Highlights
Purpose
- The paper is focused on the probability of reversal of the causes and effects of cardiovascular diseases (CVDs) and heart failure (HF).
- We proposed a methodology based on the application of the holistic principles of systems biology (SB) with the help of artificial intelligence (AI).
- This proposal requires a large amount of relative data; it employs a multi-omics approach and network analysis in order to understand the concept of clinical complexity.
- We believe this approach will help in the appreciation of the complexity of chronic heart diseases and explain the interconnection between their biological and clinical networks.
- We also believe that this methodology will enhance the prospects for research and clinical applications aimed at the reversal of clinical phenotypes.
Abstract
Recent advances in cardiology and biological sciences have improved quality of life in patients with complex cardiovascular diseases (CVDs) or heart failure (HF). Regardless of medical progress, complex cardiac diseases continue to have a prolonged clinical course with high morbidity and mortality. Interventional coronary techniques together with drug therapy improve quality and future prospects of life, but do not reverse the course of the atherosclerotic process that remains relentlessly progressive. The probability of CVDs and HF phenotypes to reverse can be supported by the advances made on the medical holistic principle of systems biology (SB) and on artificial intelligence (AI). Studies on clinical phenotypes reversal should be based on the research performed in large populations of patients following gathering and analyzing large amounts of relative data that embrace the concept of complexity. To decipher the complexity conundrum, a multiomics approach is needed with network analysis of the biological data. Only by understanding the complexity of chronic heart diseases and explaining the interrelationship between different interconnected biological networks can the probability for clinical phenotypes reversal be increased.
1. Introduction
This review paper is focused on the probability of reversal of the causes and effects of cardiovascular diseases (CVDs) and heart failure (HF) and the possibility of genetic guidance for risk prediction and primary prevention. For non-chronic human diseases such as bacterial or virus infections, usually there is full recovery. In contrast, the CVDs and HF are chronic illnesses with a progressive course and a continuously deteriorating clinical picture and high mortality. The present treatments have concentrated on disease symptoms, without rectifying the underlying cause of disease. Acute coronary syndromes (ACSs) continue to be leading causes of mortality despite changes in clinical presentation with decline in the incidence of ST-segment elevation myocardial infarction (STEMI) and increase in non-STEMI [1]. This paper also addresses the impact of the Phenotype Influencing Factors (PIFs) (comorbidities, compensatory regulatory mechanisms, environmental factors, and genes) on the development of complex cardiac disease phenotypes, and the potential of the phenotype to be reversed through therapy applied on PIFs. Comorbidities are important risk factors for clinical cardiac phenotype development and for the atherosclerotic lesion complexity. The comorbidities’ burden remains as an important clinical predictor of mortality. In HF, the most important compensatory regulatory systems, the neurohumoral and the cardiac remodeling systems, contribute to further morbidity and mortality. Reversal of CVDs and HF could be achieved with further clarification of the role played by comorbidities as well as from advances in sequencing technologies and novel computational appliances, able to build up entire genomes including genetic variations. The impact of multiomics advances on the status of the molecular mechanisms exploring and managing complex diseases is enormous, particularly at the predictive and preemptive levels. Advances made in genetic, proteomic, and metabolomic technologies can facilitate cardiovascular research, production of new biomarkers, and personalized level identification of therapeutic targets. That in conjunction with the holistic principle of systems biology (SB) and the notion of diseases’ interactive and integrative networks will help build a system able to discuss complex questions in diverse clinical and biomedical fields. Artificial intelligence (AI) application to the field of healthcare is already used in medical diagnostic and therapeutic systems such as robotic surgery or decision-making medical instruments. Integration of knowledge from both AI applications and SB understanding that the complex heart diseases are made up from interactive and integrative network systems will likely provide some answers to the question: can the heart disease calamity be reversed? To paraphrase Kuhn’s “concept of paradigm” as applied to medicine, we can point out that there is a conflict between the partial knowledge of medicine that we possess and the probability that this present knowledge is inadequate to explain the whole process of a disease; the present disease’s understanding is a temporary conceptual artifact [2]. The purpose of this review paper is to emphasize the importance of integrating all the available interactive network cardiac disease systems with help from both SB and AI in order to understand disease progression and, thus, increase the probability for clinical phenotypes reversal. In this context, the present review initially refers to recent progress made in the holistic principle of SB and its application to clinical cardiology and the potentiality of genetic guidance and PIFs for prediction and primary prevention of complex cardiac diseases, and finally emphasizing the prospects for clinical phenotypes reversal.
2. Constraints from Higher Levels of Disease (Phenotypes) towards Lower Disease Levels Can Reverse Causes and Effects of Illness?
Two interrelated holistic concepts of SB are related to clinical cardiology: the emerging properties with upward direction (upward constraints) and the constraint-based thinking with downward causation (downward constraints) (Figure 1) [3].
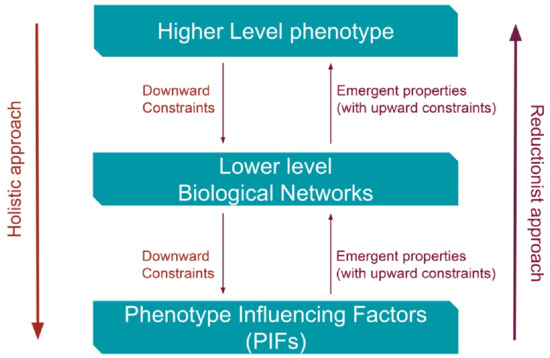
Figure 1.
Constraints and emergent properties: two concepts of systems biology (SB) explaining the interrelationship and interaction between higher and lower levels of the biological ladder. PIFs = Phenotype Influencing Factors (comorbidities, diseases’ compensatory–regulatory mechanisms (DCRM), environmental factors and deoxyribonucleic acid (DNA) sequences).
There are uncertainties regarding the existence of “recovery” constraint-based mechanisms that are originating from phenotypes and are addressed for the lower disease levels. There is uncertainty whether those constraint-based mechanisms are sufficient and compatible for a full or partial recovery. In view of the complexities of CVDs and HF illnesses, it is important to elucidate whether their progressive and clinically deteriorating nature could be reversed with implementation of new “repairing” constraints in a downward direction. The “repair” constraint-based reversal, from phenotypes to biological networks until the molecular or cellular level, should override the established robust mechanisms of the CVDs or HF. The constraint-based thinking constitutes a holistic concept with downward causation interpreting complexity in biology as well as chronic progression of human diseases. Causal reasoning constraints are robust antagonistic biological processes addressed to every evolutionary change in order to strengthen natural selection [3]. The constraint-based reasoning was also applied to strengthen robustness of some experimental biological models [4]. It is imperative to understand that the new repairing constraints should overcome the existing robust restrictions implemented by the chronic heart diseases. The robustness of biological (or disease) networks is connected to the non-random connectivity dissemination and to the hierarchical structure (disease progression) [5]. Biological (or disease) networks possess a high degree of functional overlap, and molecular or cellular networks activated in one biological level can be energized at the same time in other lower or higher levels. Thus, the whole biological functional network system is dynamically interconnected. Chronic heart diseases represent dynamic biological states that, during progression to complex clinical phenotypes, implement constraints with robust changes not only to the lower levels of disease networks but also to some PIFs such as comorbidities and environmental factors. A commonsense question is whether these lower-level changes are partially reversible with an overriding new constraint-based “repair” mechanism with downwards direction, due to the dynamic nature of the disease system or if these changes can be reversed with therapy. At the present level of knowledge, only the appropriate existing therapy seems to be available for a disease’s possible partial “repair”. Specific biological constraints from phenotype towards lower disease levels in order to completely reverse causes and effects of chronic heart diseases are inconspicuous; of course, the possibility for some unnoticeable partial “repair” constraints cannot be ruled out. Future research advances in cellular, tissue, and particularly in network level will change present chronic heart disease understanding.
3. Systems Biology
3.1. The Holistic Principle of Systems Biology
The whole idea of CVDs and HF reversal is related to the holistic principle of SB in conjunction with the notion of interactive and integrative disease networks. The basic holistic thinking explores the whole (phenotypes, biological networks, multiomics, and disease progression) rather than isolated biological parts. The holistic principle of SB presents an active and expanding scientific field founded on the principles of integrative computational analysis and produces interacting biological networks and phenotypes with wide applications to clinical medicine.
To understand the concept of the holistic principle of SB and its application to the disease reversal hypothesis, we should briefly address some recent developments in biology. Modern Synthesis (neo-Darwinist) is a theory of evolution based on the reductionist approach as a gene-centered theory of evolution, where a specific biological structure is broken down into its fractions in order to understand the chemical principles underlying molecular activities. A new theory called the theory of Biological Relativity is proposed to replace Modern Synthesis, as it is based on the integration of a variety of interacting mechanisms and networks that are dynamically functional [6]. The proposed conceptual theory of Biological Relativity or Integrative Synthesis is based on integrated systems of complex networks while the “active causation resides in the networks which include many components for which there are no deoxyribonucleic acid (DNA) templates”; and “it is the physics and chemistry of those dynamic networks that determine what happens” [7]. Genes are considered passive causes and they are activated through a variety of cellular mechanisms; rather “active causation lies with proteins, membranes, metabolites, organelles, and the dynamic functional networks they form in interaction with the environment” [8]. Dynamic analysis of cancer cellular networks “allowed biological studies to move from a reductionist approach, such as isolation of specific pathways and mechanisms, to a more integrative approach, where biological systems are seen as a network of interconnected components” [9]. The holistic principle of downward (top-down) causation in the biological ladder with an integrated systems view differs from the reductionist approach that understands biological reality as upward (bottom-up) causation [10]. The reductionist approach assumes that sufficient details are available about lower-scale processes; a fact that is untrue. The integrative network analysis and the constraint-based thinking are important tools for having a better understanding of the intrinsic complexity of CVDs and HF [11,12].
In biological research there is a problem to overcome: at first, if the biological complexity and networks can be reversed engineered, and, secondly, if design principles are fundamental to complex networks’ functional capabilities. Braun and Marom [13] disagree that in biological complexity there is a role for reverse engineering and describe “two universal characteristics of biological complexity: two-way microscopic-macroscopic degeneracy and lack of time scale separation within and between levels of organization”; also, they stress the difficulties in separating any given biological level from all other levels, and they emphasize the ability of maintaining dynamic stability that is unrelated to constraints. Green [14] clarifies “how the notion of design principles can be more broadly conceived and that reverse engineering is compatible with a dynamic view of organisms”, and makes clear the “productive role of reverse engineering that goes beyond reductionism, and, also, the regulative role of design principles to general constraints on phenotypic variation”. Modern methodology of microscopy such as the cryo-electron tomography allows the observation of the hidden cellular proteins at high resolution replacing “older methods that take individual proteins out of their niches to study them” with new techniques that “provide a holistic view of proteins and other molecules together with the cellular landscape” [15].
Artificial Intelligence
AI methodologies improve, through digital health application, the quality of life, home medical care, and daily clinical cardiology practice by upgrading medical clinical information from cardiac imaging to appropriate clinical decisions. The potential of AI to collect, analyze, and integrate electronic data from clinical and non-clinical sources is significant for understanding the complexities of chronic cardiac diseases at the individual level and, in addition, the possible reversal of the clinical process. Interpretation of complex diseases’ interactive and integrative networks (molecular and clinical) and increase in data quality and access could be achieved through AI; “promoting data quality, access, and sharing, as well as the use of both structured and unstructured data and the integration of non-clinical data is critical to developing effective AI tools” [16]. It is imperative to use AI for integrative network analysis and constraint-based thinking in order to understand the intrinsic complexity of chronic heart diseases and obtain electronic health records by incorporating diverse data references uncovering individual patient-related modes of disease progression. It seems that “while the complexities of disease at the individual level have made it difficult to utilize healthcare information in clinical decision making, some of the existing constraints have been greatly minimized by technological advancements” [17].
Accumulating a large number of relative materials and following a multiomics approach with a network analysis of the data, we can address the concept of complexity in chronic heart diseases. The interrelationship and interconnection between disease networks (risks, progression, prediction, and prevention) probably is the answer to the problem of clinical phenotype reversal (Figure 2).
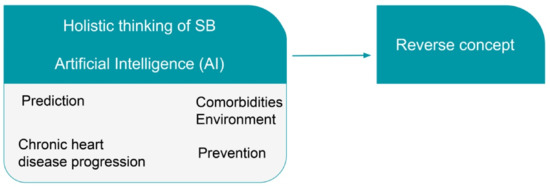
Figure 2.
The holistic thinking of Systems Biology (SB), as it deals with the disease as a whole (clinical phenotype), together with the advances made by Artificial Intelligence (AI) will help to understand and explain the possibility to reverse cardiovascular diseases (CVDs) and heart failure (HF).
3.2. Systems Biology Directions
The SB methodology explains the complex clinical phenotype using two complementary directions of research and understanding; the top-down direction and the bottom-up direction that present and analyze biological and clinical processes with a different and distinct approach (Figure 3).
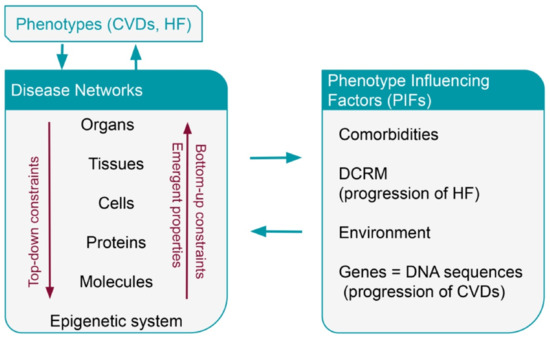
Figure 3.
Interaction between phenotypes, disease networks and Phenotype Influencing Factors (PIFs) in CVDs and HF. CVDs = cardiovascular diseases; HF = heart failure; PIFs = Phenotype Influencing Factors (PIFs); DCRM = disease compensatory regulatory mechanisms; deoxyribonucleic acid (DNA).
In clinical medicine, the hierarchical development of a complex disease demonstrates interacting connections between biological processes and clinical stages that are important to follow up disease progression until the end stage [18,19]. Biological causation is present in both directions, upward and downward, but most important for the SB approach is the downward causality. The top-down (downward direction) causation is the holistic explanation of the biological phenomenon while the reductionist interpretation is the bottom-up (upward direction) causation [10]. The concept of emergence and self-organization are important scientific tools for understanding the construction of networks with emergent properties in an upward direction. Both directions, upward and downward, are symbolic as causality in the case of organisms’ work “through many forms of circular causality” [20]. With SB approach, the complex CVDs and HF are behaving as hierarchically organized biological systems having multileveled understanding and represent a multi-stage model of disease progression. The mathematical concept of physical constraints and the concept of constraints in information theory (the degree of dependence between variables) could be conveyed to clinical cardiology to interpret some of the features of CVDs [18,21].
4. Phenotype Influencing Factors—PIFs
In biological systems including human diseases and in downward causation with application of constraints, the functional properties of higher levels (clinical phenotypes) have an impact on biological networks at the lower levels. Biological properties of organs, tissues, and cells have a significant impact on epigenetic systems, molecules, and proteins. A variety of PIFs such as comorbidities, diseases’ compensatory-regulatory mechanisms (DCRM), environmental factors, and genes (DNA sequences) play a significant functional role interacting with all biological networks. Multiple disease interacting networks, from the epigenetic domain to the intrinsic complexity of atherosclerotic heart disease or failing myocardium, are responsible for the clinical syndromes of CVDs and HF, and their progressive nature [12]. The diseases’ networks are interacting with PIFs affecting the genesis of phenotype complexity and its progressive course; this interaction between phenotype and PIFs is exercised through the disease networks [18]. Complex diseases’ active causation dwells in the dynamically functional networks of phenotypes, which are interacting with PIFs and lower-level disease networks from organs to epigenetic systems. The interaction of “DNA sequences, environment and phenotype as occurring through biological networks” is central to understanding of the genesis of phenotypes in biological entities [7,22].
4.1. Comorbidities
In medicine the term “comorbidity” means that more than one disease is present in one individual at the same time. As the average extent of life has increased with improved diagnosis, due to technological advances, and a better management and treatment is established, medical complexity therefore increases. It is often difficult to understand which medical condition is the main cause of illness, if one clinical condition is caused by another one, or if the coexistence has no causal relationship. Therefore, in the case of clinical complexity, a medical illness exists simultaneously and independently from another one or there is causal relationship between them. However, in patients experiencing increased clinical complexity with, for example, the simultaneous presence of comorbid diabetes mellitus, renal failure, and CVD, it is not as important to recognize only the primary illness but it is essential to deal with the whole medical complexity.
4.1.1. Comorbidities in Coronary Artery Disease
The 2019, the ESC guidelines for the diagnosis and management of chronic coronary syndromes (CCS) differentiated the comorbidities that are related to CCS and coronary artery disease (CAD) into two categories: the cardiovascular and the non-cardiovascular comorbidities. In the cardiovascular comorbidities, the following were included: hypertension, valvular heart disease (including planned transcatheter aortic valve implantation), and after-heart transplantation (heart transplantation is an immunological event with high morbidity and mortality). In the non-cardiovascular comorbidities, the following are included: cancer (increased occurrence of CAD in active cancer), diabetes mellitus (two-fold increased risk for CAD), chronic kidney disease (CKD; CAD is highly prevalent with CKD), elderly (high incidence and prevalence of CAD), and sex (women are underrepresented in cardiovascular studies; therefore, true sex-related differences in CAD mortality remain unclear) [23].
In a group of high-risk patients with left main disease and/or multivessel CAD, the effects of coronary anatomy, lesion complexity, and comorbidities on outcomes of elective percutaneous coronary intervention (PCI) were studied; it was found that “mortality was significantly increased in patients with multiple comorbidities” and “coronary anatomy and lesion complexity, having any four or more comorbidities was associated with significantly increased odds of dying after elective PCI”; moreover, “when adjusted for the extent of CAD and lesion complexity, comorbidities burden remains an important predictor of mortality” [24].
4.1.2. Comorbidities in Sudden Cardiac Death
In the 2017 American Heart Association (AHA)/American College of Cardiology (ACC)/Heart Rhythm Society (HRS) guidelines, sudden cardiac death (SCD) is determined as the “sudden cessation of cardiac activity so that the victim becomes unresponsive, with no normal breathing and no signs of circulation”; and “if corrective measures are not taken rapidly, this condition progresses to SCD” [25]. Ventricular arrhythmias (VA) and SCD are related to specific populations and clinical conditions which are represented in this paper as comorbidities: adult congenital heart disease, medication-induced arrhythmias, sex-related differences, valvular heart disease, CKD, athletes, older patients with other comorbidities (such as CAD and HF), and pregnancy [25]. Among the conditions predisposing SCD most prominent is CAD, and implantable cardioverter-defibrillator (ICD) is considered the principal effective mode of therapy; in patients with nonischemic cardiomyopathy risk stratification for SCD is limited [26].
Regardless of important breakthroughs in the pathophysiology and therapy of HF, morbidity and mortality remain high and four-year survival rate is lower than 50%. Patients with HF and reduced ejection fraction (HFrEF) are exposed to an increased risk of SCD but predicting which patients with HFrEF are at an increased risk of SCD is a difficult task; and numerous times, prediction “overestimates the risk in those with ejection fractions less than 35% and not identifying those at risk with ejection fractions greater than 35%”; while contemporary treatments reduce the risk of SCD, further research is needed to identify those at higher risk [27]. Patients with HF induced by CAD are regarded to be at high risk of SCD; many trials were carried out for primary and secondary prevention of SCD in patients with HF [28]. The ICD therapy in HF is a decidedly successful and cost-effective method of terminating life-threatening ventricular arrhythmias and reducing mortality [28].
It is important to identify those HF patients with an increased risk of ventricular arrhythmias in whom an ICD implantation would be advantageous to prevent SCD episodes. In the multicenter automatic defibrillator implantation trial II (MADIT II) of patients with low left ventricular ejection factor (LVEF) and an old history of myocardial infarction (MI), the all-cause annual mortality rate was 10%; in the “eplerenone post-acute myocardial infarction heart failure efficacy and survival study (EPHESUS)” of patients with symptomatic HF and LVEF <40%, the annual mortality in the placebo group was 13.6% (SCD rate was 5% in the first year of follow up); in the “sudden cardiac death in heart failure trial (SCD-HeFT)” it was found that the use of ICD in patients with very low LVEF was advantageous [29,30,31].
In the general population, older people with acquired structural heart diseases and young individuals with inherited cardiac conditions are known to be at high risk of SCD. Recently, it was found that the SCD rates decreased after the CAD decline, but there is still an increasing proportion of SCD patients, not related to CAD, with heterogeneity of pathologies, which is hard to recognize and difficult to identify. These heterogeneous pathologies represent comorbidities or risk factors for SCD and require targeted preventive strategies; thus “multifaceted preventative approaches, which address risk factors in seemingly low-risk and know high-risk populations, will be required to decrease the burden of SCD” [32]. Thus, the epidemiology of SCD includes some hidden cardiac conditions (comorbidities) with undefined and difficult recognizable pathologies. Therapy and preventive measures of malignant ventricular arrhythmias and SCD are based on antiarrhythmic drug therapy and implantable defibrillators. However, this therapeutic strategy should be complemented by searching for preventable comorbidities and other hidden risk factors; reduction in SCD continues to be a major challenge in cardiology at present.
4.1.3. Comorbidities in Heart Failure
HF is a chronic progressive disease complicated throughout its course, especially in the elderly, with cardiac and non-cardiac co-occurring chronic conditions with various coexisting pathologies: CAD, CKD, liver disease, cardiac arrhythmias such as atrial fibrillation, sleep-disordered breathing, anemia and iron deficiency, orthostatic hypotension, hypertension, hyperlipidemia, diabetes mellitus, arthritis, chronic obstructive pulmonary disease (COPD), depression, Alzheimer’s disease/dementia, and social and economic deprivation followed by chronic malnutrition [33]. It is a difficult task to describe and evaluate the complex clinical entity of HF, in view of the multiplicity of comorbidities, risk factors and hidden clinical subgroups. The coexistence of a multitude of clinical entities, initially “contribute to cardiac remodeling, sharing similar pathophysiological mechanisms”, with early myocardial interstitial changes and late development of structural changes and clinical HF [34].
For an early HF diagnosis, advances in imaging techniques such as echocardiography and magnetic resonance imaging (MRI) have allowed “tissue characterizations, cardiac motion analysis, and cardiac performance analysis under stress”; this “can potentially detect changes earlier…with clinical benefits for screening, planning preventive therapies and risk stratifying patients” [34]. For early diagnosis and follow-up evaluation of HF patients, two natriuretic peptide biomarkers (B-type natriuretic peptide (BNP) and N-terminal pro-B-type natriuretic peptide (NT-proBNP)) are used for “the diagnosis or exclusion of HF as a cause of symptoms” and “to establish the presence and severity of HF” [35]. Furthermore, “there are insufficient data to inform specific guideline recommendations related to natriuretic peptide-guided therapy or serial measurements for the purpose of reducing hospitalization or deaths” [35].
CKD often predisposes to chronic myocardial dysfunction inducing a complex cardiorenal interrelationship called cardiorenal syndrome. Different pathogenic factors are involved, with uremic toxins being the most prevalent inducing arterial stiffening, decrease in vascular compliance, and microvascular cardiac ischemia [36]. HF and liver disease frequently co-exist, having complex cardiohepatic interactions that lead to acute cardiogenic liver injury, congestive hepatopathy, and cardiac failure; often emphasized is “the emerging role of altered liver X receptor signaling in the pathogenesis of HF comorbidities” and the significance “of the intestinal microbiome and its metabolites in HF and liver disease” [37]. Comorbidities aggravate HF while their recognition and therapy decrease mortality and increase quality of life [38].
In patients with HFrEF, predisposing risk factors, basic pathological mechanisms, comorbidities, and effective therapeutic measures are known and accepted as conventional knowledge and procedures. However, HF with preserved ejection fraction (HFpEF) is a more diverse clinical entity with a variety of predisposing causes but still with an inadequate explanation of pathological mechanisms and unsuccessful therapy [39]. The HFpEF knowledge for the “natural history of clinical progression from the pre-clinical diastolic dysfunction until the final clinical stages is significantly limited…and clinical progression to some more complex clinical models with multi-organ involvement…poorly understood” [40].
The Studies of Left Ventricular Dysfunction (SOLVD) were scheduled to explore the effect of an angiotensin-converting-enzyme (ACE) inhibitor (enalapril) on survival and hospitalizations of HF patients with low ejection fraction (<35%). It was found that enalapril when added to standard therapy “significantly reduced mortality and hospitalizations of heart failure in patients with chronic congestive heart failure and low ejection fractions” [41]. In post hoc analysis from the SOLVD prevention and SOLVD treatment trials, the effects of comorbidities were assessed “on outcomes in predominantly asymptomatic populations without previous heart failure treatment of the SOLVD prevention trial compared to symptomatic heart failure patients of SOLVD treatment and to evaluate associations to the effect of enalapril”; it was found that “comorbidities increased events in asymptomatic left ventricular dysfunction and in symptomatic heart failure, but did not interfere with the effects of enalapril” [42].
There are potential interconnections and interactions between HF, atrial and ventricular arrhythmias, and sleep-disordered breathing, a conundrum difficult to interpret. It is recognized that patients with HF have an increased risk of having a high prevalence (50%) of central sleep apnea (CSA) with a close relationship between CSA and atrial fibrillation, and between CSA and ventricular tachycardia or ventricular fibrillation [43]. It is uncertain whether CSA is an independent risk factor with a poor prognosis for HF patients or if CSA is associated with an increased risk for supraventricular and ventricular arrhythmias for HF patients [43].
Moreover, there is a close interconnection between HF and arrhythmias and conductance disturbances with the arrhythmias to be a “serious complication, but also etiology of heart failure” [44]. Due to a rapid ventricular rate, supraventricular arrhythmias may be responsible for adverse hemodynamic effects with the sudden disappearance of atrial contraction and onset of ventricular pump failure [45]. Clinical coexistence of AF and HF is a frequent clinical problem as both AF and HF are common cardiac disorders with significant cardiovascular morbidity and mortality. This coexistence gives rise to multiple heterogeneous syndromes (HF phenotypes) not easily defined and it seems that often each clinical entity generates the other one. Between these phenotypes there is an ill-defined and underrated subgroup of patients with pre-existing AF that is followed by incident HF. This cohort of patients represents a discrete clinical phenotype that involves various contributing clinical comorbidities or risk factors with undefined pathophysiological mechanisms progressing to clinically significant HF [46].
4.2. Environment
In general, environmental factors are related to the start and development of human diseases. The environment is part of the conundrum and implicates chemicals, microbial infections, gut bacteria, stress, and many other noxious elements. Environmental factors are interrelated with genes and interact with them. Genetic and environmental contributions are both regarded as independent causes for the atherosclerotic process and the appearance of clinical cardiac phenotypes. Exposure to some environmental elements such as “ambient air pollution and the metals arsenic, cadmium and lead” increase the development and mortality of CVDs through “augmentation or initiation of pathophysiological processes associated with CVDs”; it seems that “environmental exposure is an important but underappreciated risk factor contributing to the development and severity of CVDs” [47]. Reduction in environmental noxious factors from present levels probably could decrease CVD morbidity and mortality. However, the effects of the environment on CVD risk has been difficult to evaluate and the involved mechanisms continue to be unclear; human environments are complex and particularly changeable “because of diversity in human ecosystems, evolutionary histories, social structures, and individual choices” [48]. Unhealthy lifestyle behaviors such as harmful nutritional habits, smoking, untreated hypertension and diabetes, is connected to the atherosclerotic plaque pathogenesis and its progression to clinical disease. In contrast, “adherence to a healthy lifestyle was associated with a substantially reduced risk of coronary artery disease” [49].
Recently, the interest has been concentrated on the complex interactions of microbiota and their metabolites with the development and progression of CVDs and HF [50]. There are preclinical and clinical suggestions “that the gut microbiota and its metabolites have the potential to be a novel therapeutic and preventative target” for CVDs, HF, and metabolic diseases [51]. CVDs demonstrate a “seasonal pattern” with reported “winter peaks and clusters” and peaks connected to “heat waves”; potential strategies have been described to counteract “peaks in cardiovascular events during cold and hot periods of the year” [52]. There is a prevailing agreement supported by large scale studies that “environmental pollution and non-chemical stressors” are leading to the increased incidence and prevalence of CVDs [53].
5. Progression, Prevention, and Risk Prediction
5.1. Progression of HF-Compensatory and Regulatory Mechanisms
The most important compensatory regulatory systems in HF are the neurohumoral and the cardiac remodeling systems. The neurohumoral regulatory systems include the later-activated vasoconstrictive systems such as the renin–angiotensin–aldosterone system (RAAS) and the sympatho-adrenal system (SAS), as well as the vasodilatory systems such as the early mobilized natriuretic peptide axis system, prostaglandins (PGE2 and PGEI2), and nitric oxide systems. The vasoconstrictive regulatory mechanisms increase and preserve cardiac output while the vasodilatory mechanisms are restricting excessive vasoconstriction. The vasodilatory mechanisms of natriuretic peptide (ANP), brain natriuretic peptide (BNP), and their prohormones (proANP and proBNP) are produced by cardiomyocytes and represent strong compensatory mechanisms to prevent cardiac dysfunction.
Left ventricular remodeling is a mechanical compensatory mechanism that restores the shape and geometry of the left ventricle (LV) and delays HF progression [18]. In the homeostatic regulation of the human body, the heart is engaged in an integrated complex and functional network system of organs, vessels, and various regulatory mechanisms [18]. The HF syndrome together with the clinical deterioration and progression should be considered on the whole as a complex adaptive system; the progressive clinical deterioration consists of a dynamical and non-linear system with chaotic behavior [54].
HF is the end result of numerous pathophysiological states with increased complexity. Clinical deterioration has the characteristics of a complex and unstable system stabilized with a self-organized positive feedback neurohumoral and left ventricular remodeling mechanisms. Actually, the progressive course of HF is regulated by the built-in compensatory mechanisms while the whole HF syndrome is represented by periods of clinical stabilization suspended by intervals of clinical instability [18]. There is no convincing therapy successful in both clinical periods, to strengthen the stable equilibrium periods and to prevent instability and deterioration to the final clinical phase [18].
Thus, the syndrome of chronic HF is characterized by persistent progression of mechanical myocardial dysfunction of LV and by recurrent episodes of clinical deterioration requiring hospitalization for further management; the clinical episodes are followed by intervals of temporary hemodynamic stability due to LV remodeling and clinical compensation. This period of clinical and hemodynamic stability “is short-lived and soon becomes maladaptive to ventricular remodeling followed by deterioration to a worse clinical and hemodynamic instability period” [55]. This way, left ventricular remodeling deteriorates “after a lengthy period of continuous clinical progression and multiorgan involvement into irreversible myocardial damage” [55]. In the progressive deterioration of HF other processes are participating such as increase in coronary resistance from neurohumoral activation and endothelial dysfunction, alterations in the extracellular matrix, cellular remodeling, programmed cell death, and cardiomyocyte regeneration [56].
The RAAS takes part in structural and electrical remodeling in patients with HF and left ventricular hypertrophy (LVH); it is also known that angiotensin II participates in cardiac cell proliferation and cardiac myocyte ion channels’ modulation. It seems likely that cardiac remodeling is responsible for the emergence of atrial and ventricular arrhythmias common in patients with HF and LVH [57]. Patients with HF and LVH benefited from RAAS blockade, after use of angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin II receptor blockers (ARBs) due to a reversal of cardiac remodeling and prevention of new-onset atrial fibrillation [57]. Treatment with ACEIs, ARBs, and β-blockers impedes deterioration of myocardial function as well as clinical deterioration caused by the deleterious impact of the compensatory systems [58,59]. Therefore, the therapy with ACEIs, ARBs, and β-blockers is the appropriate therapy to block LV remodeling and HF progression and reduce symptoms and/or mortality [55].
In general, the HF syndrome demonstrates a modular construction with predictable behavior of functional clinical phenotypes having a strong impact on biological networks from epigenetic, cellular to regulatory systems [18]. The importance of individual genes for the pathogenesis and clinical progression of the HF syndrome is restricted to the hypertrophic and dilated cardiomyopathies. It seems that some HF patients have a complex multigenic inheritance, but the importance of individual genes is limited. In contrast, the significant role of epigenetics, proteomics, and metabolomics is increased; but, the complete genetic network system and the interactions between multiomics systems are still uncertain [60]. Multimodal systems that include genetic networks, multiomics, metabolic pathways, environmental factors, and sophisticated disease-related clinical networks are required to be integrated and provide a new holistic and realistic picture.
Significant breakthroughs have been made to understand many of the pathophysiological mechanisms of HFrEF but the natural pathophysiological history and clinical progression of HFpEF still remains inadequately defined [39]. The subclinical progression of pre-clinical diastolic dysfunction (PDD) of LV “to clinical phenotype of HFpEF and the further clinical progression to some more complex clinical models with multi-organ involvement…continue to be poorly understood” [40]. Prospective studies are expected to clarify the natural history and clinical progression of HFpEF and define the LV remodeling mechanisms involved. The pathophysiology of LV systolic dysfunction is different to the diastolic dysfunction, as systolic dysfunction is considered a disease of calcium handling and diastolic dysfunction is regarded as a disease of increased myofilament sensitivity to calcium [61,62,63].
5.2. Prevention and Prediction of Heart Failure
HF requires a more preventive model for earlier diagnosis while prompt management of risk factors and establishment of healthy habits and practices could slow the HF epidemic [64]. A significant number of HF patients presented with a protracted asymptomatic period followed by a symptomatic phase with clinical manifestations. Prevention or postponement of clinical symptoms depends upon the appropriate management of the asymptomatic period. Identification of asymptomatic patients with echocardiography and natriuretic peptides measurement, followed by ACEIs and ARBs therapy, can prevent or delay the incidence of symptoms and decrease mortality [65].
A risk prediction study analytically evaluated the methods underneath incident HF risk prediction models; EMBASE and PubMed data were examined for suitable articles published in the period 1990–2016 that reported “at least one multivariable model for prediction of HF”; it was concluded that “there is an abundance of HF risk prediction models that had sufficient discriminative ability, although few are externally validated" and “methods not recommended were frequently used, and resulting algorithms should be applied with caution” [66].
Prevention and prediction of HF (as well for CVDs) are considered as major objectives of clinical cardiology. Prediction and prevention of HF are quantitative risk-based approaches taking into consideration the need for personalized decision making [67]. At a population level “HF risk prediction tools can be applied…to estimate risk of incident HF”, describe “the clinical utility of biomarkers to personalize risk estimation” and identify “low and high-risk individuals with excellent accuracy” [67]. Yet, a number of unsolvable uncertainties must be confronted and answered for a more individualized HF risk assessment and answers to uncertainties would be given after appropriate randomized trials. In a large sample of participants, risk prediction models for HF subtypes (HFpEF and HFrEF) were described with good discrimination between subtypes; it was found that “risk factors differed between HFpEF and HFrEF, supporting the notion of pathogenetic differences among HF subtypes” [68].
In elderly frail patients with HF, the increased morbidity and mortality can be ascribed to some extent to cardiac aging at the level of cells and organs while functional deterioration may predict mortality [69]. Frailty is “an independent risk factor” for incident HF in the aging population, but, also, it is “a dynamic and potentially reversible state”; clinical research should be addressed “into the pathobiology of frailty, the development of novel therapeutics and the identification of biomarkers” [69].
The ANP, BNP, and their prohormones (proANP and proBNP) are produced by cardiomyocytes after an increase in atrial and ventricular myocardial stretch or by excessive sodium intake; the ANP (mainly from atria) and the BNP (ventricles) are parallel endocrine systems, regulated by different mechanisms and are considered as compensatory procedures counteracting cardiac dysfunction [18,70]. BNP is a diagnostic biomarker of HF and, also, a prognostic factor. Genomic studies identified polymorphic variants for BNP, having an impact in the circulating levels of BNP and NT-proBNP [71]. In patients with severe HF, proteomic studies with the use of quantitative mass spectrometry have identified high molecular weight forms of BNP with reduced biological activity, but with diagnostic and prognostic significance [72].
Furthermore, proteomics studies identified other promising biomarkers in patients with acute decompensated heart failure (ADHF), such as the quiescin Q6 sulfhydryl oxidase 1 (QSOX-1); the QSOX-1 biomarker is considered as the most important predictive protein for ADHF identification [73,74]. In human and animal studies, the expression of QSOX1 correlates to the pressure overload and hypertrophy of LV and LA as well as with the ensuing development of ADHF [74]. QSOX1 expression was not evident in patients with non-cardiac dyspnea or in stable chronic compensated HF; the QSOX1 diagnostic ability was comparable to BNP and NT-proBNP, demonstrating increased diagnostic value when combined with BNP [74,75].
5.3. Progression of CAD
All sub-clinical and clinical manifestations of CAD and CVDs originate from early atherosclerotic lesions and their after-effects [76]. The atherosclerotic plaque progression is a chronic process that starts from an initial asymptomatic stage of coronary artery lumen stenosis/obstruction and gradually advances to a subclinical or clinical CAD. This gradual and usually slow progression follows a sigmoidal (S-shaped) curve with a prolonged initial development period of 30–50 years and a subsequent fast expanding asymptomatic period of 10 years followed by a final symptomatic period [60].
Heritability for CAD was supported by some studies showing an increased risk of CAD after the identification by Genome-Wide Association Studies (GWAS) of some independent loci associated with CAD or by incorporating multiple loci into CVD risk prediction or by incorporation of single nucleotide polymorphisms (SNPs) for risk prediction improvement [77,78,79]. Many independent loci are known to be associated with myocardial infarction and CAD [80]. Despite the association, the endeavor for the recruited GWAS to recognize specific genetic loci related/linked to CVDs proved inadequate to explain the whole process of genesis and progression of complex CVDs. Bjorkegren et al. are suggesting that genetic participation and environmental contribution are two independent causative factors involved in CVDs and other complex disorders [81]. They assert that GWAS identifies 153 possible CAD genetic loci with 46 of those to possess genome-wide significance able to explain only <10% of genetic variance of CAD, while the remaining 90% of CAD heritability is connected to environmental contributor factors. It is important to understand that the CAD genetic loci as they are recognized by the GWAS are linked to early atherosclerotic lesions rather than to clinical phenotypes developed later by the atherosclerotic process. Pathology and clinical course of CAD phenotypes cannot be explained only by the GWAS genetic loci identification. Large sample sizes of at least half a million participants are required in order to evaluate the clinical importance of genetic loci risks derived by GWAS [82].
Epigenetic adjustments underlie and modify gene expression throughout the atherosclerotic complex process, transmitting information independently from DNA [60]. Feinberg and Fallin explain that the term “epigenetics” refers “to information transmitted during cell division other than the DNA sequence per se” [83]. Moreover, DNA methylation, posttranslational modifications of nucleosome proteins, and the density of nucleosomes, are examples of epigenetic transmission of information [83].
With the Human Genome Project emerged the multiomics technologies, which gave rise to a variety of data related with precision to CVDs and HF diseases. The term multiomics refers to a new interdisciplinary field of biological and medical research that gives a new perspective to cell and tissue biological pathways and functions. Multiomics methodology presents a new fundamental principle to study a biological system and extract information “about what gene, protein or lipid is valuable for investigation, as well as potentially validating new biomarkers or therapeutic targets in CVDs” [73]. For personalized medicine, genomics, proteomics, and metabolomics are promising for cardiovascular research in order to explain biological mechanisms and progression of CVDs in individuals with different health backgrounds. The SB multiomics approach of CVDs is using new multiomics techniques which assemble large, multidimensional data amenable to analysis using new informatics approaches alongside established statistical methods [84]. Network analysis and machine learning evaluate and “integrate data from more than one omic technique and understanding the interactions between different omics data requires increasingly complex concepts and methods” [84]. The multiomics are novel technologies able to identify a multitude of molecules (biomarkers) connected to the pathology of atherosclerosis or provide information regarding CVDs severity and progression. In reality, in CVDs “many events remain unpredictable and to avoid them, we need more accurate, stable, easily detectable, specific and sensitive biomarkers to improve the diagnosis and prognosis of these pathologies” [73,85]. Moreover, specific biomarkers are required to provide important information regarding reversal of CVDs pathologies. The multiomics approach permits the discovery of novel molecular elements and strategies for research and therapy.
Proteomics, a systems physiology discipline, is addressed on the features and properties of proteins of all biological systems and environments particularly the structure and function of the human proteome. Proteomics examines the role of proteins in cells, tissues, and organs, in health and human diseases. Proteomics is the apparatus for the disease phenotype quantification helping us to comprehend what disease actually is in the background of health. The proteomics discipline is based on mass spectrometry and calculates the molecular mass of proteins or peptides. The proteomics research in CVDs is focused mainly on the discovery of novel biomarkers with a personalized risk profile and, also, integrates “proteomics results with clinical phenotypes, metabolite changes and genetic haplotype information” [86]. In human diseases, genetic variants with different proteomes and specific environmental features are integrated and interact. Proteomics effectively fills the knowledge gap “between traditional research in the pathophysiology field” and the full explanation of “the molecular basis of the CVD occurrence” [87]. Proteomic approaches are unambiguously powerful tools that may provide deeper knowledge into the molecular mechanisms associated with CVDs with repercussions for molecular pathophysiology and clinical management; furthermore, integration of proteomics with other multiomics data facilitates both drug and novel biomarkers development with clinical applications [88].
The methodology of recognizing new biomarkers may be achieved with various proteomic and metabolomic procedures; thus, “tissue analysis …may show the proteins that are expressed in the pathological process” while “to identify circulating biomarkers, analyzing the secretome of cultured atherosclerotic tissue, analysis of blood cells and/or plasma may be more straightforward” [89]. The American Heart Association (AHA) issued a scientific statement (2015) on the decisive effect of proteomics “on cardiovascular health and disease” and the advances made during the last two decades in proteomics analysis in CVDs [90]. Proteomics analysis in CVDs focused on recognition of circulating protein biomarkers, identification of disease pathophysiological mechanisms, and exploration for prospective therapeutic applications [91]. Research studies on prospective targets for thrombosis and novel biomarkers is concentrated “on plasma, circulating cells or thrombus tissue” aiming “to propose a prothrombic state and represent a link between genotype, environment and disease phenotype” [73,92]. A proteomic study on coronary thrombus aspirates revealed that pigment epithelium-derived factor (PDEF) processing is associated with coronary plaque rupture while other proteomic studies extracted significant facts from platelets and monocytes involved in thrombosis and atherosclerotic lesions [93,94].
Metabolites are important biological substances necessary for human metabolism; the majority of metabolites are lipids, while lipids participate in the start and progression of most CVDs [95]. In a genetic study of the human lipidome, “hierarchical clustering based on complex genetic correlation patterns identified 12 genetic clusters that characterized the plasma lipidome”; these clusters were related to risk factors predisposing CVDs and predicted cardiovascular deaths during follow-up [96]. Metabolomic technologies measuring thousands of metabolites can provide a personalized metabolic profile important for biomarkers in CVDs management; “the integration of metabolomics with other multiomics platforms will allow us to gain insight into pathophysiological interactions of metabolites, proteins, genes, and disease states, while advancing personalized medicine” [97]. Metabolic profile is a screening procedure aiming to increase the understanding of CAD pathology, to improve prediction for subclinical atherosclerosis, and identify patients at risk for early CAD. For example, in a large prospective epidemiological study for identification of novel biomarkers for incident CAD “were identified lysophosphatidylcholines 18:1, 18:2, monoglyceride 18:2 and sphingomyelin 28:1 as risk factors of CHD and suggested a causal effect for monoglyceride 18:2 on CHD” [98]. The majority of these studies concerning metabolomics were carried out in small populations, but to identify robust biomarkers large populations studies are needed [89].
There is a continuous atherosclerotic process that follows specific steps from the initial phase of atherosclerotic plaque fatty streaks to intermediate atherosclerotic lesion, and finally to the stage of plaque rupture, obstructive coronary lesions, and clinical forms of CAD [60]. Furthermore, coronary artery intraluminal rupture of a non-obstructive plaque can give rise to a thrombus having partial or complete artery blockage with the development ofACS; also, an unbroken plaque can grow and progress to an obstructive lesion inducing symptomatic chronic CAD [99]. Genesis and progression of atherosclerotic plaques are closely related to the significance of low-density lipoprotein (LDL) receptor expression on LDL blood concentrations; the increase in LDL receptor expression reduces LDL blood levels and abates the genesis and progression of atherosclerotic plaques [60]. Mutations affecting the LDL receptor lead to new LDL cholesterol-lowering therapies, an important application of lipidomics (a branch of metabolomics) [100]. Many mutations in the gene encoding the LDL receptor have been recorded in patients with familiar hypercholesterolemia which is an autosomal-dominant inherited condition, and a database has been set up on the internet for mutations in the LDLR gene [101].
Imaging and computing data analysis techniques have been developed to study and follow-up the atherosclerotic process in order to explain the progressive nature of the CVDs. Computational methods and 3-D imaging were used to identify endothelial cells covering atherosclerotic plaques and their relation to the shear stress and wall stress during blood flow; understanding mechanotransduction on endothelial cells will expand therapeutic potential [102]. In the coronary circulation, there is a strong connection between the circulating plasma LDL and the turbulent flow in areas of bifurcations and curbs; in bifurcations or trifurcations, the wall shear stress (WSS) alters endothelial cells’ gene expression and promotes arterial remodeling and atherosclerosis [19,103].
Many complex cellular and molecular mechanisms are implicated in the atherosclerotic plaque destabilization and chronic disease progression, but the responsible and precise biological network models with their interconnections have not yet been identified; it is concluded that “network models combined with the network perturbation amplitude algorithm provide a sensitive, quantitative method to follow disease progression at the molecular level” [104,105].
During episodes of ACS, the imaging of immune activity during myocardial ischemia with the form of systemic inflammatory networks is underlined [106]. With the positron emission tomography (PET), the 18F-fluorodeoxyglucose (FDG) is accumulated in metabolically active cells, labeling inflammatory networks in the myocardium; PET together with magnetic resonance imaging (MRI) findings can track down important inflammatory networks significant for the pre-clinical stage of CAD or after episodes of ACS [107].
In conclusion, the advances made in genetic, proteomic, and metabolomic technologies can facilitate cardiovascular research, production of new biomarkers related to CVDs, and at a personalized level to identify therapeutic targets.
5.4. Prevention and Prediction of Cardiovascular Diseases
Sequencing technologies and novel computational appliances facilitated the building of entire genomes including genetic variations. The impacts of these advances in medicine and clinical practice are enormous. Between medical practitioners, healthcare providers, and administrators, an important question is forged: how predictive and preemptive are the multiomics advances and the status of molecular medicine in exploring and managing complex CVDs. Furthermore, are these advances able to modify and upgrade the present healthcare system, ameliorating clinical outcomes with a reasonable cost-effective result? Paynter et al., in a review article, identified three fields connected to atherosclerosis that are being assessed for possible genetic inclusion into everyday clinical practice: familial hypercholesterolemia (FH) with established genetic testing for research, diagnosis, and successful therapy; genetic guidance for cardiovascular risk prediction and primary prevention is not currently incorporated into used risk scores; genetic interactions with treatment (pharmacogenetics) are used for some therapies today while there are prospects for future further clinical utility with therapeutic strategies [108].
FH is a genetic familial disorder with significant early atherosclerotic development and increased risk of CAD particularly in young people. The FH disorder is an autosomal-dominant inherited condition with defects in the low-density lipoprotein (LDL) receptor induced by mutations in a few genes encoding the LDL receptor. The genetic defects in LDL cholesterol metabolism lead to increased levels of LDL cholesterol. The FH disorder is inherited in heterozygous and homozygous forms, and it is considered as a classical mendelian disorder with usually low prevalence [19]. A large cohort of patients with FH and their unaffected relatives were screened to track down “the influence of different mutations of the LDL receptor gene on lipoprotein levels and the risk of CVDs”; it was concluded that “LDL receptor mutations only partly contributed to the variation of LDL cholesterol levels and cardiovascular burden in FH” and thus other “unidentified, familial risk factors must underlie the CVD risk, most likely independent of lipids and lipoproteins” [109].
Common genes involved in FH are the gene encoding the LDL receptor (LDLR) and the apolipoprotein B gene (APOB) or the proprotein convertase subtilisin kexin 9 (PCSK9) which encodes an enzyme involved in LDL receptor processing [109,110]. In many instances, the identification of clinically diagnosed FH patients may be successful due to the presence of high cholesterol levels rather than the manifestation of one of the above known defects. In some of those patients, there is probably an unidentified monogenic defect, or the presence of high cholesterol levels is polygenic. Regardless of the number of the involved genes, the genetic importance of each one of those is restricted, as subclinical forms of heterozygous FH patients are treated successfully with statin monotherapy. The use of PCSK9 inhibitors proved highly effective in “reducing LDLC of heterozygous FH with reduced LDL receptor activity and in those cases of homozygous FH with residual LDL receptor activity” [108]. The use of PCSK9 inhibitors for either heterozygous or homozygous FH patients will improve clinical outcomes [111].
Prediction of the phenotypic characteristics of complex and multileveled cardiac diseases for recognition of the underlying polymorphisms is difficult under the current technology and accurate gene interdependence is unknown [19]. A variety of CVD risk prediction models have been described: the Systematic Coronary Risk Evaluation (SCORE) [112]; the Reynolds Risk Score [113]; the QRISK®2 [114]; and the US-based Pooled Cohort [115]. The intention of the 2012 guidelines from the European Societies on Cardiovascular Disease Prevention in Clinical Practice was to update the present knowledge in preventive cardiology and provide a cardiovascular risk assessment in clinical practice [112]. The objective of The Reynolds Risk Score was “to develop and validate cardiovascular risk algorithms for women… for global cardiovascular risk prediction and reclassified 40% to 50% of women at intermediate risk into higher- or lower-risk categories” [113]. The QRISK2 CVD risk algorithm was scheduled to assess the CVD risk in patients from different ethnic groups in England and Wales with incorporation of “ethnicity, deprivation, and other clinical conditions into QRISK2 algorithm” and increase the “accuracy of identification of those at high risk in a nationally representative population” [114].
The Work Group on practice guidelines in the USA-based Pooled Cohort equation allowed for additional and novel cardiovascular risk markers in the existing risk equations; the new risk markers were evaluated for improved model performance. The Work Group concluded that none of the additional risk markers under evaluation (such as diastolic BP, family history of CVD, moderate or severe chronic kidney disease, and body mass index) “significantly improved discrimination for 10-year hard CVD risk prediction when added to the final base models” [115]. Further research using state-of-the art statistical techniques are needed to examine the utility of novel biomarkers added to these new Pooled Cohort Equations in different populations and patient subgroups; new “randomized clinical trials demonstrating the utility of screening with novel risk markers would represent the best evidence for their inclusion in future risk assessment algorithms” [115]. Any new biomarker should change risk assessment for CVDs having an impact on clinical practice and should be based on well-controlled randomized trials.
Spatial biology is the new frontier of systems and molecular biology, studying the 2D or 3D context of tissues and the spatial distributions of cells, genes, proteins, ribosomes, and miRNAs. The techniques involved have high sensitivity and precision and a wide variety of applications in basic research, clinical medicine, and biomarker discovery: single-molecule fluorescence techniques give “high resolution spatial distribution of ribosomes and RNA polymerase in live, rapidly growing Escherichia coli…and…new information about the spatial organization of the E. coli” [116]; single-cell sequencing techniques increase our understanding of the “heterogeneous regulatory landscape encoded in complex diseases, by inferring various biological networks” [117]; the multiplex immunofluorescence technique is a new type of biomarker assay in cancer patients and examines the expression of many proteins in individual tumor cells within the tumor microenvironment, predicting response to immunotherapy [118]. The application of spatial biology techniques to spatial gene or molecular expression and regulation may prove important for complex CVDs, facilitating biomarker discovery and validation. Generally, there has been a general discouragement to include genetic markers in the existing CVDs risk equations; exemption to this suggestion is the FH, a genetic familiar disorder. Genetic risk prediction factors or identified SNPs related to the FH disorder are not adequate to rationalize predictive genetic testing for CVDs in everyday clinical practice.
Genetic information, treating patients with CVDs, encompasses two pharmacological areas of interest: risk prediction for adverse events (safety) and drug efficacy (treatment outcomes). Pharmacogenetic outcomes are restricted “to the primary effects on the drug target (pharmacodynamics) and aspects of drug absorption, distribution, metabolism, and elimination (pharmacokinetics)” [108]. Safety and efficacy are pharmacological properties closely related to pharmacodynamic and pharmacokinetic genetic factors. Nevertheless, “despite substantial progress in identifying the genetic etiology of pharmacological response, current biomarker panels still largely rely on single gene tests with a large portion of the genetic effects remaining to be discovered” [119]. It seems that genetic factors are important for drug responses, and precise biomarker tests for the dominant genetic components may affect therapeutic success at an individual level. Although currently, genetic testing for prediction and management of atherosclerosis proceeds with caution, the inclusion of genetic information in clinical practice relies also on general availability and reduced cost [108]. A central clinical genomic information database is required to accumulate and interpret genetic results such as high-penetrance traits and carrier states; instead of considering a genome sequence as a test, it may be used as a health asset to reassess the atherosclerotic process over the lifetime of the patient [120].
In CVD patients, identification of genetic variants for aspirin use or pharmacogenetic tests for managing hypertension, statin therapy, or antithrombotic treatment, are not considered necessary preventive procedures [121,122,123]. Furthermore, genetic tests for safety and efficacy are not required to guide statin use for low-density lipoprotein cholesterol (LDLC) lowering (efficacy) or for safety reasons [124,125].
Clopidogrel, a P2Y12 platelet inhibitor, is activated by the intermediate metabolizing enzyme CYP2C19. The genetic variation in CYP2C19 in many forms created the need for genetic testing. The Tailored Antiplatelet Initiation to Lessen Outcomes due to Decreased Clopidogrel Response After Percutaneous Coronary Intervention (TAILOR-PCI) trial “failed to show that a genotype-guided strategy was superior at reducing adverse cardiovascular events compared with standard therapy after PCI” [126]. Warfarin has a large interpatient variability in the required dose due to genetic variants in the cytochrome P450-2C9 (CYP2C9) and vitamin K-epoxide reductase complex (VKORC1) enzymes, responsible for ~50% of warfarin dose variability; but without appropriate trials to confirm clinical benefit, the routine clinical use of genetic testing for warfarin dosing is not required [127].
In conclusion, genetic information in sequencing technologies, novel computational appliances, and CVD risk prediction models are promising important scientific tools with a significant impact on clinical practice.
6. Reversal of Coronary Artery Disease with Statins and Coronary Aggressive Therapy
From a clinical point of view, the current cardiovascular therapeutic approach clinically improves the majority of the patients with CAD, but progression of the atherosclerotic process persists, and the epidemic of CVDs continues relentlessly (Figure 4).
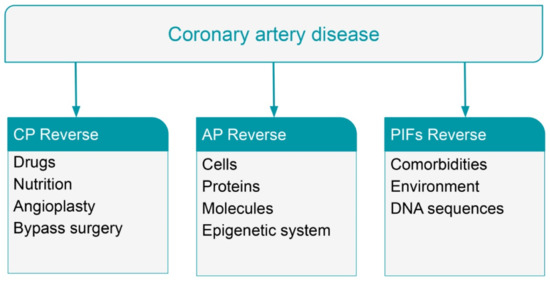
Figure 4.
Potential reverse (partial or complete) of coronary artery disease (CAD) at the level of clinical phenotype (CP) or atherosclerotic plaque (AP) or Phenotype Influencing Factors (PIFs).
Dyslipidemias are changes in the plasma lipid consistency which are related to a bigger risk of developing CVDs. The term dyslipidemia includes many forms such as the elevated levels of LDL, the hypertriglyceridemia, and the composite lipid appearance of high triglyceride levels with low HDL levels. With the decrease in those lipid abnormalities the risk of CVDs is diminished [128].
A comprehensive personalized approach to the problem of CAD requires amendment of a patient’s lifestyle habits, adherence to nutritional changes, and modern pharmacological therapy, if needed. It was stated, in a podcast by Catapano A and Sackier J, in a discussion about primary prevention and treatment of atherosclerotic CVDs that “multiple primary and secondary prevention trials have shown a significant reduction of 25–35% in the risk of cardiovascular events with statin therapy that leaves a residual risk of 65–75% despite the achievement of target low-density lipoprotein cholesterol levels and it is critical to escalate treatment in these patients” [129]. Possible damaging effects of statin therapy such as the risk of new-onset diabetes mellitus, hemorrhagic stroke, and cognitive impairment in addition to the damage on muscles and the liver, unfortunately, lead to discontinuation or to a behavior of nonadherence to statin therapy; however, the safety and efficacy of statin therapy are undisputable and “the benefits of statin therapy far outweigh any real or perceived risks” [130]. A combined use of statins and exercise decreased “the incidence of diabetes mellitus, improved insulin sensitivity and inflammation with no change in lipid profile compared to statins alone” [131]. A prospective meta-analysis of data in 14 randomized trials of statins was undertaken, aiming to detect different clinical outcomes per 1.0 mmol/L reduction in LDL cholesterol; it was found that “statin therapy can safely reduce the 5-year incidence of major coronary events, coronary revascularization, and stroke by about one fifth per mmol/L reduction in LDL cholesterol” [132].
In our clinical thinking, HDL concentrations are correlated in a negative way to the development of CVDs. Recently a “reverse cholesterol transport model” was developed, as “several large studies have revealed an inverse correlation between macrophage cholesterol efflux to plasma HDL and CVDs” [133]. The 2019 European Society of Cardiology (ESC)/European Atherosclerosis Society (EAS) guidelines for the management of dyslipidaemias and lipid modification to reduce cardiovascular risk suggest: “throughout the range of LDL-C levels, ‘lower is better’ with no lower threshold” and “lowering LDL-C depends on the absolute reduction in LDL-C, with each 1 mmol/L reduction corresponding to a reduction of about one-fifth in CVD”; in risk stratification the cardiac imaging (coronary artery calcification score assessment with computer tomography) and the measurement of apolipoprotein B (ApoB) and lipoprotein (a) (Lp(a)) are important; the guidelines support that “both a minimum percentage LDL-C reduction (50%) and an absolute LDL-C treatment goal of <1.4 mmol/L (<55 mg/dL) for very-high risk patients, and <1.8 mmol/L (<70 mg/dL) for high-risk patients” are needed [134].
Current management of CVDs includes long aggressive drug therapy and/or interventional and surgical approaches. Those medical approaches are effective and lifesaving in patients with AMI or other ACS [135]. Regardless of interventional success immediately after the procedure and clinical improvement in the foreseeable future, these methods carry a morbidity and mortality risk, and require patient hospitalization with significant expenditure for the insurer [136].
Classically, it was believed that “atherosclerotic plaque rupture of the fibrous cap is the main culprit in ACS”, but “plaque erosion with an intact fibrous cap” is considered now “responsible for about one third of ACS and up to two thirds of non-STEMI… since the introduction of statins”; in a “potential major shift in the management” probably patients with plaque erosion could be managed conservatively without coronary intervention [1].
In conclusion, these techniques together with drug therapy improve quality and future prospects of life, but do not reverse the course of the atherosclerotic process that remains chronically progressive and relentless. Therefore, the current aggressive therapy may improve symptoms and well-being but the possibility for future cardiac events remains.
7. Reversal of Coronary Artery Disease with Nutritional Intervention
There are some indications that plant-based nutrition can interfere with the atherosclerotic process improving vascular function. Some important cohort epidemiologic studies substantiate the nutritional intervention to arrest, reverse, and prevent primary and secondary advances of CVDs.
As indicators of CVDs severity and, also, in order to unravel the pathology of early atherosclerotic changes, “three physiological parameters, endothelial function, arterial stiffness, and autonomic nervous system function, have been extensively studied in relation to cardiovascular disease”; it is proposed that these parameters “should be monitored routinely, both to reduce the impact of their injury and as indicators of cardiovascular health and response to treatment” [137].
Vascular endothelial dysfunction is a crucial pathological event starting the progressive process of atherosclerosis most important for CVDs/CAD development. The endothelial normal function influences a number of activities; participates in the anticoagulant process, platelet adhesion, immune function, and regulation of volume and electrolyte content of both intravascular and extravascular spaces [138]. Coronary endothelial dysfunction is considered the earliest stage of coronary atherosclerosis and it is associated with progressive endothelial injury and inflammatory oxidative stress [139]. Endothelial dysfunction is characterized by reduced endothelium-dependent vasodilator function with vasoconstrictive reaction to vasodilator acetylcholine, and by impaired production of nitric oxide. Thus, the endothelium presented as a powerful protective impediment to vascular atherosclerosis while endothelial dysfunction, marked by decreased endothelium-dependent vasodilation, became the first step towards cardiovascular disease [140].
Increased dietary fat is related to postprandial lipemia that transiently impairs endothelial function, and comprises a potential atherogenic process independent of low-density lipoprotein [141,142]. Rathnayake et al. determined the impact of postprandial lipemia on vascular function in postmenopausal women; they concluded that “there is urgent requirement for suitably powered robust randomized controlled trials to investigate the impact of meal fat composition on postprandial novel and established CVD risk markers in postmenopausal women” [143]. Suwaidi et al., in a long-term study on patients with mild CAD and endothelial dysfunction, suggested that “severe endothelial dysfunction in the absence of obstructive coronary artery disease is associated with increased cardiac events” and that “coronary endothelial dysfunction may play a role in the progression of coronary atherosclerosis” [144].
Esselstyn et al. proposed that “the persistent results in treating the cause of vascular disease by whole-food plant-based nutrition offer a paradigm shift from existing practice. We think the time is right for a controlled trial” [145]. Plant-based nutrition was advised to 198 consecutive patients with CAD for a mean of 3.7 years; in 177 (89%) adherent participants CAD arrest and reversal was achieved with a recurrent event rate of 0.6%, while 13 of 21 (62%) nonadherent participants experienced adverse events; it seems that “plant-based nutrition has the potential for a large effect on the CVD epidemic” and “Western diet injures or impairs endothelial function after each ingestion, making food choices a major, if not the major, cause of CAD” [145]. Dehghan et al. followed a large number (31,546) of patients with CVDs or diabetes, for 4.5 years, aiming to find the “relationship between healthy diet and risk of CVD among patients on drug therapies for secondary prevention”; a reduction in CVD-related risk was found in patients with the healthiest diet [146]. Crowe et al. followed 44,561 people, aiming to figure out the “risk of hospitalization or death from ischemic heart disease among British vegetarians and nonvegetarians”; during a 11.6-year follow-up, the vegetarians had a 32% lower risk of having ischemic heart disease [147]. The nutritional state in old (over 65 years) patients with angiographic-documented CAD is predictive for all-cause death independently of comorbidities with “no interaction between nutritional risk and clinical characteristics with regard to all-cause death” [148].
However, to adhere to a very strict diet is not an easy task: For the US Preventive Services Task Force, a systematic review based on publications of various data sources was undertaken to “review the evidence on the benefits and terms of behavioral counseling for the primary prevention of cardiovascular disease in adults without known cardiovascular risk factors”; it was found that diet and physical activity “result in consistent modest benefits across a variety of important intermediate health outcomes” and that “there is very limited evidence on longer-term intermediate and health outcomes or on harmful effects of these interventions” [149].
Furthermore, the Heart Disease Reversal Program (HDRP) was developed as an interdisciplinary and multicomponent lifestyle program at the USA Department of Veterans Affairs with “a streamlined adaptation of behavioral and lifestyle interventions aimed at promoting partial reversal (regression) of atherosclerotic heart disease and achievement of comprehensive cardiovascular risk reduction”; and with this program a “significant value and benefit added to the health care system” and “to reverse a disease can be considered the highest aim of medicine and health care” [150]. Tilinin, in a thesis for the reverse risk assessment of diet-related chronic diseases such as CVDs, stated that the “proposed solution is based on a mathematical model which relates diet risk factors with the probability of contracting a chronic disease and a software system capable of generating a large number of possible food intake patterns” [151].
In conclusion, it seems that plant-based nutrition can correct injurious fat-rich nutritional effects on the endothelium with consequence for the arrest and reversal of the cardiovascular atherosclerotic process. Probable mechanisms are the restoration of nitric oxide production by the recovered endothelium, and reduction in the vasoconstricting molecules of endothelin and thromboxane by the injured endothelium.
8. Reversal of Heart Failure
HF treatment can ameliorate clinical signs and symptoms but only occasionally cures (myocardial restoration) after addressing the primary cause. In the majority of patients with HF, myocardial damage is permanent; notwithstanding the limited improvement in myocardial function (Figure 5).
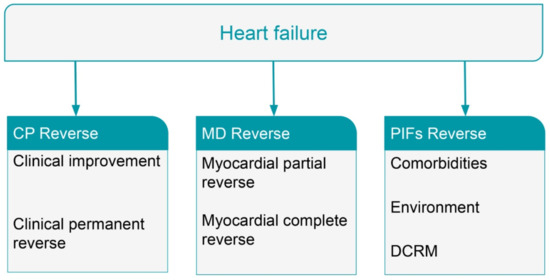
Figure 5.
Potential reverse (partial or complete) of heart failure (HF) at the level of clinical phenotype (CP) or myocardial dysfunction (MD) or Phenotype Influencing Factors (PIFs). DCRM = disease compensatory regulatory mechanisms.
The ESC Guidelines for the diagnosis and treatment of acute and chronic HF (2016) in the chapter for “Terminology related to the time course of HF” state that “Although symptoms and signs of HF may resolve, the underlying cardiac dysfunction may not, and patients remain at the risk of recurrent decompensation” [152]. Recently, it was found that deficiency of the Hippo pathway, in mouse hearts, reverses systolic HF after myocardial infarction; it was shown that “deletion of the Hippo pathway component Salvador (Salv) in mouse hearts with established ischemic heart failure…induces a reparative genetic program with increased scar border vascularity, reduced fibrosis, and recovery of pumping function” and those “findings indicate that the failing heart has a previously unrecognized reparative capacity involving more than cardiomyocyte renewal” [153].
The JACC Scientific Expert Panel reviewed the natural history of patients with HF recovered ejection fraction (HFreEF), as appropriate guidelines for managing and treating these patients do not exist. It was concluded that HFrEF patients with reverse LV remodeling and recovery of LV function are improved clinically, but “even among patients who experience a complete normalization of LV ejection fraction (LVEF), a significant proportion will develop recurrent LV dysfunction”; also, this panel reviewed “the biology of reverse LV remodeling and the clinical course of patients with HFreEF” and issued guidelines “for defining, diagnosing, and managing patients with HFreEF” [154]. In the CARDIOCHUS-CHOP registry the “clinical characteristics and prognosis of patients with HFreEF in an HF population” were evaluated; follow-up of patients with HFreEF showed a significantly lower mortality rate and hospitalization rate compared to HFrEF and HFpEF patients [155]. It seems that patients with HFreEF are a “distinct population of HF patients with different underlying etiologies, comorbidities, response to therapies, and outcomes compared with HF patients with persistent reduced or preserved EF”; but, the “mortality and morbidity in HFreEF patients remain higher than those in the normal population” while “data to guide the management of HFreEF patients are lacking” [156].
In conclusion, reversal of chronic HF with permanent restoration of myocardial integrity and complete normalization of LV ejection fraction without further episodes of LV dysfunction is a remote possibility. The HFreEF patients are characterized by a partial recovery of myocardial dysfunction but this improvement is not a permanent cure.
9. Methods for Network Analysis
Bioinformatics and computational approaches to biological and medical phenomena concurrently with the emergence of multiomics technologies provided a variety of disease data. Multiomics technologies based on mathematics, biochemistry, and physics enabled scientists to reconsider the nature of complex diseases from a reductionist to a holistic status demonstrating chronic quantitative and qualitative progressively deteriorating changes. Cellular large networks of genes, molecules, cells, or tissues are cooperating, producing emergent properties and disease phenotypes. A multiomics approach with network analysis of biological and medical data is required to decode the complexity of CVDs and HF. Cellular network behaviors with feedback mechanisms are in need of tools of feedback control theory able to investigate and explore self-regulating systems. These tools should be able to analyze complex data acquired from multiomics research. Intracellular biochemical processes induced by reaction to diverse stimuli are studied with specific mathematical models which delineate the dynamics of these processes supporting “experimental work in analysis of pathways and regulatory networks” [157]. As complementary mathematical methods for comprehensive analysis, two different modeling methods are used, the ordinary differential equations (ODEs) and the Petri nets; ODEs are applied “to find parameters, whose change results in the greatest quantitative and qualitative changes in the model response” while the Petri nets is “a structural analysis and allows indicating most important subprocesses in terms of information flow in Petri net” [157]. Costa et al. [158] summarize: “the bioinformatic tools and strategies that could be used for the characterization and functional analysis of non-coding RNAs, with special emphasis in their applications to the cardiovascular field”. Subramanian et al. [159], in a review paper, “collected the tools and methods that adopt integrative approach to analyze multiple omics data and summarized their ability to address applications such as disease subtyping, biomarker prediction, and deriving insights into the data”. Worheide et al. [160] provided an “overview over a typical multi-omics workflow, focusing on integration methods that have the potential to combine metabolomics data with two or more omics…and discuss multiple integration concepts including data-driven, knowledge-based, simultaneous and step-wise approaches”.
10. Conclusions
In patients with CVDs, the current invasive coronary interventions and pharmaceutical therapy improve quality and future prospects of life, but do not reverse the course of the atherosclerotic process or the development of CHD; both of which remain chronically progressive and relentless. The reversal of chronic HF with permanent recovery of myocardial integrity along with complete normalization of LV ejection fraction and without further episodes of LV dysfunction is a remote possibility.
This paper proposes and endorses that progression towards reversal of CVDs and HF phenotypes should be based on the application of a more holistic medical approach such as the holistic principle of SB with the help of AI trained on human medical data (Figure 6).
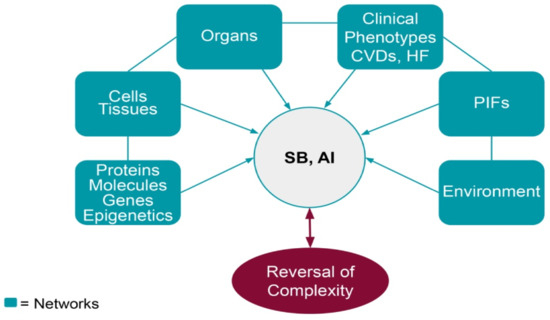
Figure 6.
Schematic diagram based on the SB concept of communication (links and data transmission) between sub-clinical and clinical stages of chronic complex cardiac diseases (CVDs and HF). The reversal of CVDs and HF phenotypes depends on data transmission (SB holistic principle) and AI (focused on human medical data). SB = systems biology; AI = artificial intelligence; CVDs = cardiovascular diseases; HF = heart failure; PIFs = phenotype influencing factors.
Clinical phenotypes reversal is a potentiality that depends on research carried out in large populations of CVD and HF patients. Such an approach requires a large amount of relative data using a multiomics approach and network analysis in order to embrace the concept of complexity. Analysis of this data will help to understand the complexity of chronic heart diseases and explain the interrelationship and interconnection between their biological networks in order to achieve improvement in the prospects for reversal of clinical phenotypes.
Author Contributions
K.G.L. and G.E.L. contributed equally to the preparation and submission of the research. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors report that they have no financial or other relations that could lead to a conflict of interest.
References
- Fahed, A.C.; Jang, I.K. Plaque erosion and acute coronary syndromes: Phenotype, molecular characteristics and future directions. Nat. Rev. Cardiol. 2021, 18, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Johansson, I.; Lynoe, N. Medicine and Philosophy. A Twenty-First Century Introduction; Boston De Gruyter: Berlin, Germany, 2008. [Google Scholar] [CrossRef]
- Lourida, K.G.; Louridas, G.E. Constraints in clinical cardiology and personalized medicine: Interrelated concepts in clinical cardiology. Cardiogenetics 2021, 11, 7. [Google Scholar] [CrossRef]
- Chen, C.; Cui, J.; Zhang, W.; Shen, P. Robustness analysis identifies the plausible model of the bcl-2 apoptotic switch. FEBS Lett. 2007, 581, 5143–5150. [Google Scholar] [CrossRef]
- Steinacher, A.; Soyer, O. Evolutionary principles underlying structure and response dynamics of cellular networks. In Evolutionary Systems Biology; Soyer, O., Ed.; Springer: London, UK, 2012; pp. 225–247. [Google Scholar]
- Noble, D. Physiology is rocking the foundations of evolutionary biology. Exp. Physiol. 2013, 98, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Noble, D. Evolution beyond neo-Darwinism: A new conceptual framework. J. Exp. Biol. 2015, 218, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Noble, D. Genes and causation. Philos. Trans. R. Soc. 2008, 366, 3001–3015. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, M.; Wang, E. Dynamic modeling and analysis of cancer cellular network motifs. Integr. Biol. 2011, 3, 724–732. [Google Scholar] [CrossRef]
- Ellis, G.F.R.; Noble, D.; O’Connor, T. Top-down causation: An integrating theme within and across the sciences. Interface Focus 2012, 2, 1–3. [Google Scholar] [CrossRef]
- Kitano, H. Systems biology: A brief overview. Science 2002, 295, 1662–1664. [Google Scholar] [CrossRef]
- Louridas, G.E.; Lourida, K.G. Heart failure: A complex clinical process interpreted by systems biology approach and network medicine. Anadolou Kardiyol. Derg. 2014, 14, 178–185. [Google Scholar] [CrossRef]
- Braun, E.; Marom, S. Universality, Complexity and the Praxis of Biology: Two Case Studies. Stud. Hist. Philos. Biol. Biomed. Sci. 2015, 53, 68–72. [Google Scholar] [CrossRef]
- Green, S. Can biological complexity be reverse engineered? Stud. Hist. Philos. Biol. Biomed. Sci. 2015, 53, 73–83. [Google Scholar] [CrossRef]
- Kwon, D. Proteins in their natural habitats. Nature 2021, 598, 558–560. [Google Scholar] [CrossRef]
- Matheny, M.; Israni, S.T.; Ahmed, M.; Whicher, D. (Eds.) Artificial Intelligence in Health Care: The Hope, the Hype, the Promise, the Peril; National Academy of Medicine; NAM Special Publication: Washington, DC, USA, 2019. [Google Scholar]
- Ahmed, Z.; Mohamed, K.; Zeeshan, S.; Dong, X. Artificial intelligence with multi-functional machine learning platform development for better healthcare and precision medicine. Database 2020, 2020, baaa010. [Google Scholar] [CrossRef]
- Louridas, G.E.; Lourida, K.G. A conceptual paradigm of heart failure and systems biology approach. Intern. J. Cardiol. 2012, 159, 5–13. [Google Scholar] [CrossRef]
- Louridas, G.E.; Lourida, K.G. The complex cardiac atherosclerotic disorder: The elusive role of genetics and the new consensus of systems biology approach. J. Adv. Ther. Med. Innov. Sci. 2017, 2, 10–17. [Google Scholar] [CrossRef][Green Version]
- Noble, D. A theory of biological relativity: No privileged level of causation. Interface Focus 2012, 2, 55–64. [Google Scholar] [CrossRef]
- Louridas, G.E.; Lourida, K.G. Systems biology and constraint-based downward causation in medical clinical practice: A perspective concept for cardiology. In Advances in Medicine and Biology; Berhardt, K.V., Ed.; Nova Science Publishers Inc.: New York, NY, USA, 2020; ISBN 978-1-53617-491-5. [Google Scholar]
- Kohl, P.; Crampin, E.J.; Quinn, T.A.; Noble, D. Systems Biology: An Approach. Clin. Pharmacol. Ther. 2010, 88, 25–33. [Google Scholar] [CrossRef]
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes: The Task Force for the diagnosis and management of chronic coronary syndromes of the European Society of Cardiology (ESC). Eur. Heart J. 2020, 41, 407–477. [Google Scholar] [CrossRef]
- Yager, N.; Schulman-Marcus, J.; Torosoff, M. Coronary anatomy and comorbidities impact on elective PCI outcomes in left main and multivessel coronary artery disease. Catheter. Cardiovasc. Interv. 2021, 98, 436–444. [Google Scholar] [CrossRef]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Field, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Circulation 2018, 138, e272–e391. [Google Scholar] [CrossRef] [PubMed]
- Morin, D.P.; Homoud, M.K.; Mark Estes, N.A., 3rd. Prediction and prevention of sudden cardiac death. Card. Electrophysiol. Clin. 2017, 9, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Sapp, J. The risk and prevention of sudden death in patients with heart failure with reduced ejection fraction. Curr. Opin. Cardiol. 2020, 35, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.E.; Cowie, M.R.; Chow, A.W.C. Prediction and prevention of sudden cardiac death in heart failure. Heart 2005, 91, 674–680. [Google Scholar] [CrossRef]
- Moss, A.J.; Zareba, W.; Hall, W.J.; Klein, H.; Wilber, D.J.; Cannom, D.S.; Daubert, J.P.; Higgins, S.L.; Brown, M.W.; Andrews, M.L. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N. Engl. J. Med. 2002, 346, 877–883. [Google Scholar] [CrossRef]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef]
- Bardy, G.H.; Lee, K.L.; Mark, D.B.; Poole, J.E.; Packer, D.L.; Boineau, R.; Domanski, M.; Troutman, C.; Anderson, J.; Johnson, G.; et al. Amiodarone or an implantable defibrillator for congestive heart failure. N. Engl. J. Med. 2005, 352, 225–237. [Google Scholar] [CrossRef]
- Hayashi, M.; Shimizu, W.; Albert, C.M. The spectrum of epidemiology underlying sudden cardiac death. Circ. Res. 2015, 116, 1887–1906. [Google Scholar] [CrossRef]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2013, 62, e147–e239. [Google Scholar] [CrossRef]
- Wong, C.; Chen, S.; Iyngkaran, P. Cardiac imaging in heart failure with comorbidities. Curr. Cardiol. Rev. 2017, 13, 63–75. [Google Scholar] [CrossRef]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey Jr, D.E.; Colvin, M.M.; Drazner, M.H.; Filippatos, G.S.; Fonarow, G.C.; Givertz, M.M.; et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure. J. Am. Coll. Cardiol. 2017, 70, 776–803. [Google Scholar] [CrossRef]
- Zanoli, L.; Lentini, P.; Briet, M.; Castellino, P.; House, A.A.; London, G.M.; Malatino, L.; McCullough, P.A.; Mikhailidis, D.P.; Boutouyrie, P. Arterial stiffness in the heart disease of CKD. J. Am. Soc. Nephrol. 2019, 30, 918–928. [Google Scholar] [CrossRef]
- Xanthopoulos, A.; Starling, R.C.; Kitai, T.; Triposkiadis, F. Heart failure and liver disease: Cardiohepatic interactions. JACC Heart Fail. 2019, 7, 87–97. [Google Scholar] [CrossRef]
- Boully, C.; Hanon, O. Heart failure and comorbidities. Geriatr. Psychol. Neuropsychiatr. Vieil 2015, 13, 13–22. [Google Scholar] [CrossRef]
- Del Buono, M.G.; Iannaccone, G.; Scacciavillani, R.; Carbone, S.; Camilli, M.; Niccoli, G.; Borlaug, B.A.; Lavie, C.J.; Arena, R.; Crea, F.; et al. Heart failure with preserved ejection fraction diagnosis and treatment: An updated review of the evidence. Prog. Cardiovasc. Dis. 2020, 63, 570–584. [Google Scholar] [CrossRef]
- Louridas, G.E.; Lourida, K.G. Heart failure in patients with preserved ejection fraction: Questions concerning clinical progression. J. Cardiovasc. Dev. Dis. 2016, 3, 27. [Google Scholar] [CrossRef]
- The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N. Engl. J. Med. 1991, 325, 293–302. [Google Scholar] [CrossRef]
- Bohm, M.; Pogue, J.; Kindermann, I.; Pöss, J.; Koon, T.; Yusuf, S. Effect of comorbidities on outcomes and angiotensin converting enzyme inhibitor effects in patients with predominantly left ventricular dysfunction and heart failure. Eur. J. Heart Fail. 2014, 16, 325–333. [Google Scholar] [CrossRef]
- Grimm, W.; Koehler, U. Cardiac arrhythmias and sleep-disordered breathing in patients with heart failure. Int. J. Mol. Sci. 2014, 15, 18693–18705. [Google Scholar] [CrossRef]
- Vitovec, J.; Spinar, J.; Spinarova, L. Arrhythmias and conductance disturbances and heart failure. Vnitr. Lek. 2018, 64, 874–877. [Google Scholar] [CrossRef]
- Faber, T.S.; Zehender, M. Antiarrhythmic therapy in patients with heart failure. Ther. Umsch. 2000, 57, 324–332. [Google Scholar] [CrossRef]
- Louridas, G.; Lourida, K. Atrial fibrillation as a cause of incident heart failure: A discrete clinical phenotype. J. Atr. Fibrillation 2018, 10, 1807. [Google Scholar] [CrossRef]
- Cosselman, K.; Navas-Acien, A.; Kaufman, J. Environmental factors in cardiovascular disease. Nat. Rev. Cardiol. 2015, 12, 627–642. [Google Scholar] [CrossRef]
- Bhatnagar, A. Environmental Determinants of Cardiovascular Disease. Circ. Res. 2017, 121, 162–180. [Google Scholar] [CrossRef]
- Khera, A.V.; Emdin, C.A.; Drake, I.; Natarajan, P.; Bick, A.G.; Cook, N.R.; Chasman, D.I.; Baber, U.; Mehran, R.; Rader, D.J.; et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N. Engl. J. Med. 2016, 375, 2349–2358. [Google Scholar] [CrossRef]
- Tang, W.H.; Kitai, T.; Hazen, S.L. Gut Microbiota in Cardiovascular Health and Disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef]
- Kitai, T.; Tang, W.H.W. Gut microbiota in cardiovascular disease and heart failure. Clin. Sci. 2018, 132, 85–91. [Google Scholar] [CrossRef]
- Stewart, S.; Keates, A.; Redfern, A.; McMurray, J.J.V. Seasonal variations in cardiovascular disease. Nat. Rev. Cardiol. 2017, 14, 654–664. [Google Scholar] [CrossRef]
- Daiber, A.; Lelieveld, J.; Steven, S.; Oelze, M.; Kroller-Scxhon, S.; Sorensen, M.; Munzel, T. The "exposome" concept—how environmental risk factors influence cardiovascular health. Acta Biochim. Pol. 2019, 66, 269–283. [Google Scholar] [CrossRef]
- Louridas, G.E.; Louridas, A.G. Impact of chaos in the progression of heart failure. Int. J. Appl. Sci. Technol. 2012, 2, 24–30. [Google Scholar] [CrossRef]
- Louridas, G.E.; Lourida, K.G. Progressive nature of heart failure and systems biology. Int. Cardiovasc. Forum J. 2015, 3, 5–9. [Google Scholar] [CrossRef][Green Version]
- Fedak, P.W.; Verma, S.; Weisel, R.D.; Li, R.K. Cardiac remodeling and failure: From molecules to man (part 1). Cardiovasc. Path. 2005, 14, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Makkar, K.M.; Sanoski, C.A.; Spinler, S.A. Role of angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, and aldosterone antagonists in the prevention of atrial and ventricular arrhythmias. Pharmacotherapy 2009, 29, 31–48. [Google Scholar] [CrossRef] [PubMed]
- Konstam, M.A.; Rousseau, M.F.; Kronenberg, M.W.; Udelson, J.E.; Melin, J.; Stewart, D.; Dolan, N.; Edens, T.R.; Ahn, S.; Kinan, D.; et al. Effects of the angiotensin converting enzyme inhibitor enalapril on the long-term progression of left ventricular dysfunction in patients with heart failure. SOLVD Investigators. Circulation 1992, 86, 431–438. [Google Scholar] [CrossRef]
- Doughty, R.N.; Whalley, G.A.; Gamble, G.; MacMahon, S.; Sharpe, N. Left ventricular remodeling with carvedilol in patients with congestive heart failure due to ischemic heart disease. Australia-New Zealand Heart Failure Research Collaborative Group. J. Am. Coll. Cardiol. 1997, 29, 1060–1066. [Google Scholar] [CrossRef]
- Louridas, G.E.; Lourida, K.G. Conceptual foundations of systems biology explaining complex cardiac diseases. Healthcare 2017, 5, 10. [Google Scholar] [CrossRef]
- Wan, S.H.; Vogel, M.W.; Chen, H.H. Pre-Clinical Diastolic Dysfunction. J. Am. Coll. Cardiol. 2014, 63, 407–416. [Google Scholar] [CrossRef]
- Hamdani, N.; Bishu, K.G.; von Frieling-Salewsky, M.; Redfield, M.M.; Linke, W.A. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc. Res. 2013, 97, 464–471. [Google Scholar] [CrossRef]
- Jeong, E.M.; Monasky, M.M.; Gu, L.; Taglieri, D.M.; Patel, B.G.; Liu, H.; Wang, Q.; Greener, I.; Dudley, S.C., Jr.; Solaro, R.J. Tetrahydrobiopterin improves diastolic dysfunction by reversing changes in myofilament properties. J. Mol. Cell. Cardiol. 2013, 56, 44–54. [Google Scholar] [CrossRef]
- O’Meara, E.; Thibodeau-Jarry, N.; Ducharme, A.; Rouleau, J.L. The epidemic of heart failure: A lucid approach to stemming the rising tide. Can. J. Cardiol. 2014, 30, S442–S454. [Google Scholar] [CrossRef]
- Frigerio, M.; Oliva, F.; Turazza, F.M.; Bonow, R.O. Prevention and management of chronic heart failure in management of asymptomatic patients. Am. J. Cardiol. 2003, 91, 4F–9F. [Google Scholar] [CrossRef]
- Sahle, B.W.; Owen, A.J.; Chin, K.L.; Reid, C.M. Risk prediction models for incident heart failure: A systematic review of methodology and model performance. J. Card. Fail. 2017, 23, 680–687. [Google Scholar] [CrossRef]
- Sinha, A.; Gupta, D.K.; Yancy, C.W.; Shah, S.J.; Rasmussen-Torvik, L.J.; McNally, E.M.; Greenland, P.; Lloyd-Jones, D.M.; Sadiya, S.; Khan, S.S. Risk-based approach for the prediction and prevention of heart failure. Circ. Heart Fail. 2021, 14, e007761. [Google Scholar] [CrossRef]
- Ho, J.E.; Enserro, D.; Brouwers, F.P.; Kizer, J.R.; Shah, S.J.; Psaty, B.M.; Bartz, T.M.; Santhanakrishnan, R.; Lee, D.S.; Chan, C.; et al. Predicting heart failure with preserved and reduced ejection fraction: The international collaboration on heart failure subtypes. Circ. Heart Fail. 2016, 9, e003116. [Google Scholar] [CrossRef]
- Shinmura, K. Cardiac senescence, heart failure, and frailty: A triangle in elderly people. Keio J. Med. 2016, 65, 25–32. [Google Scholar] [CrossRef]
- Clerico, A.; Recchia, F.A.; Passino, C.; Emdin, M. Cardiac endocrine function is an essential component of the homeostatic regulation network: Physiological and clinical implications. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H17–H29. [Google Scholar] [CrossRef]
- Lanfear, D.E. Genetic variation in the natriuretic peptide system and heart failure. Heart Fail. Rev. 2010, 15, 219–228. [Google Scholar] [CrossRef]
- Hawkridge, A.M.; Heublein, D.M.; Bergen, H.R., III.; Cataliotti, A.; Burnett, J.C., Jr. Quantitative mass spectral evidence for the absence of circulating brain natriuretic peptide (BNP-32) in severe human heart failure. Proc. Natl. Acad. Sci. USA 2005, 102, 17442–17447. [Google Scholar] [CrossRef]
- Rabani, V.; Davani, S. Translational Approaches In Cardiovascular Diseases by “Omics”. Curr. Issues Mol. Biol. 2018, 28, 1–14. [Google Scholar] [CrossRef]
- Mebazaa, A.; Vanpoucke, G.; Thomas, G.; Verleysen, K.; Cohen-Solal, A.; Vanderheyden, M.; Bartunek, J.; Mueller, C.; Launay, J.M.; Van Landuyt, N.; et al. Unbiased plasma proteomics for novel diagnostic biomarkers in cardiovascular disease: Identification of quiescin Q6 as a candidate biomarker of acutely decompensated heart failure. Eur. Heart J. 2012, 33, 2317–2324. [Google Scholar] [CrossRef]
- Doehner, W. Diagnostic biomarkers in cardiovascular disease: The proteomics approach. Eur. Heart J. 2012, 33, 2249–2251. [Google Scholar] [CrossRef][Green Version]
- Stary, H.C. Natural history and histological classification of atherosclerotic lesions: An update. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1177–1178. [Google Scholar] [CrossRef]
- Nikpay, M.; Goel, A.; Won, H.H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Saleheen, D.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C.; et al. CARDIoGRAMplusC4D Consortium. A comprehensive 1000 genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015, 47, 1121–1130. [Google Scholar] [CrossRef]
- Palomaki, G.E.; Melillo, S.; Bradley, L.A. Association between 9p21 genomic markers and heart disease: A meta-analysis. JAMA 2010, 303, 648–656. [Google Scholar] [CrossRef]
- Krarup, N.T.; Borglykke, A.; Allin, K.H.; Sandholt, C.H.; Justesen, J.M.; Andersson, E.A.; Grarup, N.; Jørgensen, T.; Pedersen, O.; Hansen, T. A genetic risk score of 45 coronary artery disease risk variants associates with increased risk of myocardial infarction in 6041 Danish individuals. Atherosclerosis 2015, 240, 305–310. [Google Scholar] [CrossRef]
- O’Donnell, C.J.; Nabel, E.G. Genomics of Cardiovascular Disease. N. Engl. J. Med. 2011, 365, 2098–2109. [Google Scholar] [CrossRef]
- Bjorkegren, J.L.M.; Kovacic, J.C.; Dudley, J.T.; Schadt, E.E. Genome-wide significant loci: How important are they? Systems genetics to understand heritability of coronary artery disease and other common complex disorders. J. Am. Coll.Cardiol. 2015, 65, 830–845. [Google Scholar] [CrossRef]
- Assimes, T.L.; Roberts, R. Genetics: Implications for prevention and management of coronary artery disease. J. Am. Coll. Cardiol. 2016, 68, 2797–2818. [Google Scholar] [CrossRef][Green Version]
- Feinberg, A.P.; Fallin, M.D. Epigenetics at the crossroads of genes and the environment. JAMA 2015, 314, 1129–1130. [Google Scholar] [CrossRef]
- Joshi, A.; Rienks, M.; Theofilatos, K.; Mayr, M. Systems biology in cardiovascular disease: A multiomics approach. Nat. Rev. Cardiol. 2021, 18, 313–330. [Google Scholar] [CrossRef]
- Napoli, C.; Zullo, A.; Picascia, A.; Infante, T.; Francesco Paolo Mancini, F.P. Recent advances in proteomic technologies applied to cardiovascular disease. J. Cell. Biochem. 2013, 114, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Balestrieri, M.L.; Giovane, A.; Mancini, F.P. Proteomics and cardiovascular disease: An update. Curr. Med. Chem. 2008, 15, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Zuk, N.; Dubis, J.; Witkiewicz, W. The role of proteomics in prognosis and treatment of cardiovascular diseases. Przegl Lek. 2013, 70, 143–148. [Google Scholar] [PubMed]
- Mokou, M.; Lygirou, V.; Vlahou, A.; Mischak, H. Proteomics in cardiovascular disease: Recent progress and clinical implication and implementation. Expert Rev. Proteom. 2017, 14, 117–136. [Google Scholar] [CrossRef] [PubMed]
- Tunon, J.; Barbas, C.; Blanco-Colio, L.; Burillo, E.; Lorenzo, O.; Ventura, J.L.M.; Más, S.; Rupérez, F.J.; Egido, J. Proteomics and metabolomics in biomarker discovery for cardiovascular diseases: Progress and potential. Expert Rev. Proteom. 2016, 13, 857–871. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, M.L.; Mayr, M.; Gomes, A.V.; Delles, C.; Arrell, D.K.; Murphy, A.M.; Lange, R.A.; Costello, C.E.; Jin, Y.F.; Laskowitz, D.T.; et al. Transformative Impact of Proteomics on Cardiovascular Health and Disease. Circulation 2015, 132, 852–872. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.P.Y.; Ping, P.; Murphy, E. Proteomics research in cardiovascular medicine and biomarker discovery. J. Am. Coll. Cardiol. 2016, 68, 2819–2830. [Google Scholar] [CrossRef]
- Howes, J.M.; Keen, J.N.; Findlay, J.B.; Carter, A.M. The application of proteomics technology to thrombosis research: The identification of potential therapeutic targets in cardiovascular disease. Diabetes Vasc. Dis. Res. 2008, 5, 205–212. [Google Scholar] [CrossRef]
- Distelmaier, K.; Adlbrecht, C.; Jakowitsch, J.; Wagner, O.; Gerner, C.; Lang, I.M.; Kubicek, M. Proteomic profiling of acute coronary thrombosis reveals a local decrease in pigment epithelium-derived factor in acute myocardial infarction. Clin. Sci. 2012, 123, 111–119. [Google Scholar] [CrossRef]
- Tuñón, J.; Martín-Ventura, J.L.; Blanco-Colio, L.M.; Lorenzo, O.; López, J.A.; Egido, J. Proteomic strategies in the search of new biomarkers in atherothrombosis. J. Am. Coll. Cardiol. 2010, 55, 2009–2016. [Google Scholar] [CrossRef]
- Haase, T.; Börnigen, D.; Müller, C.; Tanja Zeller, T. Systems Medicine as an Emerging Tool for Cardiovascular Genetics. Front. Cardiovasc. Med. 2016, 3, 27. [Google Scholar] [CrossRef]
- Bellis, C.; Kulkarni, H.; Mamtani, M.; Kent, J.W., Jr.; Wong, G.; Weir, J.M.; Barlow, C.K.; Diego, V.; Almeida, M.; Dyer, T.D.; et al. Human plasma lipidome is pleiotropically associated with cardiovascular risk factors and death. Circulation. Cardiovasc. Genet. 2014, 7, 854–863. [Google Scholar] [CrossRef]
- Ussher, J.R.; Elmariah, S.; Gerszten, R.E.; Dyck, J.R.B. The emerging role of metabolomics in the diagnosis and prognosis of cardiovascular disease. J. Am. Coll. Cardiol. 2016, 68, 2850–2870. [Google Scholar] [CrossRef]
- Ganna, A.; Salihovic, S.; Sundstrom, J.; Broeckling, C.D.; Hedman, A.K.; Magnusson, P.K.E.; Pedersen, N.L.; Larsson, A.; Siegbahn, A.; Zilmer, M.; et al. Large-scale metabolomic profiling identifies novel biomarkers for incident coronary heart disease. PLoS Genet. 2014, 10, e1004801. [Google Scholar] [CrossRef]
- Rader, D.J.; Daugherty, A. Translating molecular discoveries into new therapies for atherosclerosis. Nature 2008, 451, 904–913. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The LDL Receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef]
- Wilson, D.J.; Gahan, M.; Haddad, L. A World Wide Web site for low-density lipoprotein receptor gene mutations in familial hypercholesterolemia: Sequence-based, tabular, and direct submission data handling. Am. J. Cardiol. 1998, 81, 1509–1511. [Google Scholar] [CrossRef]
- Frueh, J.; Maimari, N.; Lui, Y.; Kis, Z.; Mehta, V.; Pormehr, N.; Grant, C.; Chalkias, E.; Falck-Hansen, M.; Bovens, S.; et al. Systems and synthetic biology of the vessel wall. FEBS Lett. 2012, 586, 2164–2170. [Google Scholar] [CrossRef]
- Giannoglou, G.D.; Soulis, J.V.; Farmakis, T.M.; Farmakis, D.M.; Louridas, G.E. Haemodynamic factors and the important role of local low static pressure in coronary wall thickening. Int. J. Cardiol. 2002, 86, 27–40. [Google Scholar] [CrossRef]
- Szostak, J.; Martin, F.; Talikka, M.; Peitsch, M.C.; Hoeng, J. Semi-Automated curation allows causal network model building for the quantification of age-dependent plaque progression in ApoE-/- mouse. Gene Regul. Syst. Biol. 2016, 10, 95–103. [Google Scholar] [CrossRef]
- Cho, D.Y.; Kim, Y.A.; Przytycka, T.M. Chapter 5: Network biology approach to complex diseases. PLoS Comput. Biol. 2012, 8, e1002820. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Frantz, S.; Swirski, F.K.; Mulder, W.J.M.; Randolph, G.; Ertl, G.; Ntziachristos, V.; Piek, J.P.; Stroes, E.S.; Schwaiger, M.; et al. Imaging systemic inflammatory networks in ischemic heart disease. J. Am. Cardiol. 2015, 65, 1583–1591. [Google Scholar] [CrossRef]
- Wollenweber, T.; Roentgen, P.; Schäfer, A.; Schatka, I.; Zwadlo, C.; Brunkhorst, T.; Berding, G.; Bauersachs, J.; Bengel, F.M. Characterizing the inflammatory tissue response to acute myocardial infarction by clinical multi-modality noninvasive imaging. Circ. Cardiovasc. Imaging 2014, 7, 811–818. [Google Scholar] [CrossRef]
- Paynter, N.P.; Ridker, P.M.; Chasman, D.I. Are genetic tests for atherosclerosis ready for routine clinical use? Circ. Res. 2016, 118, 607–619. [Google Scholar] [CrossRef]
- Umans-Eckenhausen, M.A.; Sijbrands, E.J.; Kastelein, J.J.; Defesche, J.C. Low-density lipoprotein receptor gene mutations and cardiovascular risk in a large genetic cascade screening population. Circulation 2002, 106, 3031–3036. [Google Scholar] [CrossRef][Green Version]
- Usifo, E.; Leigh, S.E.; Whittall, R.A.; Lench, N.; Alison Taylor, A.; Yeats, C.; Orengo, C.A.; Martin, A.C.R.; Celli, J.; Humphries, S.E. Low-density lipoprotein receptor gene familial hypercholesterolemia variant database: Update and pathological assessment. Ann. Hum. Genet. 2012, 76, 387–401. [Google Scholar] [CrossRef]
- Ridker, P.M. LDL cholesterol: Controversies and future therapeutic directions. Lancet 2014, 384, 607–617. [Google Scholar] [CrossRef]
- Perk, J.; De Backer, G.; Gohlke, H.; Graham, I.; Reiner, Z.; Verschuren, M.; Albus, C.; Benlian, P.; Boysen, G.; Cifkova, R.; et al. European Guidelines on cardiovascular disease prevention in clinical practice (version 2012). The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts). Eur. Heart J. 2012, 33, 1635–1701. [Google Scholar] [CrossRef]
- Ridker, P.M.; Buring, J.E.; Rifai, N.; Cook, N.R. Development and validation of improved algorithms for the assessment of global cardiovascular risk in women: The Reynolds Risk Score. JAMA 2007, 297, 611–619. [Google Scholar] [CrossRef]
- Hippisley-Cox, J.; Coupland, C.; Vinogradova, Y.; Robson, J.; Minhas, R.; Sheikh, A.; Peter Brindle, P. Predicting cardiovascular risk in England and Wales: Prospective derivation and validation of QRISK2. BMJ 2008, 336, 1475–1482. [Google Scholar] [CrossRef]
- Goff, D.C., Jr.; Lloyd-Jones, D.M.; Bennett, G.; Coady, S.; D’Agostino, R.B.; Gibbons, R.; Greenland, P.; Lackland, D.T.; Levy, D.; O’Donnell, C.J.; et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2014, 63, 2935–2959. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, S.; Choi, H.C.; Weisshaar, J.C. The spatial biology of transcription and translation in rapidly growing Escherichia coli. Front. Microbiol. 2015, 6, 636. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, A.; Mathe, E.A.; Li, L.; Liu, B.; Ma, Q. Elucidation of biological networks across complex diseases using single-cell omics. Trends Genet. 2020, 36, 951–966. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Stein, J.E.; Rimm, D.L.; Wang, D.W.; Bell, J.M.; Johnson, D.B.; Sosman, J.A.; Schalper, K.A.; Anders, R.A.; Wang, H.; et al. Comparison of Biomarker Modalities for Predicting Response to PD-1/PD-L1 Checkpoint Blockade: A Systematic Review and Meta-analysis. JAMA Oncol. 2019, 5, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Chhibber, A.; Kroetz, D.L.; Tantisira, K.G.; McGeachie, M.; Cheng, C.; Plenge, R.; Stahl, E.; Sadee, W.; Ritchie, M.D.; Sarah A Pendergrass, S.A. Genomic architecture of pharmacological efficacy and adverse events. Pharmacogenomics 2014, 15, 2025–2048. [Google Scholar] [CrossRef] [PubMed]
- Biesecker, L.G. Opportunities and challenges for the integration of massively parallel genomic sequencing into clinical practice: Lessons from the ClinSeq project. Genet. Med. 2012, 14, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.T.; Boerwinkle, S.E.; O’Connell, J.R.; Bailey, K.R.; Gong, Y.; Chapman, A.B.; McDonough, C.W.; Beitelshees, A.L.; Schwartz, G.L.; Gums, J.G.; et al. Genomic association analysis of common variants influencing antihypertensive response to hydrochlorothiazide. Hypertension 2013, 62, 391–397. [Google Scholar] [CrossRef]
- Ross, S.; Eikelboom, J.; Anand, S.S.; Eriksson, N.; Gerstein, H.C.; Mehta, S.; Connolly, S.J.; Rose, L.; Ridker, P.M.; Wallentin, L.; et al. Association of cyclooxygenase-2 genetic variant with cardiovascular disease. Eur. Heart J. 2014, 35, 2242–2248. [Google Scholar] [CrossRef]
- Polisecki, E.; Muallem, H.; Maeda, N.; Peter, I.; Robertson, M.; McMahon, A.D.; Ford, I.; Packard, C.; Shepherd, J.; Jukema, J.W.; et al. Prospective Study of Pravastatin in the Elderly at Risk (PROSPER) Investigators. Genetic variation at the LDL receptor and HMG-CoA reductase gene loci, lipid levels, statin response, and cardiovascular disease incidence in PROSPER. Atherosclerosis 2008, 200, 109–114. [Google Scholar] [CrossRef]
- Postmus, I.; Trompet, S.; Deshmukh, H.A.; Barnes, M.R.; Li, X.; Warren, H.R.; Chasman, D.I.; Zhou, K.; Arsenault, B.J.; Donnelly, L.A.; et al. Welcome Trust Case Control Consortium. Pharmacogenetic meta-analysis of genome-wide association studies of LDL cholesterol response to statins. Nat. Commun. 2014, 5, 5068. [Google Scholar] [CrossRef]
- Stewart, A. SLCO1B1 polymorphisms and statin-induced myopathy. PLoS Curr. 2013, 5. [Google Scholar] [CrossRef]
- Pereira, N.L.; Bavry, A.A.; Eagle, K.A. Tailored Antiplatelet Initiation to Lessen Outcomes due to Decreased Clopidogrel Response after Percutaneous Coronary Intervention-TAILOR PCI. In Proceedings of the ACC.21 Meeting, Virtual, 17 May 2021. [Google Scholar]
- Johnson, J.A.; Gong, L.; Whirl-Carrillo, M.; Gage, B.F.; Scott, S.A.; Stein, C.M.; Anderson, J.L.; Kimmel, S.E.; Lee, M.T.M.; Pirmohamed, M.; et al. Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin. Pharmacol. Ther. 2011, 90, 625–629. [Google Scholar] [CrossRef]
- Pirillo, A.; Casula, M.; Olmastroni, E.; Norata, G.D.; Catapano, A.L. Global epidemiology of dyslipidaemias. Nat. Rev. Cardiol. 2021, 18, 689–700. [Google Scholar] [CrossRef]
- EMG-Health Podcast: Unmet Need among Patients with Atherosclerotic Cardiovascular Disease Receiving Statin Therapy. Primary Prevention and Treatment of Atherosclerotic Cardiovascular Disease. Available online: https://www.emg-health.com/omnipresent/emg-health-podcast-unmet-needs-among-patients-with-atherosclerotic-cardiovascular-disease-receiving-statin/ (accessed on 1 December 2021).
- Adhyaru, B.B.; Jacobson, T.A. Safety and efficacy of statin therapy. Nat. Rev. Cardiol. 2018, 15, 757–769. [Google Scholar] [CrossRef]
- Gui, Y.J.; Liao, C.X.; Liu, Q.; Guo, Y.; Yang, T.; Chen, J.Y.; Wang, Y.T.; Hu, J.H.; Xu, D.Y. Efficacy and safety of statins and exercise combination therapy compared to statin monotherapy in patients with dyslipidaemia: A systematic review and meta-analysis. Eur. J. Prev. Cardiol. 2017, 24, 907–916. [Google Scholar] [CrossRef]
- Baigent, C.; Keech, A.; Kearney, P.M.; Blackwell, L.; Buck, G.; Pollicino, C.; Kirby, A.; Sourjina, T.; Peto, R.; Collins, R.; et al. Efficacy and safety of cholesterol-lowering treatment: Prospective meta-analysis of data from 90,056 participants in 14 randomized trials of statins. Lancet 2005, 366, 1267–1278. [Google Scholar] [CrossRef]
- Pownall, H.J.; Rosales, C.; Gillard, B.K.; Gotto, A.M., Jr. High-density lipoproteins, reverse cholesterol transport and atherogenesis. Nat. Rev. Cardiol. 2021, 18, 712–723. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk: The task force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Boden, W.E.; O’Rourke, R.A.; Teo, K.K.; Hartigan, P.M.; Maron, D.J.; Kostuk, W.J.; Knudtson, M.; Dada, M.; Paul Casperson, P.; Harris, C.L.; et al. Optimal medical therapy with or without PCI for stable coronary disease. N. Engl. J. Med. 2007, 356, 1503–1516. [Google Scholar] [CrossRef]
- Trogdon, J.G.; Finkelstein, E.A.; Nwaise, I.A.; Tangka, F.K.; Orenstein, D. The economic burden of chronic cardiovascular disease for major insurers. Health Promot. Pract. 2007, 8, 234–242. [Google Scholar] [CrossRef]
- Amitzur, G.; Einav, S. Three Cardiovascular Physiological Parameters, Endothelial Function, Arterial Stiffness and Autonomic System Function, Represent Unique Complementary Properties. Tel Aviv: Academia, Tel Aviv University. 2007. Available online: https://telaviv.academia.edu/GioraAmitzur (accessed on 1 March 2021).
- Yamaoka-Tojo, M. Endothelial function for cardiovascular disease prevention and management. Int. J. Clin. Cardiol. 2017, 4, 103. [Google Scholar] [CrossRef]
- Oikonomou, E.; Siasos, G.; Tsigkou, V.; Bletsa, E.; Panoilia, M.E.; Oikonomou, I.N.; Simanidis, I.; Spinou, M.; Papastavrou, A.; Kokosias, G.; et al. Coronary artery disease and endothelial dysfunction: Novel diagnostic and therapeutic approaches. Curr. Med. Chem. 2020, 27, 1052–1080. [Google Scholar] [CrossRef]
- de Koning, E.J.; Rabelink, T.J. Endothelial function in the post-prandial state. Atheroscler. Suppl. 2002, 3, 11–16. [Google Scholar] [CrossRef]
- Vogel, R.A.; Corretti, M.C.; Plotnick, G.D. Effect of a single high-fat meal on endothelial function in healthy subjects. Am. J. Cardiol. 1997, 79, 350–354. [Google Scholar] [CrossRef]
- Giannattasio, C.; Zoppo, A.; Gentile, G.; Failla, M.; Capra, A.; Maggi, F.M.; Catapano, A.; Mancia, G. Acute effect of high-fat meal on endothelial function in moderately dyslipidemic subjects. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 406–410. [Google Scholar] [CrossRef]
- Rathnayake, K.M.; Weech, M.; Jackson, K.G.; Lovegrove, J.A. Impact of meal fatty acid composition on postprandial lipaemia, vascular function and blood pressure in postmenopausal women. Nutr. Res. Rev. 2018, 31, 193–203. [Google Scholar] [CrossRef]
- Suwaidi, J.A.; Hamasaki, S.; Higano, S.T.; Nishimura, R.A.; Holmes, D.R., Jr.; A Lerman, A. Long-term follow-up of patients with mild coronary artery disease and endothelial dysfunction. Circulation 2000, 101, 948–954. [Google Scholar] [CrossRef]
- Esselstyn Jr, C.B.; Gendy, G.; Doyle, J.; Golubic, M.; Roizen, M.F. A way to reverse CAD? J. Fam. Pract. 2014, 63, 356–364. [Google Scholar]
- Dehghan, M.; Mente, A.; Teo, K.K.; Gao, P.; Sleight, P.; Dagenais, G.; Avezum, A.; Probstfield, J.L.; Dans, T.; Yusuf, S. Ongoing Telmisartan Alone and in Combination With Ramipril Global End Point Trial (ON TARGET)/Telmisartan Randomized ASSESSMENT Study in ACEI Intolerant Subjects With Cardiovascular Disease (TRANSCEND) Trial Investigators. Relationship between healthy diet and risk of cardiovascular disease among patients on drug therapies for secondary prevention: A prospective cohort study of 31,546 high-risk individuals from 40 countries. Circulation 2012, 126, 2705–2712. [Google Scholar] [CrossRef]
- Crowe, F.L.; Appleby, P.N.; Travis, R.C.; Key, T.J. Risk of hospitalization or death from ischemic heart disease among British vegetarians and nonvegetarians: Results from the EPIC-Oxford cohort study. Am. J. Clin. Nutr. 2013, 97, 597–603. [Google Scholar] [CrossRef]
- Huang, B.T.; Peng, Y.; Liu, W.; Zhang, C.; Chai, H.; Huang, F.Y.; Zuo, Z.L.; Liao, Y.B.; Xia, T.L.; Chen, M. Nutritional state predicts all-cause death independent of comorbidities in geriatric patients with coronary artery disease. J. Nutr. Health Aging 2016, 20, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Patnode, C.D.; Evans, C.V.; Senger, C.A.; Redmond, N.; Lin, G.S. Behavioral counseling to promote a healthful diet and physical activity for cardiovascular disease prevention in adults without know cardiovascular disease risk factors: Updated evidence report and systematic review for the US preventive services task force. JAMA 2017, 318, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Yiaslas, T.A.; Sood, A.; Ono, G.; Rogers-Soeder, T.S.; Kitazono, R.E.; Embree, J.; Spann, C.; Caputo, C.A.; Taylor, J.; Schaefer, S. The Design and Implementation of a Heart Disease Reversal Program in the Veterans Health Administration: Before and During the COVID-19 Pandemic. Fed. Pract. 2020, 37, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Tilinin, I.S. Design and Implementation of Reverse Risk Assessment Software. Master’s Theses, San Jose State University, San Jose, CA, USA, 2008. [Google Scholar]
- Ponikowski, P.; Voors, A.A.; Anker, S.D. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef] [PubMed]
- Leach, J.; Heallen, T.; Zhang, M.; Rahmani, M.; Morikawa, Y.; Hill, M.C.; Segura, A.; Willerson, J.T.; Martin, J.F. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 2017, 550, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, J.E.; Fang, J.C.; Margulies, K.B.; Mann, D.L. Heart failure with recovered left ventricular ejection fraction: JACC Scientific Expert Panel. J. Am. Coll. Cardiol. 2020, 76, 719–734. [Google Scholar] [CrossRef]
- Bermejo, R.A.; Babarro, E.G.; Canoa, J.N.L.; Román, A.V.; Otero, I.G.; Ayude, M.O.; Vazquez, P.P.; Rodríguez, I.G.; Castro, O.D.; Juanatey, J.R.G. Heart failure with recovered ejection fraction: Clinical characteristics, determinants and prognosis. CARDIOCHUS-CHOP registry. Cardiol. J. 2018, 25, 353–362. [Google Scholar] [CrossRef]
- Tanabe, K.; Sakamoto, T. Heart failure with recovered ejection fraction. J. Echocardiogr. 2019, 17, 5–9. [Google Scholar] [CrossRef]
- Gutowska, K.; Kogut, D.; Kardynska, M.; Formanowicz, P.; Smieja, J.; Puszynski, K. Petri nets and ODEs as complementary methods for comprehensive analysis on an example of the ATM-p53-NF-[Formula: See text]B signaling pathways. Sci. Rep. 2022, 12, 1135. [Google Scholar] [CrossRef]
- Costa, M.C.; Gabriel, A.F.; Enguita, F.J. Bioinformatics Research Methodology of Non-coding RNAs in Cardiovascular Diseases. Adv. Exp. Med. Biol. 2020, 1229, 49–64. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef]
- Wörheide, M.A.; Krumsiek, J.; Kastenmüller, G.; Arnold, M. Multi-omics integration in biomedical research—A metabolomics-centric review. Anal. Chim. Acta 2021, 1141, 144–162. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).