1. Introduction
Fabry disease is a rare X-linked lysosomal storage disorder (MIM 301500). It is an inborn error of glycosphingolipid metabolism, resulting from a deficiency or total absence of the alpha-galactosidase A (α-Gal A) enzyme [
1]. This enzyme deficiency results in the accumulation of globotriaosylceramide (Gb3) in a number of cell types, leading to complex, progressive, multisystem disease. In affected males, the usual presentation is of episodic crises in childhood of severe pain in the extremities (acroparesthesia), angiokeratomas (vascular cutaneous lesions), sweating abnormalities, and characteristic corneal and lenticular opacities [
2,
3]. Gradual deterioration of renal function to end-stage renal disease (ESRD) usually occurs in the third to fifth decade [
2]. Cardiac involvement in Fabry disease is common, and typically cardiac hypertrophy develops, often with a concentric non-obstructive pattern mimicking hypertrophic cardiomyopathy (HCM) due to variants in genes encoding components of the sarcomere [
4]. The precise mechanism resulting in the degree of cardiac hypertrophy is unknown. Estimates of incidence range from as high as 1 in 40,000 to 1 in 117,000 live male births [
5,
6].
Males with Fabry disease are hemizygous for a loss of function
GLA allele [
7]. Females are heterozygous for a loss of function allele and are cellular mosaics due to random X-chromosome inactivation. This mosaicism results in a wide spectrum of phenotype with a later age at the onset of symptoms, and frequently milder disease [
8]. Often manifesting carrier females present with cardiac involvement [
8].
Males with greater than 1% α-Gal A activity may have different clinical presentations with the development of left ventricular hypertrophy and arrhythmia later in life [
2,
3].
The case presented here is of a middle aged male presenting with arm pain, which triggered an electrocardiogram (ECG) that ultimately revealed co-incidental cardiac hypertrophy and normal α-Gal A enzyme levels. Genetic studies revealed a mosaic pathogenic variant in GLA consistent with Fabry disease. The mosaicism explains the late onset milder disease presentation and low to normal enzyme activity in blood spot and plasma, and this suggests that α-Gal A activity in the normal range does not exclude a diagnosis of Fabry disease.
2. Clinical Report
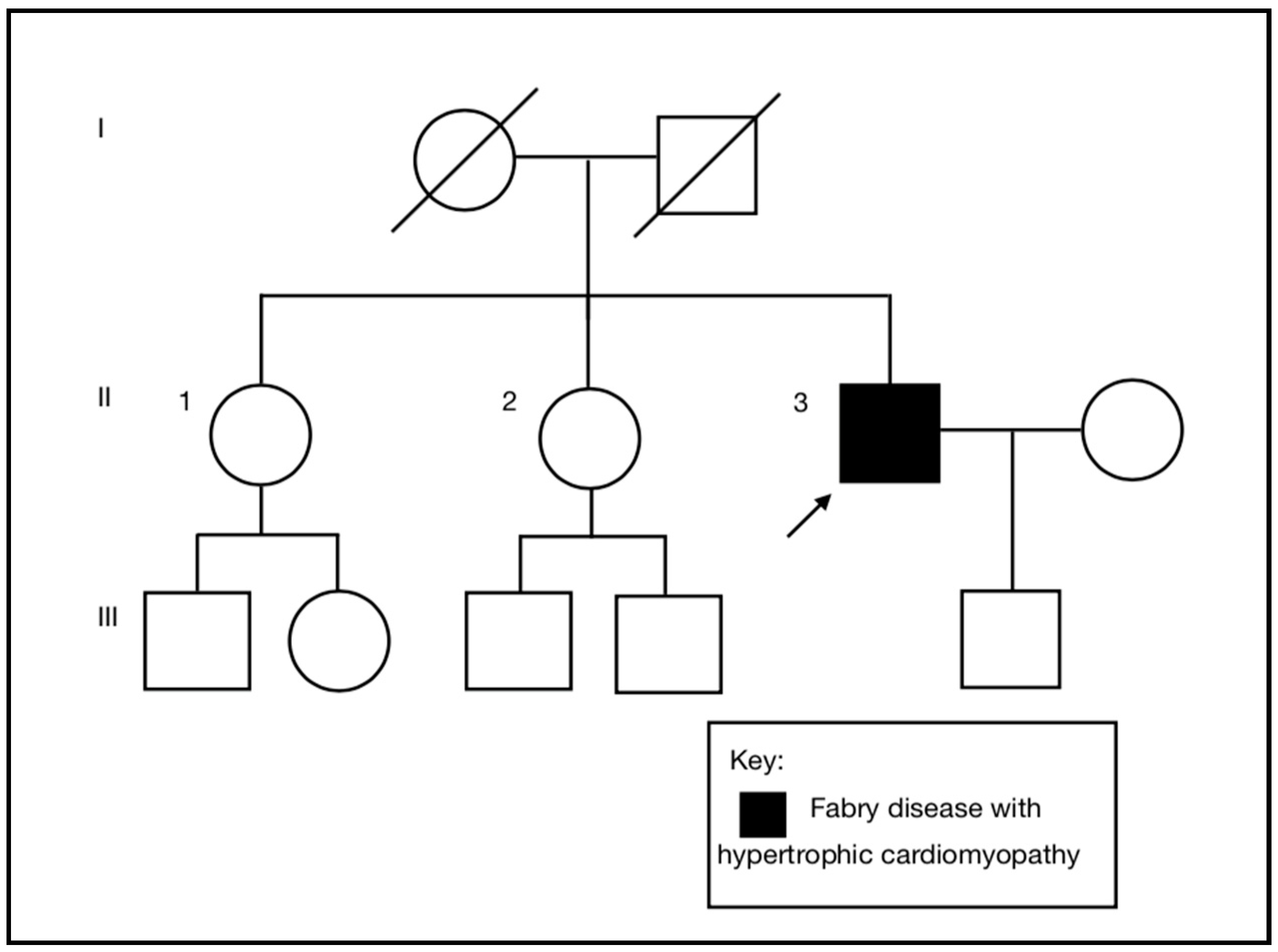
A 55-year-old (II-3), previously a fit, healthy, and normotensive male, presented to the emergency department with an episode of sudden onset left arm pain. He was an active smoker, but had no other past medical history or cardiac risk factors. There was no family history of cardiac disease (
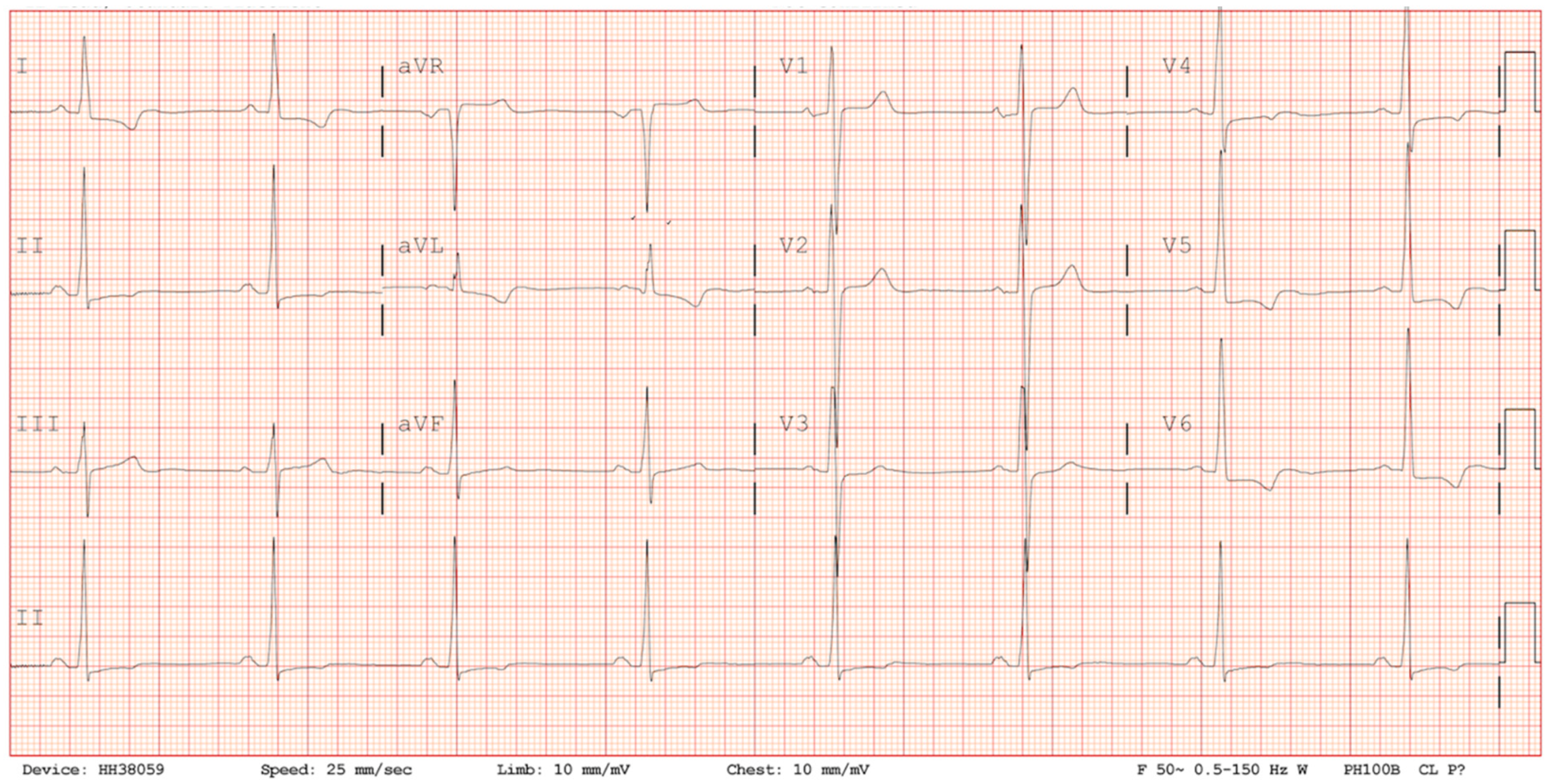
Figure 1). On arrival to the emergency department, his ECG demonstrated sinus rhythm, normal axis, preterminal negative T waves in I, avL, V4-V6, RS conversion in V3/V4 and ST-segment depression in V5 and V6 reflecting anterolateral repolarisation abnormalities (
Figure 2). There was no abnormality of conduction and no dynamic changes on serial ECGs (
Figure 2). Serial cardiac troponin levels were borderline elevated at 0.13 ng/mL and 0.14 ng/mL (normal < 0.03 ng/mL) and so an echocardiogram was performed.
The echocardiogram demonstrated overall moderate, concentric left ventricular hypertrophy (LVH) with septum predisposition. The interventricular septal diameter was 2.2 cm (normal ≤ 1.2 cm, severe hypertrophy ≥ 2.0 cm), and the posterior wall diameter was 1.5 cm (normal ≤ 1.2 cm, mild hypertrophy 1.3–1.5 cm). Left ventricular mass was calculated at 315 g (normal range 96–200 g, severely elevated ≥ 254 g) and mass index at 158.4 g/m2 (normal range 50–102 g/m2, severely elevated ≥ 130 g/m2). The right ventricle had normal size and function and was reported as showing no clear evidence of right ventricular hypertrophy (RVH). Diastolic function was impaired, but left ventricular size and systolic function were within normal limits. There was no evidence of left ventricular outflow tract obstruction at rest, and no evidence of systolic anterior motion of the mitral valve.
In view of a mild troponin rise, he underwent a coronary angiogram, via the right radial approach demonstrating smooth coronary arteries in a conventional distribution. The provisional diagnosis in the absence of a past medical history of hypertension was hypertrophic cardiomyopathy (HCM) pending further investigation.
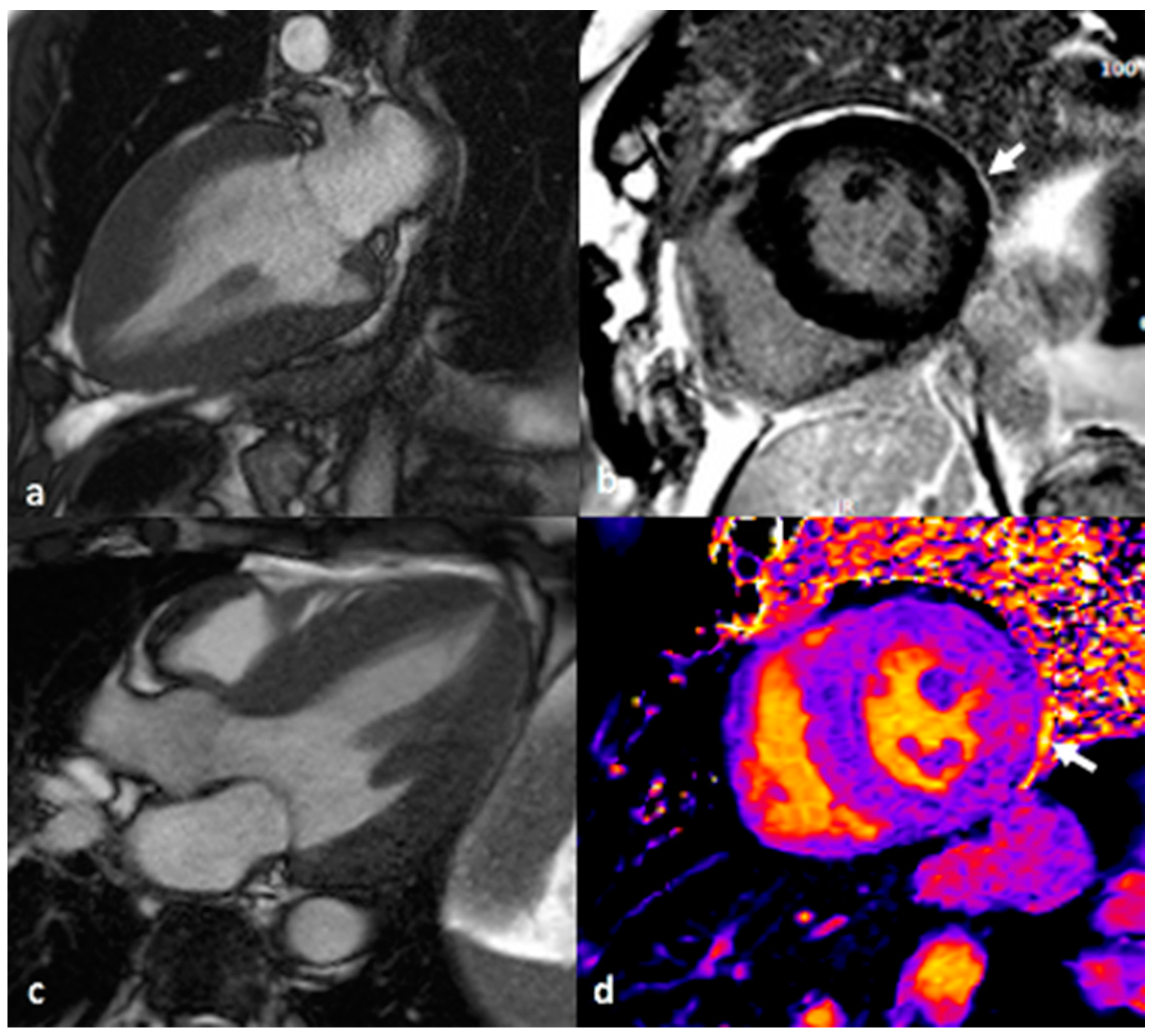
A cardiac MRI scan was performed on a 1.5 Tesla MAGNETOM Avanto Siemens (Erlangen, Germany) MRI scanner using 0.1 mmol/kg gadobutrol (Gadovist, Bayer) as a contrast agent to facilitate late enhancement imaging in order to determine the nature of the myocardial injury responsible for the small troponin leak. In addition to standard volumetric analysis and late gadolinium enhancement imaging, native, and post-contrast T1 mapping was performed (
Figure 3). This demonstrated bi-ventricular hypertrophy with concentric LVH and a maximal left ventricular wall thickness diameter of 2.3 cm. There was evidence of RVH with a maximal right ventricular free wall diameter of 7.5 mm. Late gadolinium enhancement imaging demonstrated patchy hyperenhancement in the basal/mid-lateral and inferior and inferolateral wall segments as well as the RV insertion point and apex. Volumetric analysis indicated a normal 4-chamber size and normal aortic dimensions. Biventricular function was normal; left ventricular ejection fraction was 72% (normal 58–76%). Native T1 mapping highlighted markedly reduced T1 levels in the inferoseptum (basal segment 880 ms, midsegment 870 ms, which was against local reference values of 950–1030 ms). The triad of concentric hypertrophy, typical pattern of late gadolinium enhancement, and low native septal T1 is characteristic of advanced Fabry disease cardiomyopathy [
4].
Following an initial phenotype based diagnosis of HCM, further evaluation of underlying aetiology included enzyme assays for Fabry disease, all of which were within normal limits. The patient had normal dried blood spot α-Gal A enzyme activity of 7.84 picomol/punch/h (range 6.3–47). His plasma α-Gal A level was 3.0 micromol/L/h (range 3.0–20), not indicative of a diagnosis of Fabry disease.
He provided written consent (ref 11/H10003/3) to undergo genetic testing by next generation sequencing (NGS) of a panel of 21 genes associated with hypertrophic cardiomyopathy (
Supplemental Table S1). Enrichment was performed with a custom designed SureSelect target enrichment kit (Agilent) following the manufacturer’s protocols. The target enrichment design consists of the coding region of transcripts, including the immediate splice sites (+/−5 bases) for genes associated with inherited cardiac disorders. The samples were sequenced using a NextSeq 500 (Illumina), according to the manufacturer’s protocols. Sequence data were aligned to hg19 human genome using BWA-AIn vO.6.2 and BWA-Sampe vO.6.2. For genes associated with HCM, variant calling was completed using GenomeAnalysisToolKitUte-v2.0.39 (GATK) (SNVs and indels), Pindel v0.2.4.t (large indels), and DeCON v1.0.1 to detect copy number variation (CNV) (in-house bioinformatics pipeline version 1.15.1). Analytical validation of variants detected by NGS was confirmed by Sanger sequencing. Variant classification was performed according to ACGS guidelines [
9]. Here, 99.9% of the target coding region of the genes was covered to a minimum read depth of 50×.
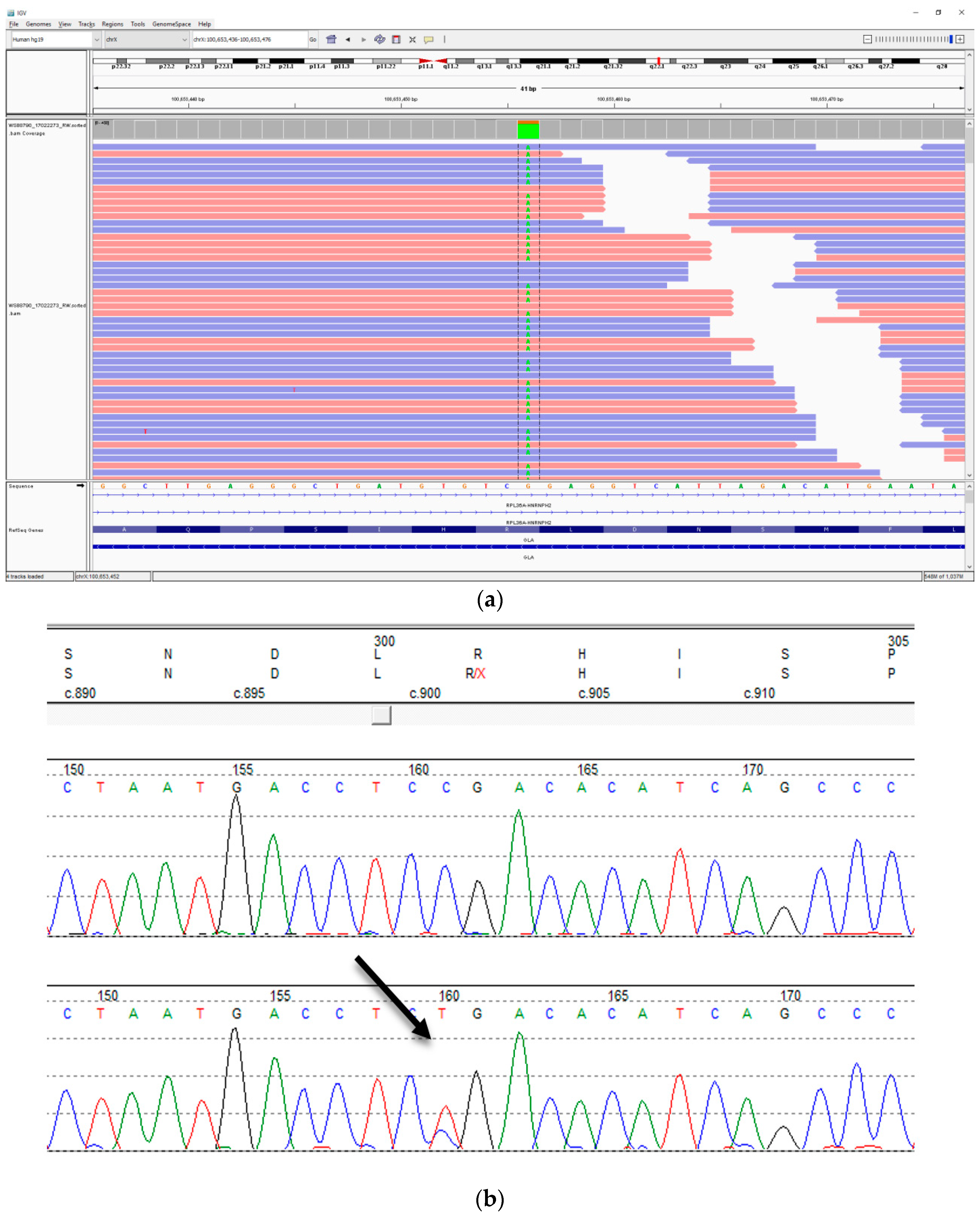
NGS testing identified that he had evidence of both a wild type (25% of reads) and the variant c.901C>T, p.(Arg301Ter) allele (75%) in
GLA (
Table 1,
Figure 4). This variant is predicted to result in a premature termination of translation of
GLA at a position expected to result in nonsense mediated decay. The variant has been reported multiple times to be associated with Fabry disease [
10,
11,
12,
13,
14,
15]. A multiplex quantitative fluorescence-PCR (QF-PCR) assay was used to exclude an X chromosome aneuploidy. This assay uses primer sets to target a mixture of polymorphic and nonpolymorphic microsatellite markers [
16]. The primers target six X-specific markers, two Y-specific markers, an Xp22 marker located within the pseudoautosomal region (PAR2) of the X and Y chromosomes that indicated the number of either sex chromosome, a marker within the
AMELX at Xp22.2 (105 bp) and Yp11.2 (110 bp) indicating the ratio of X to Y chromosomes, and a
TAF9 marker at Xq21.1 and 3p24.2 indicating the ratio of X chromosomes to autosomes. Analysis was undertaken according to the ACGS best practice guidelines [
17].
A subsequent genetic analysis was undertaken on samples of DNA extracted from blood, urine, and saliva to examine tissues of different embryonic origin to determine whether somatic mosaicism was present or whether the mosaicism was confined to blood. The analysis by an NGS and Sanger sequencing of
GLA (
Table 1) determined that the wild type (cytosine, C) allele was present at a minor allele frequency of between 25 and 41% in the samples of a different origin. Lower peak heights in the Sanger sequencing traces for the C nucleotide at position 901 compared to 900 of
GLA in the patient samples, and a lower height in position 901 between the control and patient samples were consistent with mosaicism (
Table 2).
To determine more fully the biochemical effects of the genotype, the biochemical testing was repeated. Plasma α-Gal A level was below the normal range at 1.4 micromol/L/h, as was the leukocyte level at 1.4 micromol/L/h (range 10–50). Urine globotriaosylceramide (Gb3) 0.86 mg/mmol creatinine (normal 0–0.03) and plasma globotriaosylsphingosine (lyso-Gb3) 20.7 ng/mL (normal 0–1.8) were measured. Both were elevated, consistent with a diagnosis of Fabry disease.
A reverse phenotyping of the proband established that he had experienced occasional episodes of non-limiting acroparaesthesia in his left hand. These had been attributed to his occupation as a truck driver. There was no evidence of angiokeratoma. His renal function revealed CKD stage 2 with a glomerular filtration rate (GFR) of 86 mL/min/1.73 m2, a normal urine albumin creatinine ratio (urine albumin < 3.0 mg/L, urine creatinine 5.4 mmol/L, ratio < 0.56 g/mol normal reference range 0.00–2.5 g/mol) and an elevated baseline troponin of 338 ng/L (normal reference range < 40 ng/L). Cerebral MRI demonstrated widespread deep white matter hyperdensities but no infarction or space occupying lesion. Ophthalmological and audiological assessments were unremarkable.
In the presence of the now-established Fabry cardiomyopathy, a decision was made to treat with enzyme replacement therapy (ERT). Chaperone therapy was not an option as the
GLA c.901C>T, p.(Arg301Ter) variant is not amenable to this approach [
18]. ERT was well tolerated, and no infusion related complications were reported over the next 18 months. At the six month follow-up, multiple clinical parameters showed improvement (
Table 3). Lyso-Gb3 levels had declined from 20.7 to 6.2 ng/mL, and the estimated GFR had decreased from 86 to 68 mL/min/1.73 m
2; urine albumin creatinine ratio was within normal limits at 0.31 g/mol (urine albumin 4.0 mg/L, urine creatinine 13 mmol/L, ratio 0.31 g/mol, normal reference range 0.00–2.5 g/mol). Consequently a low dose ACE-inhibitor was commenced to manage early renal complications of Fabry disease. A repeat echocardiogram at 18 months demonstrated a reduction in left ventricular hypertrophy. The interventricular septal diameter had reduced in thickness from 2.2 to 1.9 cm, while the posterior wall diameter had reduced from 1.5 to 1.4 cm (normal ≤ 1.2 cm, mild hypertrophy 1.3–1.5 cm). Left ventricular mass reduced from 315 to 293 g (normal range 96–200 g, severely elevated 254 g) and mass index had reduced from 158.4 to 146.8 g/m
2 (normal range 50–102 g/m
2, severely elevated ≥ 130 g/m
2). As seen previously, the left ventricular size and ejection fraction was within normal limits, but the left atrium appeared mildly dilated, and right ventricular hypertrophy was noted on visual assessment. Furthermore, a repeat ECG was performed demonstrating new first degree heart block (PR interval increased from 179 to 213 ms), and a subsequent 24-h tape demonstrated an isolated salvo of four ventricular ectopics as well as infrequent ventricular and supra ventricular ectopics. The clinical decision was therefore made to implant a loop recorder for further monitoring. ERT was continued and continues to be well tolerated.
3. Discussion
Here, we describe the diagnosis of Fabry disease due to a somatic pathogenic variant in GLA in a man with low, but normal levels of alpha-galactosidase. The diagnosis in the affected individual is vital, both in terms of effective treatment and determining the level of risk to his close relatives. He commenced ERT following the genetic diagnosis which would not have been available to him based on his enzyme assay results, with improvement to his cardiac function 18 months since commencing therapy. The accurate diagnosis facilitated renal, ophthalmological and skin screening, and early stage asymptomatic renal disease has been discovered and treated.
The
GLA c.901C>T pathogenic variant has been reported previously [
10,
11,
12,
13,
14,
15] and on a number of occasions in a public sequence variant database [
19]. Where clinical information has been provided, individuals hemizygous for this variant have been affected with multisystem features of Fabry disease, including joint pain and kidney disease [
11], skin, ophthalmological, renal, neuropathic and cardiac involvement [
12], and a predominantly renal disease presentation with cardiac involvement [
14]. One male with this variant who presented with renal disease had symptomatic heterozygote female relatives [
15]. His mother experienced acroparesthesia and neuropathic pain in her thirties, with angiokeratoma, and cornea verticillata and significant left ventricular hypertrophy, without renal involvement. His sister had more extensive features with skin, ophthalmological, renal and neurological evidence of disease and marked left ventricular hypertrophy [
15]. This series of clinical reports illustrates the broad phenotypic variability expressed in both male and females with the
GLA c.901C>T variant. There is no evidence to indicate that this variant results in a predominantly or exclusively cardiac phenotype as has been shown for some
GLA variants. The predominant cardiac presentation in the proband is more similar to the females reported to be symptomatic heterozygotes for this variant [
15] and parallels the cellular mosaicism present in him that is present in heterozygote females.
Somatic mosaicism for a known pathogenic loss of function
GLA variant was reported previously in a 58 year old male who presented with features consistent with hypertrophic cardiomyopathy, proteinuria and mild renal impairment [
20]. The similarity in the presentation and age of onset in this individual highlights that somatic mosaicism for pathogenic
GLA variants may preferentially present with hypertrophic cardiomyopathy. In this mosaic individual, the alpha-galactosidase level in blood was below the normal range at 0.7 mmol/L/h (2.0–11.7) [
20].
The diagnosis of Fabry disease in the proband excludes any increased risk for his son and the need for clinical or genetic testing in him. It is important to note that if he had a daughter she would have been at increased—but not at obligate—risk of inheriting the variant allele as he is mosaic for this. The absence of a family history of Fabry disease is consistent with the identification of mosaicism, a de novo post zygotic event, in the proband. We excluded the proband being a chimera (a single organism composed of two or more different populations of genetically distinct cells that originated in different zygotes) compared to being a mosaic (a mixture of two cell lines in one organism originating in one zygote), as there was no evidence of biallelic variants of LAMP2 and FHL1, the other X-linked genes on the gene panel.
We considered the possibility of revertant mosaicism, i.e., that the proband inherited the pathogenic variant from his mother, but that the variant allele was replaced by the wild type allele in a proportion of cells following fertilization [
21]. To date, revertant mosaicism has not been reported in Fabry disease. Despite no history of heart problems or clinical features consistent with Fabry disease in the mother, we undertook genetic testing in one of the proband’s female siblings (II-2), which was a wild type for the variant. An alternative explanation for the identification of two
GLA alleles in a male resulting in a milder clinical phenotype would be sex chromosome aneuploidy, but this was excluded by QF-PCR.
Generally, the preferred approach for diagnosis of Fabry disease in males if there is no previous family history, is enzyme and biomarker analysis followed by DNA sequencing of
GLA if high clinical suspicion remains. However, if there is a known familial genotype, then a DNA analysis of
GLA is recommended as the first line test. Enzyme level analysis is not the preferred approach to diagnose Fabry disease in females, as the levels can often be normal despite obligate affected status. This may be due to variable X-inactivation in some tissues. Our proband exhibits a phenotype more in keeping with a heterozygote female where single organ disease is more common, compared to male hemizygotes who exhibit younger age of symptom onset and a broader spectrum of organ system involvement [
2,
8].
We recommend that males with cardiomyopathy, especially with imaging changes characteristic of Fabry disease, should not have the diagnosis excluded entirely on the basis of a normal enzyme assay, but should consider more detailed biochemical testing, including lyso-Gb3 or genetic testing to determine if mosaicism is present. Such diagnoses will have a profound impact for the affected individuals and their family members.






{kind=link}
{kind=link}
{kind=link}
{kind=link}