. Sci. Rep. 2022, 12, 1135. [Google Scholar] [CrossRef]
- Costa, M.C.; Gabriel, A.F.; Enguita, F.J. Bioinformatics Research Methodology of Non-coding RNAs in Cardiovascular Diseases. Adv. Exp. Med. Biol. 2020, 1229, 49–64. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef]
- Wörheide, M.A.; Krumsiek, J.; Kastenmüller, G.; Arnold, M. Multi-omics integration in biomedical research—A metabolomics-centric review. Anal. Chim. Acta 2021, 1141, 144–162. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lourida, K.G.; Louridas, G.E. Clinical Phenotypes of Cardiovascular and Heart Failure Diseases Can Be Reversed? The Holistic Principle of Systems Biology in Multifaceted Heart Diseases. Cardiogenetics 2022, 12, 142-169. https://doi.org/10.3390/cardiogenetics12020015
Lourida KG, Louridas GE. Clinical Phenotypes of Cardiovascular and Heart Failure Diseases Can Be Reversed? The Holistic Principle of Systems Biology in Multifaceted Heart Diseases. Cardiogenetics. 2022; 12(2):142-169. https://doi.org/10.3390/cardiogenetics12020015
Chicago/Turabian StyleLourida, Katerina G., and George E. Louridas. 2022. "Clinical Phenotypes of Cardiovascular and Heart Failure Diseases Can Be Reversed? The Holistic Principle of Systems Biology in Multifaceted Heart Diseases" Cardiogenetics 12, no. 2: 142-169. https://doi.org/10.3390/cardiogenetics12020015
APA StyleLourida, K. G., & Louridas, G. E. (2022). Clinical Phenotypes of Cardiovascular and Heart Failure Diseases Can Be Reversed? The Holistic Principle of Systems Biology in Multifaceted Heart Diseases. Cardiogenetics, 12(2), 142-169. https://doi.org/10.3390/cardiogenetics12020015