Intranasal DNA Vaccine for Protection against Respiratory Infectious Diseases: The Delivery Perspectives

Abstract
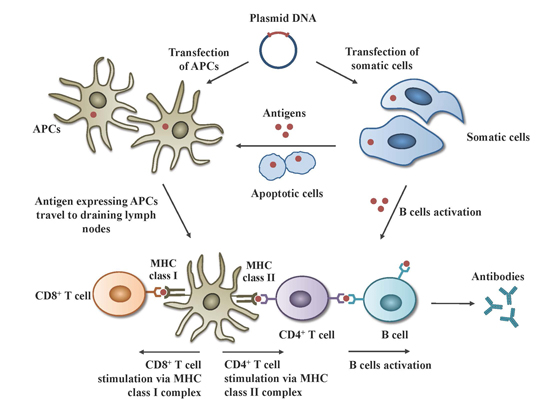
:
1. Introduction

{kind=link}
{kind=link}
{kind=link}
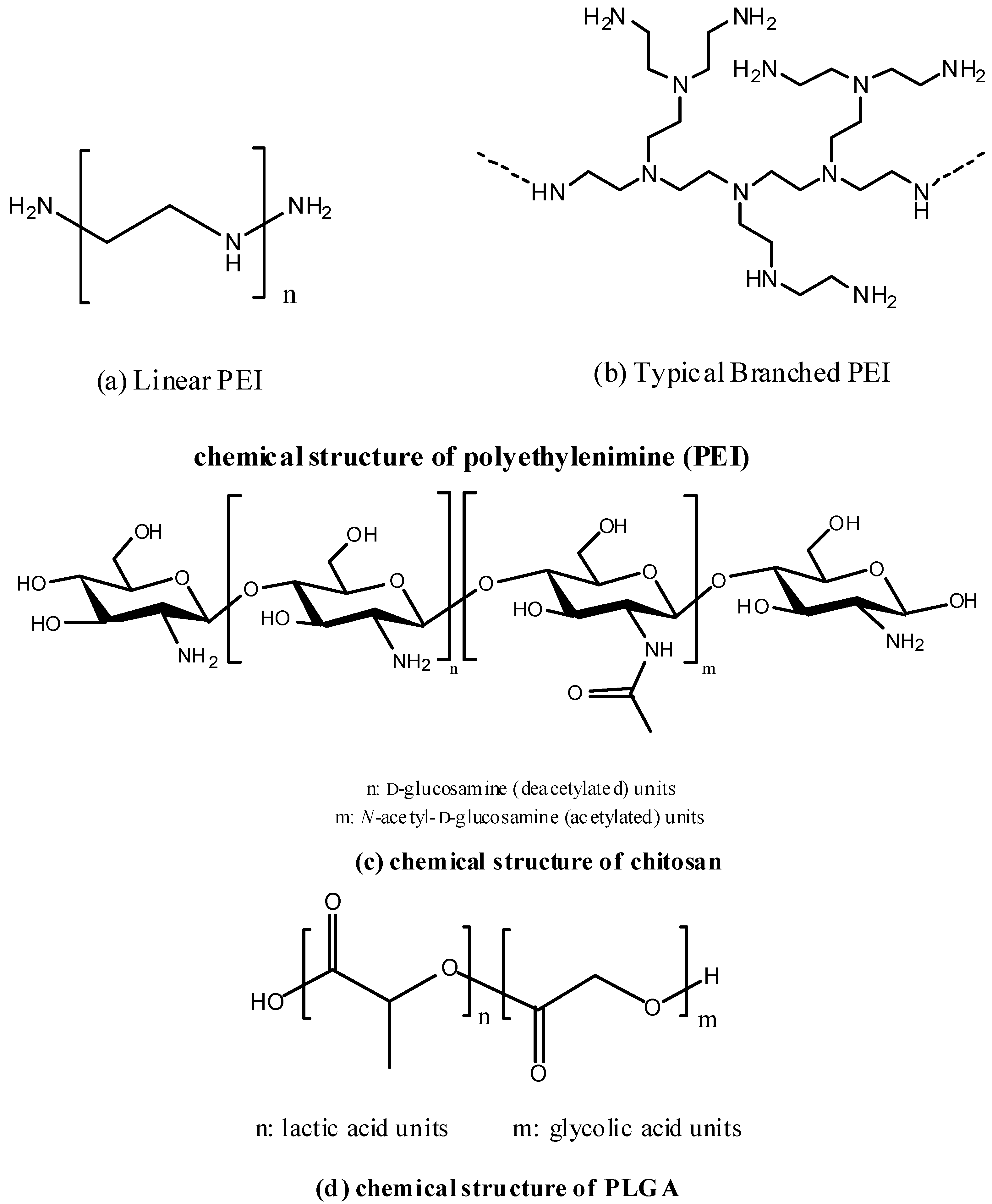{kind=link}
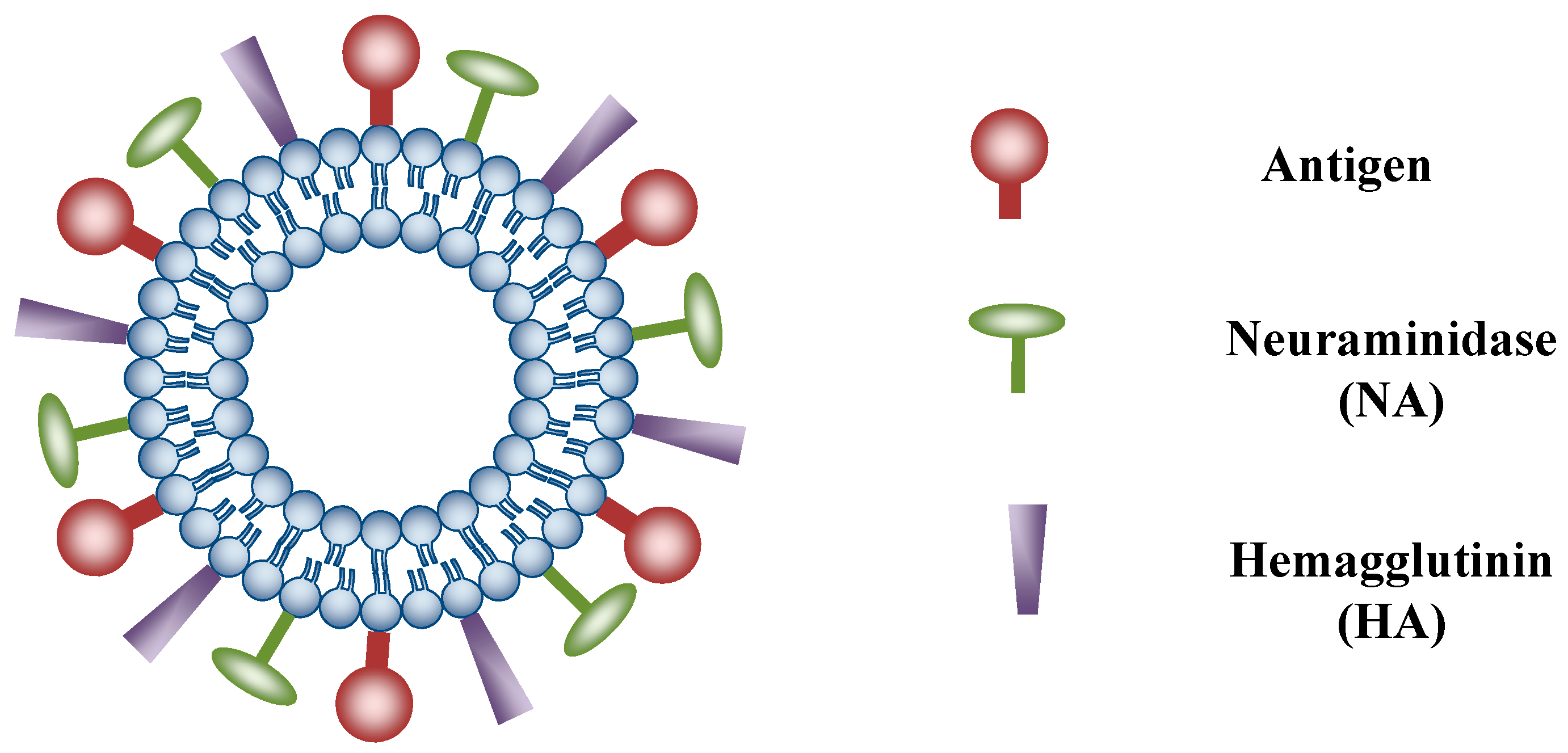{kind=link}
| Category | Characteristics |
|---|---|
| Design | Rapid design |
| Vaccine can be developed for multiple agents in a single formulation | |
| Production | Rapid and reproducible |
| Large-scale production is relatively cheap | |
| Proteins are produced by host cells to ensure proper folding | |
| Stability | Higher stability than proteins or live-attenuated microorganisms |
| Ease of storage and transportation | |
| Safety | Do not require cultivation of dangerous infectious agents |
| No risk of reverting back to virulent forms | |
| Good safety profile in clinical trials | |
| Immune responses | Induce both cellular and humoral immune responses similar to live-attenuated vaccines |
2. Principles of DNA Vaccines
2.1. Mechanisms of Action
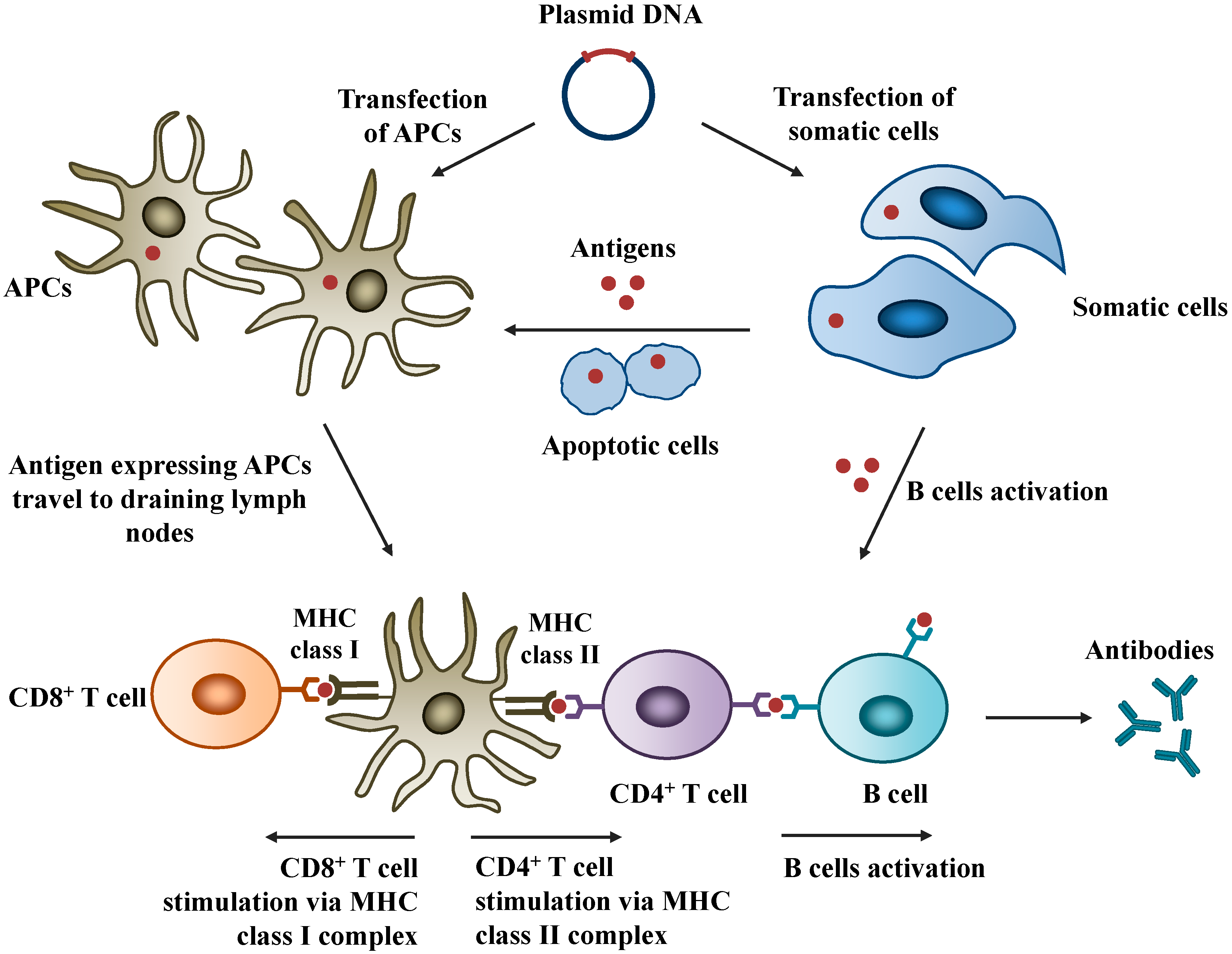
2.2. Safety of DNA Vaccines
3. Intranasal Vaccines
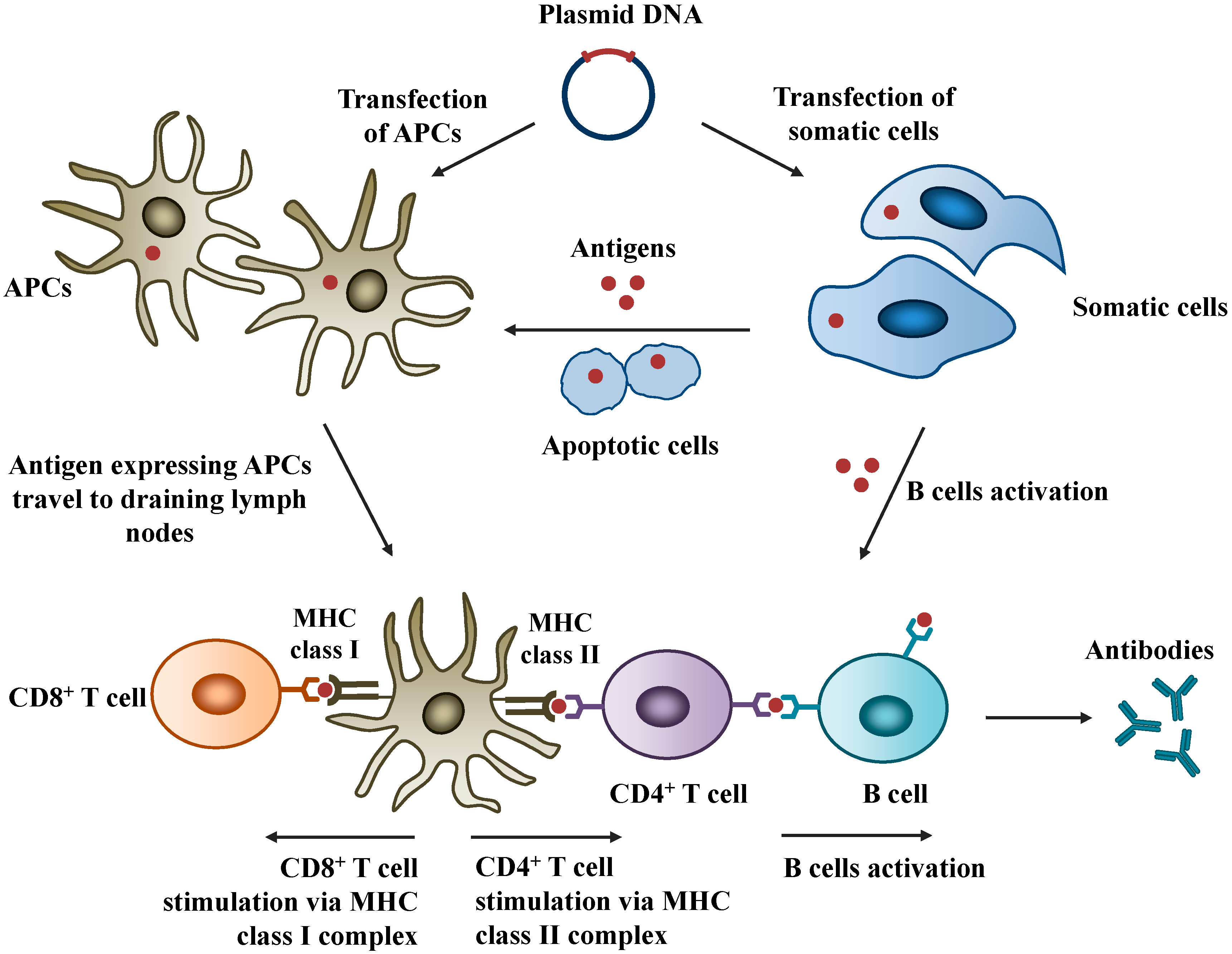
3.1. Intranasal Route of Administration
3.2. Mechanisms of Nasal Mucosal Immune Protection
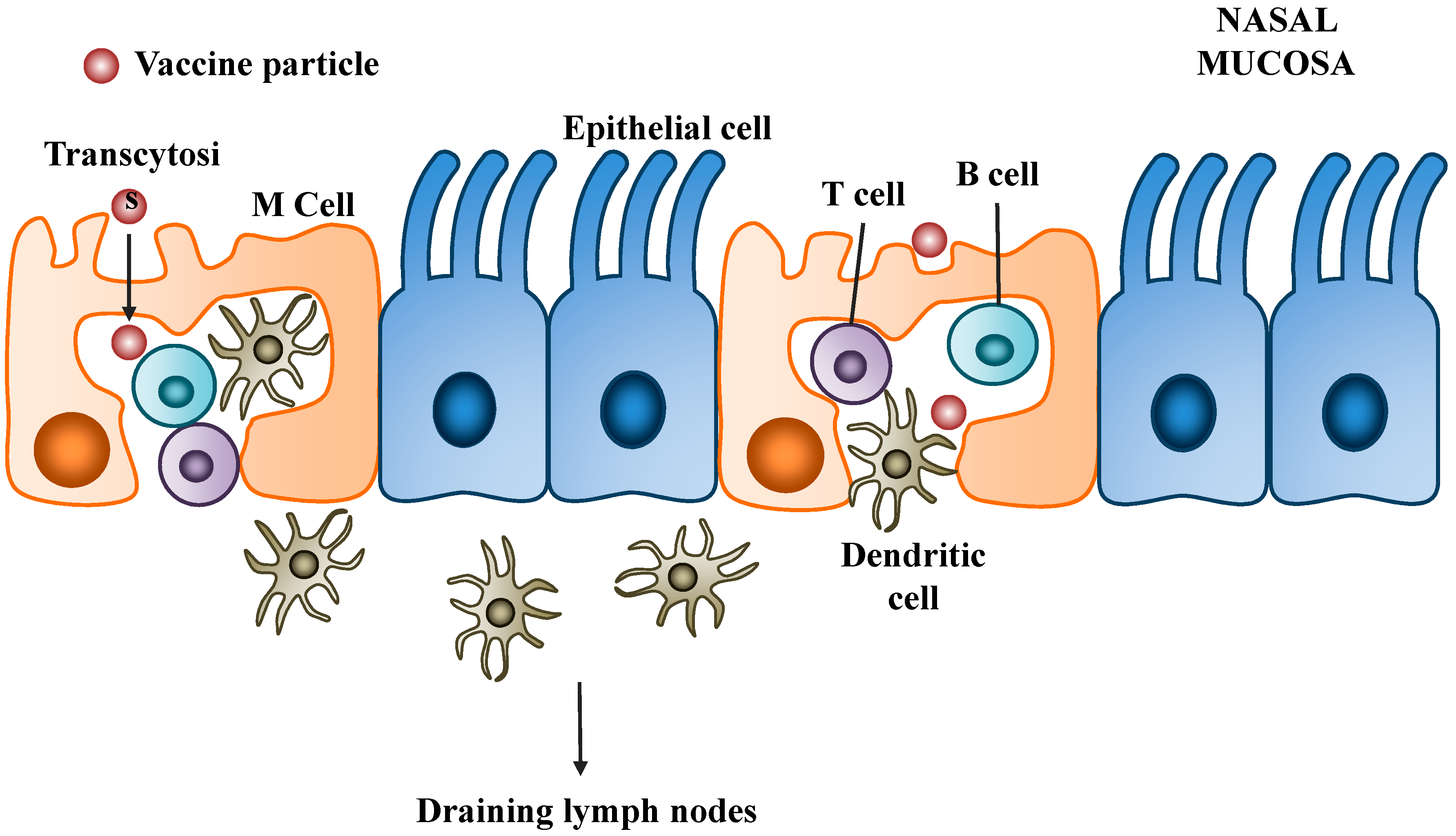
3.3. Barriers to Intranasal DNA Vaccines
3.4. Potential Risks of Intranasal Vaccine to the Central Nervous System
4. Clinical Applications of DNA Vaccines against Respiratory Infections
4.1. Tuberculosis (TB)
4.3. Influenza
4.4. Respiratory Syncytial Virus (RSV)
5. DNA Vaccine Delivery System for Intranasal Administration
5.1. Cell-Specific Targeting
5.2. Polymers
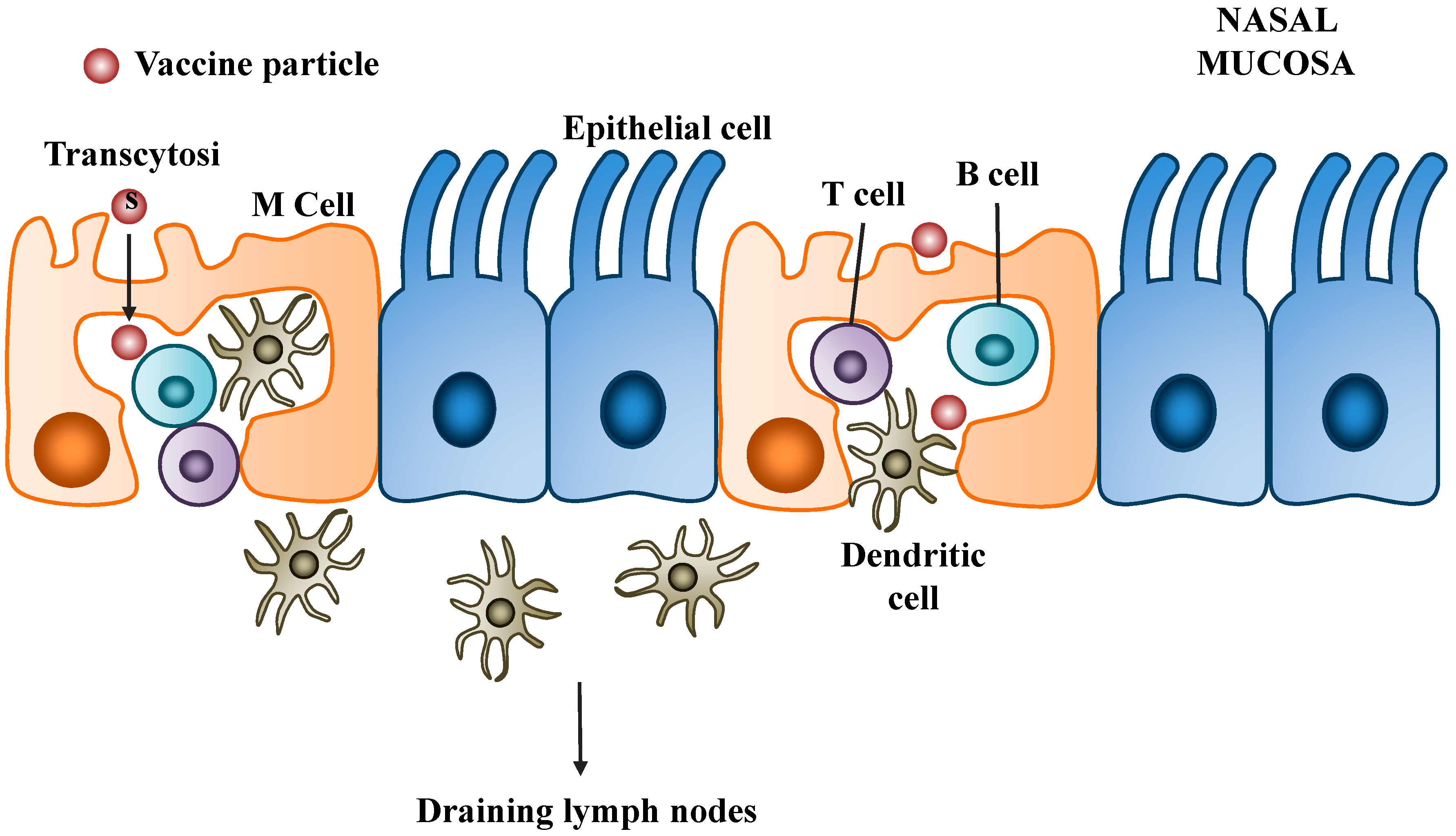
5.2.1. Polyethylenimine (PEI)
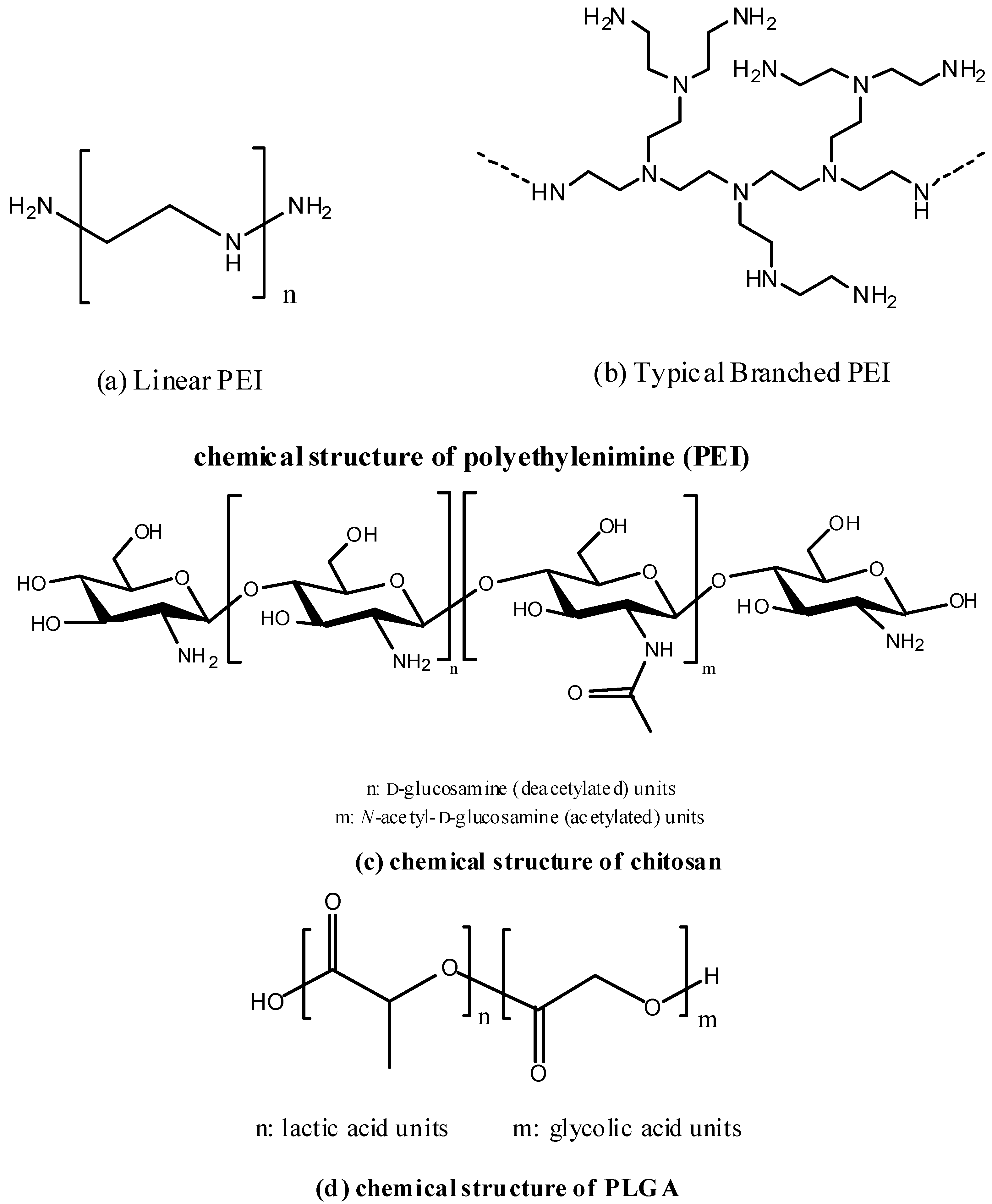
5.2.2. Chitosan
5.2.3. Poly(lactic-co-glycolic acid) (PLGA)
5.3. Liposomes
5.4. Mucosal Adjuvants
| Types | Examples | Proposed Target or Mechanisms of Action | Reference |
|---|---|---|---|
| Enterotoxins and toxin-based derivatives | Mutants of heat-labile enterotoxin and cholera toxin | Increase antigen presentation by APCs | [187,189,190] |
| LPS derivatives | MLA | TLR4 | [191] |
| Cytokines and chemokines | IL-2, IL-6, IL-7, IL-12, IL-15, GM-CSF, MCP-1, MIP-1α, RANTES | T cells stimulation Recruit and activate APCs | [192,193,194,195,196,197,198,199,200,201,202,203,204,205,206] |
| Oligonucleotides | CpG motifs | TLR9 | [21] |
| Targeting ligands | Flt3 ligand DEC-205 antibody protein sigma-1 | APC targeting DC targeting M cell targeting | [65,120,122,129,207,208] |
| Polymers | PEI | Improve DNA delivery | [146,147,148,209] |
| PLGA | Improve DNA delivery | [132,133,134,166,167,210] | |
| Chitosan | Improve DNA delivery, mucoadhesion and immunostimulating effect | [110,155,157,158,163,211] | |
| Liposomes | DOPE/DOTAP/PC; DOPE/PC/Chol | Improve DNA delivery plus immunostimulating effect | [175,176,212,213,214,215] |
6. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Gurunathan, S.; Klinman, D.M.; Seder, R.A. DNA vaccines: Immunology, application, and optimization. Annu. Rev. Immunol. 2000, 18, 927–974. [Google Scholar] [CrossRef]
- Bivas-Benita, M.; Ottenhoff, T.H.; Junginger, H.E.; Borchard, G. Pulmonary DNA vaccination: Concepts, possibilities and perspectives. J. Control. Release 2005, 107, 1–29. [Google Scholar] [CrossRef]
- Donnelly, J.J.; Wahren, B.; Liu, M.A. DNA vaccines: Progress and challenges. J. Immunol. 2005, 175, 633–639. [Google Scholar] [CrossRef]
- Villarreal, D.O.; Talbott, K.T.; Choo, D.K.; Shedlock, D.J.; Weiner, D.B. Synthetic DNA vaccine strategies against persistent viral infections. Expert Rev. Vaccines 2013, 12, 537–554. [Google Scholar] [CrossRef]
- Kutzler, M.A.; Weiner, D.B. DNA vaccines: Ready for prime time? Nat. Rev. Genet. 2008, 9, 776–788. [Google Scholar] [CrossRef]
- Redding, L.; Weiner, D.B. DNA vaccines in veterinary use. Expert Rev. Vaccines 2009, 8, 1251–1276. [Google Scholar] [CrossRef]
- Davidson, A.H.; Traub-Dargatz, J.L.; Rodeheaver, R.M.; Ostlund, E.N.; Pedersen, D.D.; Moorhead, R.G.; Stricklin, J.B.; Dewell, R.D.; Roach, S.D.; Long, R.E.; et al. Immunologic responses to West Nile virus in vaccinated and clinically affected horses. J. Am. Vet. Med. Assoc. 2005, 226, 240–245. [Google Scholar] [CrossRef]
- Garver, K.A.; LaPatra, S.E.; Kurath, G. Efficacy of an infectious hematopoietic necrosis (IHN) virus DNA vaccine in Chinook Oncorhynchus tshawytscha and sockeye O. nerka salmon. Dis. Aquat. Organ 2005, 64, 13–22. [Google Scholar] [CrossRef]
- Leitner, W.W.; Ying, H.; Restifo, N.P. DNA and RNA-based vaccines: Principles, progress and prospects. Vaccine 1999, 18, 765–777. [Google Scholar] [CrossRef]
- Manickan, E.; Karem, K.L.; Rouse, B.T. DNA vaccines—A modern gimmick or a boon to vaccinology? Crit. Rev. Immunol. 1997, 17, 135–154. [Google Scholar]
- Kalams, S.A.; Parker, S.; Jin, X.; Elizaga, M.; Metch, B.; Wang, M.; Hural, J.; Lubeck, M.; Eldridge, J.; Cardinali, M.; et al. Safety and immunogenicity of an HIV-1 gag DNA vaccine with or without IL-12 and/or IL-15 plasmid cytokine adjuvant in healthy, HIV-1 uninfected adults. PLoS One 2012, 7, e29231. [Google Scholar]
- McConkey, S.J.; Reece, W.H.; Moorthy, V.S.; Webster, D.; Dunachie, S.; Butcher, G.; Vuola, J.M.; Blanchard, T.J.; Gothard, P.; Watkins, K. Enhanced T-cell immunogenicity of plasmid DNA vaccines boosted by recombinant modified vaccinia virus Ankara in humans. Nat. Med. 2003, 9, 729–735. [Google Scholar] [CrossRef]
- Mwau, M.; Cebere, I.; Sutton, J.; Chikoti, P.; Winstone, N.; Wee, E.G.-T.; Beattie, T.; Chen, Y.-H.; Dorrell, L.; McShane, H. A human immunodeficiency virus 1 (HIV-1) clade A vaccine in clinical trials: Stimulation of HIV-specific T-cell responses by DNA and recombinant modified vaccinia virus Ankara (MVA) vaccines in humans. J. Gen. Virol. 2004, 85, 911–919. [Google Scholar] [CrossRef]
- Jaoko, W.; Nakwagala, F.N.; Anzala, O.; Manyonyi, G.O.; Birungi, J.; Nanvubya, A.; Bashir, F.; Bhatt, K.; Ogutu, H.; Wakasiaka, S.; et al. Safety and immunogenicity of recombinant low-dosage HIV-1 A vaccine candidates vectored by plasmid pTHr DNA or modified vaccinia virus Ankara (MVA) in humans in East Africa. Vaccine 2008, 26, 2788–2795. [Google Scholar] [CrossRef]
- McCormack, S.; Stohr, W.; Barber, T.; Bart, P.A.; Harari, A.; Moog, C.; Ciuffreda, D.; Cellerai, C.; Cowen, M.; Gamboni, R.; et al. EV02: A Phase I trial to compare the safety and immunogenicity of HIV DNA-C prime-NYVAC-C boost to NYVAC-C alone. Vaccine 2008, 26, 3162–3174. [Google Scholar] [CrossRef]
- Li, L.; Saade, F.; Petrovsky, N. The future of human DNA vaccines. J. Biotechnol. 2012, 162, 171–182. [Google Scholar] [CrossRef]
- Leifert, J.A.; Whitton, J.L. Immune responses to DNA vaccines: Induction of CD8 T Cells. In DNA Vaccines; Landes Bioscience: Austin, TX, USA, 2000; pp. 82–104. [Google Scholar]
- Timares, L.; Takashima, A.; Johnston, S.A. Quantitative analysis of the immunopotency of genetically transfected dendritic cells. Proc. Natl. Acad. Sci. USA 1998, 95, 13147–13152. [Google Scholar]
- Torres, C.; Iwasaki, A.; Barber, B.H.; Robinson, H.L. Differential dependence on target site tissue for gene gun and intramuscular DNA immunizations. J. Immunol. 1997, 158, 4529–4532. [Google Scholar]
- Dalpke, A.; Zimmermann, S.; Heeg, K. CpG-oligonucleotides in vaccination: Signaling and mechanisms of action. Immunobiology 2001, 204, 667–676. [Google Scholar] [CrossRef]
- Bode, C.; Zhao, G.; Steinhagen, F.; Kinjo, T.; Klinman, D.M. CpG DNA as a vaccine adjuvant. Expert Rev. Vaccines 2011, 10, 499–511. [Google Scholar]
- Babiuk, S.; Mookherjee, N.; Pontarollo, R.; Griebel, P.; van Drunen Littel-van den Hurk, S.; Hecker, R.; Babiuk, L. TLR9−/− and TLR9+/+ mice display similar immune responses to a DNA vaccine. Immunology 2004, 113, 114–120. [Google Scholar] [CrossRef]
- Tudor, D.; Dubuquoy, C.; Gaboriau, V.; Lefevre, F.; Charley, B.; Riffault, S. TLR9 pathway is involved in adjuvant effects of plasmid DNA-based vaccines. Vaccine 2005, 23, 1258–1264. [Google Scholar] [CrossRef]
- Ishii, K.J.; Kawagoe, T.; Koyama, S.; Matsui, K.; Kumar, H.; Kawai, T.; Uematsu, S.; Takeuchi, O.; Takeshita, F.; Coban, C. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 2008, 451, 725–729. [Google Scholar] [CrossRef]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef]
- Wilson, E.B.; Yamada, D.H.; Elsaesser, H.; Herskovitz, J.; Deng, J.; Cheng, G.; Aronow, B.J.; Karp, C.L.; Brooks, D.G. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013, 340, 202–207. [Google Scholar] [CrossRef]
- Swain, S.L. Class II-independent generation of CD4 memory T cells from effectors. Science 1999, 286, 1381–1383. [Google Scholar] [CrossRef]
- Ducatez, M.F.; Bahl, J.; Griffin, Y.; Stigger-Rosser, E.; Franks, J.; Barman, S.; Vijaykrishna, D.; Webb, A.; Guan, Y.; Webster, R.G. Feasibility of reconstructed ancestral H5N1 influenza viruses for cross-clade protective vaccine development. Proc. Natl. Acad. Sci. USA 2011, 108, 349–354. [Google Scholar]
- Ogunremi, O.; Pasick, J.; Kobinger, G.P.; Hannaman, D.; Berhane, Y.; Clavijo, A. A single electroporation delivery of a DNA vaccine containing the hemagglutinin gene of Asian H5N1 avian influenza virus generated a protective antibody response in chickens against a North American virus strain. Clin. Vaccine Immunol. 2013, 20, 491–500. [Google Scholar] [CrossRef]
- Belshe, R.B.; Mendelman, P.M.; Treanor, J.; King, J.; Gruber, W.C.; Piedra, P.; Bernstein, D.I.; Hayden, F.G.; Kotloff, K.; Zangwill, K. The efficacy of live attenuated, cold-adapted, trivalent, intranasal influenzavirus vaccine in children. N. Engl. J. Med. 1998, 338, 1405–1412. [Google Scholar] [CrossRef]
- Gruber, W.C. The role of live influenza vaccines in children. Vaccine 2002, 20, S66–S73. [Google Scholar] [CrossRef]
- Jones, S.; Evans, K.; McElwaine-Johnn, H.; Sharpe, M.; Oxford, J.; Lambkin-Williams, R.; Mant, T.; Nolan, A.; Zambon, M.; Ellis, J.; et al. DNA vaccination protects against an influenza challenge in a double-blind randomised placebo-controlled phase 1b clinical trial. Vaccine 2009, 27, 2506–2512. [Google Scholar] [CrossRef]
- Nichols, W.W.; Ledwith, B.J.; Manam, S.V.; Troilo, P.J. Potential DNA vaccine integration into host cell genome. Ann. N. Y. Acad. Sci. 1995, 772, 30–39. [Google Scholar] [CrossRef]
- Sheets, R.L.; Stein, J.; Manetz, T.S.; Duffy, C.; Nason, M.; Andrews, C.; Kong, W.P.; Nabel, G.J.; Gomez, P.L. Biodistribution of DNA plasmid vaccines against HIV-1, Ebola, Severe Acute Respiratory Syndrome, or West Nile virus is similar, without integration, despite differing plasmid backbones or gene inserts. Toxicol. Sci. 2006, 91, 610–619. [Google Scholar] [CrossRef]
- Ledwith, B.J.; Manam, S.; Troilo, P.J.; Barnum, A.B.; Pauley, C.J.; Griffiths, T.G., II; Harper, L.B.; Schock, H.B.; Zhang, H.; Faris, J.E.; et al. Plasmid DNA vaccines: Assay for integration into host genomic DNA. Dev. Biol. 2000, 104, 33–43. [Google Scholar]
- Wang, Z.; Troilo, P.J.; Wang, X.; Griffiths, T.G.; Pacchione, S.J.; Barnum, A.B.; Harper, L.B.; Pauley, C.J.; Niu, Z.; Denisova, L.; et al. Detection of integration of plasmid DNA into host genomic DNA following intramuscular injection and electroporation. Gene Ther. 2004, 11, 711–721. [Google Scholar] [CrossRef]
- Tavel, J.A.; Martin, J.E.; Kelly, G.G.; Enama, M.E.; Shen, J.M.; Gomez, P.L.; Andrews, C.A.; Koup, R.A.; Bailer, R.T.; Stein, J.A.; et al. Safety and immunogenicity of a Gag-Pol candidate HIV-1 DNA vaccine administered by a needle-free device in HIV-1-seronegative subjects. J. Acquir. Immune Defic. Syndr. 2007, 44, 601–605. [Google Scholar] [CrossRef]
- MacGregor, R.R.; Boyer, J.D.; Ugen, K.E.; Lacy, K.E.; Gluckman, S.J.; Bagarazzi, M.L.; Chattergoon, M.A.; Baine, Y.; Higgins, T.J.; Ciccarelli, R.B.; et al. First human trial of a DNA-based vaccine for treatment of human immunodeficiency virus type 1 infection: Safety and host response. J. Infect. Dis. 1998, 178, 92–100. [Google Scholar] [CrossRef]
- Le, T.P.; Coonan, K.M.; Hedstrom, R.C.; Charoenvit, Y.; Sedegah, M.; Epstein, J.E.; Kumar, S.; Wang, R.; Doolan, D.L.; Maguire, J.D.; et al. Safety, tolerability and humoral immune responses after intramuscular administration of a malaria DNA vaccine to healthy adult volunteers. Vaccine 2000, 18, 1893–1901. [Google Scholar] [CrossRef]
- Xiang, Z.Q.; Spitalnik, S.L.; Cheng, J.; Erikson, J.; Wojczyk, B.; Ertl, H.C. Immune responses to nucleic acid vaccines to rabies virus. Virology 1995, 209, 569–579. [Google Scholar] [CrossRef]
- Mairhofer, J.; Pfaffenzeller, I.; Merz, D.; Grabherr, R. A novel antibiotic free plasmid selection system: Advances in safe and efficient DNA therapy. Biotechnol. J. 2008, 3, 83–89. [Google Scholar] [CrossRef]
- Silverstein, A.M.; Segal, S. The ontogeny of antigen-specific T cells. J. Exp. Med. 1975, 142, 802–804. [Google Scholar] [CrossRef]
- Prince, A.M.; Whalen, R.; Brotman, B. Successful nucleic acid based immunization of newborn chimpanzees against hepatitis B virus. Vaccine 1997, 15, 916–919. [Google Scholar] [CrossRef]
- Bot, A.; Bot, S.; Bona, C. Enhanced protection against influenza virus of mice immunized as newborns with a mixture of plasmids expressing hemagglutinin and nucleoprotein. Vaccine 1998, 16, 1675–1682. [Google Scholar] [CrossRef]
- Gherardi, R.K.; Authier, F.J. Macrophagic myofasciitis: Characterization and pathophysiology. Lupus 2012, 21, 184–189. [Google Scholar] [CrossRef]
- Lanza, G.A.; Barone, L.; Scalone, G.; Pitocco, D.; Sgueglia, G.A.; Mollo, R.; Nerla, R.; Zaccardi, F.; Ghirlanda, G.; Crea, F. Inflammation-related effects of adjuvant influenza A vaccination on platelet activation and cardiac autonomic function. J. Intern. Med. 2011, 269, 118–125. [Google Scholar] [CrossRef]
- Christian, L.M.; Iams, J.D.; Porter, K.; Glaser, R. Inflammatory responses to trivalent influenza virus vaccine among pregnant women. Vaccine 2011, 29, 8982–8987. [Google Scholar] [CrossRef]
- Liuba, P.; Aburawi, E.H.; Pesonen, E.; Andersson, S.; Truedsson, L.; Yla-Herttuala, S.; Holmberg, L. Residual adverse changes in arterial endothelial function and LDL oxidation after a mild systemic inflammation induced by influenza vaccination. Ann. Med. 2007, 39, 392–399. [Google Scholar] [CrossRef]
- Ramakrishnan, A.; Althoff, K.N.; Lopez, J.A.; Coles, C.L.; Bream, J.H. Differential serum cytokine responses to inactivated and live attenuated seasonal influenza vaccines. Cytokine 2012, 60, 661–666. [Google Scholar] [CrossRef]
- Babiuk, S.; Baca-Estrada, M.E.; Foldvari, M.; Middleton, D.M.; Rabussay, D.; Widera, G.; Babiuk, L.A. Increased gene expression and inflammatory cell infiltration caused by electroporation are both important for improving the efficacy of DNA vaccines. J. Biotechnol. 2004, 110, 1–10. [Google Scholar] [CrossRef]
- Goepfert, P.A.; Elizaga, M.L.; Sato, A.; Qin, L.; Cardinali, M.; Hay, C.M.; Hural, J.; DeRosa, S.C.; DeFawe, O.D.; Tomaras, G.D.; et al. Phase 1 safety and immunogenicity testing of DNA and recombinant modified vaccinia Ankara vaccines expressing HIV-1 virus-like particles. J. Infect. Dis. 2011, 203, 610–619. [Google Scholar]
- Smith, L.R.; Wloch, M.K.; Ye, M.; Reyes, L.R.; Boutsaboualoy, S.; Dunne, C.E.; Chaplin, J.A.; Rusalov, D.; Rolland, A.P.; Fisher, C.L.; et al. Phase 1 clinical trials of the safety and immunogenicity of adjuvanted plasmid DNA vaccines encoding influenza A virus H5 hemagglutinin. Vaccine 2010, 28, 2565–2572. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Yuan, J.; Houghton, A.N.; Gallardo, H.F.; Rasalan, T.S.; Wang, J.; Zhang, Y.; Ranganathan, R.; Chapman, P.B.; Krown, S.E.; et al. Safety and immunogenicity of tyrosinase DNA vaccines in patients with melanoma. Mol. Ther. 2007, 15, 2044–2050. [Google Scholar] [CrossRef]
- Ledgerwood, J.E.; Wei, C.J.; Hu, Z.; Gordon, I.J.; Enama, M.E.; Hendel, C.S.; McTamney, P.M.; Pearce, M.B.; Yassine, H.M.; Boyington, J.C. DNA priming and influenza vaccine immunogenicity: Two phase 1 open label randomised clinical trials. Lancet Infect. Dis. 2011, 11, 916–924. [Google Scholar] [CrossRef]
- Neutra, M.R.; Pringault, E.; Kraehenbuhl, J.P. Antigen sampling across epithelial barriers and induction of mucosal immune responses. Annu. Rev. Immunol. 1996, 14, 275–300. [Google Scholar] [CrossRef]
- Neutra, M.R.; Kozlowski, P.A. Mucosal vaccines: The promise and the challenge. Nat. Rev. Immunol. 2006, 6, 148–158. [Google Scholar] [CrossRef]
- Brandtzaeg, P. Function of mucosa-associated lymphoid tissue in antibody formation. Immunol. Invest. 2010, 39, 303–355. [Google Scholar] [CrossRef]
- Lycke, N. Recent progress in mucosal vaccine development: Potential and limitations. Nat. Rev. Immunol. 2012, 12, 592–605. [Google Scholar] [CrossRef]
- Carter, N.J.; Curran, M.P. Live attenuated influenza vaccine (FluMist(R); Fluenz): A review of its use in the prevention of seasonal influenza in children and adults. Drugs 2011, 71, 1591–1622. [Google Scholar] [CrossRef]
- Dhere, R.; Yeolekar, L.; Kulkarni, P.; Menon, R.; Vaidya, V.; Ganguly, M.; Tyagi, P.; Barde, P.; Jadhav, S. A pandemic influenza vaccine in India: From strain to sale within 12 months. Vaccine 2011, 29, A16–A21. [Google Scholar] [CrossRef]
- Briles, D.E.; Ades, E.; Paton, J.C.; Sampson, J.S.; Carlone, G.M.; Huebner, R.C.; Virolainen, A.; Swiatlo, E.; Hollingshead, S.K. Intranasal immunization of mice with a mixture of the pneumococcal proteins PsaA and PspA is highly protective against nasopharyngeal carriage of Streptococcus pneumoniae. Infect. Immun. 2000, 68, 796–800. [Google Scholar] [CrossRef]
- Rigter, A.; Widjaja, I.; Versantvoort, H.; Coenjaerts, F.E.; van Roosmalen, M.; Leenhouts, K.; Rottier, P.J.; Haijema, B.J.; de Haan, C.A. A protective and safe intranasal RSV vaccine based on a recombinant prefusion-like form of the F protein bound to bacterium-like particles. PLoS One 2013, 8, e71072. [Google Scholar] [CrossRef]
- Csaba, N.; Garcia-Fuentes, M.; Alonso, M.J. Nanoparticles for nasal vaccination. Adv. Drug Deliv. Rev. 2009, 61, 140–157. [Google Scholar] [CrossRef]
- Bienenstock, J.; McDermott, M.R. Bronchus- and nasal-associated lymphoid tissues. Immunol. Rev. 2005, 206, 22–31. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, X.; Csencsits, K.L.; Haddad, A.; Walters, N.; Pascual, D.W. M cell-targeted DNA vaccination. Proc. Natl. Acad. Sci. USA 2001, 98, 9318–9323. [Google Scholar] [CrossRef]
- Woodrow, K.A.; Bennett, K.M.; Lo, D.D. Mucosal vaccine design and delivery. Annu. Rev. Biomed. Eng. 2012, 14, 17–46. [Google Scholar] [CrossRef]
- Lamm, M.E. Interaction of antigens and antibodies at mucosal surfaces. Annu. Rev. Microbiol. 1997, 51, 311–340. [Google Scholar] [CrossRef]
- Moldoveanu, Z.; Clements, M.L.; Prince, S.J.; Murphy, B.R.; Mestecky, J. Human immune responses to influenza virus vaccines administered by systemic or mucosal routes. Vaccine 1995, 13, 1006–1012. [Google Scholar] [CrossRef]
- Kaliner, M.; Shelhamer, J.H.; Borson, B.; Nadel, J.; Patow, C.; Marom, Z. Human respiratory mucus. Am. Rev. Respir. Dis. 1986, 134, 612–621. [Google Scholar]
- Couch, R.B. Nasal vaccination, Escherichia coli enterotoxin, and Bell’s palsy. N. Engl. J. Med. 2004, 350, 860–861. [Google Scholar] [CrossRef]
- Lewis, D.J.; Huo, Z.; Barnett, S.; Kromann, I.; Giemza, R.; Galiza, E.; Woodrow, M.; Thierry-Carstensen, B.; Andersen, P.; Novicki, D.; et al. Transient facial nerve paralysis (Bell’s palsy) following intranasal delivery of a genetically detoxified mutant of Escherichia coli heat labile toxin. PLoS One 2009, 4, e6999. [Google Scholar] [CrossRef]
- Eriksson, A.M.; Schon, K.M.; Lycke, N.Y. The cholera toxin-derived CTA1-DD vaccine adjuvant administered intranasally does not cause inflammation or accumulate in the nervous tissues. J. Immunol. 2004, 173, 3310–3319. [Google Scholar] [CrossRef]
- Hagiwara, Y.; Kawamura, Y.I.; Kataoka, K.; Rahima, B.; Jackson, R.J.; Komase, K.; Dohi, T.; Boyaka, P.N.; Takeda, Y.; Kiyono, H.; et al. A second generation of double mutant cholera toxin adjuvants: Enhanced immunity without intracellular trafficking. J. Immunol. 2006, 177, 3045–3054. [Google Scholar] [CrossRef]
- Gupta, U.D.; Katoch, V.M.; McMurray, D.N. Current status of TB vaccines. Vaccine 2007, 25, 3742–3751. [Google Scholar] [CrossRef]
- Stenger, S.; Modlin, R.L. T cell mediated immunity to Mycobacterium tuberculosis. Curr. Opin. Microbiol. 1999, 2, 89–93. [Google Scholar] [CrossRef]
- McShane, H.; Behboudi, S.; Goonetilleke, N.; Brookes, R.; Hill, A.V. Protective immunity against Mycobacterium tuberculosis induced by dendritic cells pulsed with both CD8+- and CD4+-T-cell epitopes from antigen 85A. Infect. Immun. 2002, 70, 1623–1626. [Google Scholar] [CrossRef]
- Okada, M.; Kita, Y. Tuberculosis vaccine development: The development of a novel (preclinical) DNA vaccine. Hum. Vaccines 2010, 6, 297–308. [Google Scholar] [CrossRef]
- Huygen, K.; Content, J.; Denis, O.; Montgomery, D.L.; Yawman, A.M.; Deck, R.R.; DeWitt, C.M.; Orme, I.M.; Baldwin, S.; D’Souza, C.; et al. Immunogenicity and protective efficacy of a tuberculosis DNA vaccine. Nat. Med. 1996, 2, 893–898. [Google Scholar] [CrossRef]
- Tascon, R.E.; Colston, M.J.; Ragno, S.; Stavropoulos, E.; Gregory, D.; Lowrie, D.B. Vaccination against tuberculosis by DNA injection. Nat. Med. 1996, 2, 888–892. [Google Scholar] [CrossRef]
- Lozes, E.; Huygen, K.; Content, J.; Denis, O.; Montgomery, D.L.; Yawman, A.M.; Vandenbussche, P.; van Vooren, J.P.; Drowart, A.; Ulmer, J.B.; et al. Immunogenicity and efficacy of a tuberculosis DNA vaccine encoding the components of the secreted antigen 85 complex. Vaccine 1997, 15, 830–833. [Google Scholar] [CrossRef]
- Tian, X.; Cai, H.; Zhu, Y.X. Protection of mice with a divalent tuberculosis DNA vaccine encoding antigens Ag85B and MPT64. Acta Biochim. Biophys. Sin. 2004, 36, 269–276. [Google Scholar] [CrossRef]
- Derrick, S.C.; Yang, A.L.; Morris, S.L. A polyvalent DNA vaccine expressing an ESAT6–Ag85B fusion protein protects mice against a primary infection with Mycobacterium tuberculosis and boosts BCG-induced protective immunity. Vaccine 2004, 23, 780–788. [Google Scholar] [CrossRef]
- Zhu, D.; Jiang, S.; Luo, X. Therapeutic effects of Ag85B and MPT64 DNA vaccines in a murine model of Mycobacterium tuberculosis infection. Vaccine 2005, 23, 4619–4624. [Google Scholar] [CrossRef]
- Wozniak, T.M.; Ryan, A.A.; Triccas, J.A.; Britton, W.J. Plasmid interleukin-23 (IL-23), but not plasmid IL-27, enhances the protective efficacy of a DNA vaccine against Mycobacterium tuberculosis infection. Infect. Immun. 2006, 74, 557–565. [Google Scholar] [CrossRef]
- Yuan, W.; Dong, N.; Zhang, L.; Liu, J.; Lin, S.; Xiang, Z.; Qiao, H.; Tong, W.; Qin, C. Immunogenicity and protective efficacy of a tuberculosis DNA vaccine expressing a fusion protein of Ag85B–Esat6–HspX in mice. Vaccine 2012, 30, 2490–2497. [Google Scholar] [CrossRef]
- Graham, R.L.; Donaldson, E.F.; Baric, R.S. A decade after SARS: Strategies for controlling emerging coronaviruses. Nat. Rev. Microbiol. 2013, 11, 836–848. [Google Scholar]
- Bolles, M.; Deming, D.; Long, K.; Agnihothram, S.; Whitmore, A.; Ferris, M.; Funkhouser, W.; Gralinski, L.; Totura, A.; Heise, M.; et al. A double-inactivated severe acute respiratory syndrome coronavirus vaccine provides incomplete protection in mice and induces increased eosinophilic proinflammatory pulmonary response upon challenge. J. Virol. 2011, 85, 12201–12215. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Tsoi, H.W.; Chen, Z.W.; Wong, B.H.; Zhang, L.; Chan, J.K.; Wong, L.P.; He, W.; Ma, C.; et al. SARS coronavirus spike polypeptide DNA vaccine priming with recombinant spike polypeptide from Escherichia coli as booster induces high titer of neutralizing antibody against SARS coronavirus. Vaccine 2005, 23, 4959–4968. [Google Scholar] [CrossRef]
- Yang, Z.Y.; Kong, W.P.; Huang, Y.; Roberts, A.; Murphy, B.R.; Subbarao, K.; Nabel, G.J. A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature 2004, 428, 561–564. [Google Scholar] [CrossRef]
- Gupta, V.; Tabiin, T.M.; Sun, K.; Chandrasekaran, A.; Anwar, A.; Yang, K.; Chikhlikar, P.; Salmon, J.; Brusic, V.; Marques, E.T.; et al. SARS coronavirus nucleocapsid immunodominant T-cell epitope cluster is common to both exogenous recombinant and endogenous DNA-encoded immunogens. Virology 2006, 347, 127–139. [Google Scholar] [CrossRef]
- Zhao, P.; Cao, J.; Zhao, L.J.; Qin, Z.L.; Ke, J.S.; Pan, W.; Ren, H.; Yu, J.G.; Qi, Z.T. Immune responses against SARS-coronavirus nucleocapsid protein induced by DNA vaccine. Virology 2005, 331, 128–135. [Google Scholar] [CrossRef]
- Garten, R.J.; Davis, C.T.; Russell, C.A.; Shu, B.; Lindstrom, S.; Balish, A.; Sessions, W.M.; Xu, X.; Skepner, E.; Deyde, V.; et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 2009, 325, 197–201. [Google Scholar] [CrossRef]
- Abdel-Ghafar, A.N.; Chotpitayasunondh, T.; Gao, Z.; Hayden, F.G.; Nguyen, D.H.; de Jong, M.D.; Naghdaliyev, A.; Peiris, J.S.; Shindo, N.; Soeroso, S.; et al. Update on avian influenza A (H5N1) virus infection in humans. N. Engl. J. Med. 2008, 358, 261–273. [Google Scholar] [CrossRef]
- Epstein, S.L.; Tumpey, T.M.; Misplon, J.A.; Lo, C.Y.; Cooper, L.A.; Subbarao, K.; Renshaw, M.; Sambhara, S.; Katz, J.M. DNA vaccine expressing conserved influenza virus proteins protective against H5N1 challenge infection in mice. Emerg. Infect. Dis. 2002, 8, 796–801. [Google Scholar] [CrossRef]
- Epstein, S.L.; Kong, W.P.; Misplon, J.A.; Lo, C.Y.; Tumpey, T.M.; Xu, L.; Nabel, G.J. Protection against multiple influenza A subtypes by vaccination with highly conserved nucleoprotein. Vaccine 2005, 23, 5404–5410. [Google Scholar] [CrossRef]
- Ferraro, B.; Morrow, M.P.; Hutnick, N.A.; Shin, T.H.; Lucke, C.E.; Weiner, D.B. Clinical applications of DNA vaccines: Current progress. Clin. Infect. Dis. 2011, 53, 296–302. [Google Scholar] [CrossRef]
- Chen, Z.; Matsuo, K.; Asanuma, H.; Takahashi, H.; Iwasaki, T.; Suzuki, Y.; Aizawa, C.; Kurata, T.; Tamura, S. Enhanced protection against a lethal influenza virus challenge by immunization with both hemagglutinin- and neuraminidase-expressing DNAs. Vaccine 1999, 17, 653–659. [Google Scholar] [CrossRef]
- Donnelly, J.J.; Friedman, A.; Martinez, D.; Montgomery, D.L.; Shiver, J.W.; Motzel, S.L.; Ulmer, J.B.; Liu, M.A. Preclinical efficacy of a prototype DNA vaccine: Enhanced protection against antigenic drift in influenza virus. Nat. Med. 1995, 1, 583–587. [Google Scholar] [CrossRef]
- Donnelly, J.J.; Friedman, A.; Ulmer, J.B.; Liu, M.A. Further protection against antigenic drift of influenza virus in a ferret model by DNA vaccination. Vaccine 1997, 15, 865–868. [Google Scholar] [CrossRef]
- Chen, Q.; Kuang, H.; Wang, H.; Fang, F.; Yang, Z.; Zhang, Z.; Zhang, X.; Chen, Z. Comparing the ability of a series of viral protein-expressing plasmid DNAs to protect against H5N1 influenza virus. Virus Genes 2009, 38, 30–38. [Google Scholar] [CrossRef]
- Drape, R.J.; Macklin, M.D.; Barr, L.J.; Jones, S.; Haynes, J.R.; Dean, H.J. Epidermal DNA vaccine for influenza is immunogenic in humans. Vaccine 2006, 24, 4475–4481. [Google Scholar] [CrossRef]
- Collins, P.L.; Hill, M.G.; Johnson, P.R. The two open reading frames of the 22k mRNA of human respiratory syncytial virus: Sequence comparison of antigenic subgroups A and B and expression in vitro. J. Gen. Virol. 1990, 71, 3015–3020. [Google Scholar] [CrossRef]
- Falsey, A.R.; Hennessey, P.A.; Formica, M.A.; Cox, C.; Walsh, E.E. Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 2005, 352, 1749–1759. [Google Scholar] [CrossRef]
- Martinez, X.; Li, X.; Kovarik, J.; Klein, M.; Lambert, P.H.; Siegrist, C.A. Combining DNA and protein vaccines for early life immunization against respiratory syncytial virus in mice. Eur. J. Immunol. 1999, 29, 3390–3400. [Google Scholar] [CrossRef]
- Johnson, P.R.; Collins, P.L. The fusion glycoproteins of human respiratory syncytial virus of subgroups A and B: Sequence conservation provides a structural basis for antigenic relatedness. J. Gen. Virol. 1988, 69, 2623–2628. [Google Scholar] [CrossRef]
- Li, X.; Sambhara, S.; Li, C.X.; Ewasyshyn, M.; Parrington, M.; Caterini, J.; James, O.; Cates, G.; Du, R.P.; Klein, M. Protection against respiratory syncytial virus infection by DNA immunization. J. Exp. Med. 1998, 188, 681–688. [Google Scholar] [CrossRef]
- Ternette, N.; Tippler, B.; Uberla, K.; Grunwald, T. Immunogenicity and efficacy of codon optimized DNA vaccines encoding the F-protein of respiratory syncytial virus. Vaccine 2007, 25, 7271–7279. [Google Scholar] [CrossRef]
- Li, X.; Sambhara, S.; Li, C.X.; Ettorre, L.; Switzer, I.; Cates, G.; James, O.; Parrington, M.; Oomen, R.; Du, R.P.; et al. Plasmid DNA encoding the respiratory syncytial virus G protein is a promising vaccine candidate. Virology 2000, 269, 54–65. [Google Scholar] [CrossRef]
- Connors, M.; Kulkarni, A.B.; Collins, P.L.; Firestone, C.Y.; Holmes, K.L.; Morse, H.C., III; Murphy, B.R. Resistance to respiratory syncytial virus (RSV) challenge induced by infection with a vaccinia virus recombinant expressing the RSV M2 protein (Vac-M2) is mediated by CD8+ T cells, while that induced by Vac-F or Vac-G recombinants is mediated by antibodies. J. Virol. 1992, 66, 1277–1281. [Google Scholar]
- Boyoglu, S.; Vig, K.; Pillai, S.; Rangari, V.; Dennis, V.A.; Khazi, F.; Singh, S.R. Enhanced delivery and expression of a nanoencapsulated DNA vaccine vector for respiratory syncytial virus. Nanomedicine 2009, 5, 463–472. [Google Scholar] [CrossRef]
- Barnum, S.; Subbarayan, P.; Vig, K.; Pillai, S.; Dennis, V. Nano-encapsulated DNA and/or protein boost immunizations increase efficiency of DNA vaccine protection against RSV. J. Nanomed. Nanotechnol. 2012, 3, 1000132. [Google Scholar]
- Dupuis, M.; Denis-Mize, K.; Woo, C.; Goldbeck, C.; Selby, M.J.; Chen, M.; Otten, G.R.; Ulmer, J.B.; Donnelly, J.J.; Ott, G. Distribution of DNA vaccines determines their immunogenicity after intramuscular injection in mice. J. Immunol. 2000, 165, 2850–2858. [Google Scholar] [CrossRef]
- Fynan, E.F.; Webster, R.G.; Fuller, D.H.; Haynes, J.R.; Santoro, J.C.; Robinson, H.L. DNA vaccines: Protective immunizations by parenteral, mucosal, and gene-gun inoculations. Proc. Natl. Acad. Sci. USA 1993, 90, 11478–11482. [Google Scholar] [CrossRef]
- Pertmer, T.M.; Eisenbraun, M.D.; McCabe, D.; Prayaga, S.K.; Fuller, D.H.; Haynes, J.R. Gene gun-based nucleic acid immunization: Elicitation of humoral and cytotoxic T lymphocyte responses following epidermal delivery of nanogram quantities of DNA. Vaccine 1995, 13, 1427–1430. [Google Scholar] [CrossRef]
- Yoshida, A.; Nagata, T.; Uchijima, M.; Higashi, T.; Koide, Y. Advantage of gene gun-mediated over intramuscular inoculation of plasmid DNA vaccine in reproducible induction of specific immune responses. Vaccine 2000, 18, 1725–1729. [Google Scholar]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef]
- Mellman, I.; Steinman, R.M. Dendritic cells: Specialized and regulated antigen processing machines. Cell 2001, 106, 255–258. [Google Scholar] [CrossRef]
- Steinman, R.M. Dendritic cells in vivo: A key target for a new vaccine science. Immunity 2008, 29, 319–324. [Google Scholar] [CrossRef]
- Foged, C.; Sundblad, A.; Hovgaard, L. Targeting vaccines to dendritic cells. Pharm. Res. 2002, 19, 229–238. [Google Scholar] [CrossRef]
- Wang, W.W.; Das, D.; Suresh, M.R. A versatile bifunctional dendritic cell targeting vaccine vector. Mol. Pharm. 2009, 6, 158–172. [Google Scholar] [CrossRef]
- Nchinda, G.; Amadu, D.; Trumpfheller, C.; Mizenina, O.; Uberla, K.; Steinman, R.M. Dendritic cell targeted HIV gag protein vaccine provides help to a DNA vaccine including mobilization of protective CD8+ T cells. Proc. Natl. Acad. Sci. USA 2010, 107, 4281–4286. [Google Scholar] [CrossRef]
- Raghuwanshi, D.; Mishra, V.; Das, D.; Kaur, K.; Suresh, M.R. Dendritic cell targeted chitosan nanoparticles for nasal DNA immunization against SARS CoV nucleocapsid protein. Mol. Pharm. 2012, 9, 946–956. [Google Scholar] [CrossRef]
- Caminschi, I.; Proietto, A.I.; Ahmet, F.; Kitsoulis, S.; Shin Teh, J.; Lo, J.C.; Rizzitelli, A.; Wu, L.; Vremec, D.; van Dommelen, S.L.; et al. The dendritic cell subtype-restricted C-type lectin Clec9A is a target for vaccine enhancement. Blood 2008, 112, 3264–3273. [Google Scholar] [CrossRef]
- Shortman, K.; Lahoud, M.H.; Caminschi, I. Improving vaccines by targeting antigens to dendritic cells. Exp. Mol. Med. 2009, 41, 61–66. [Google Scholar] [CrossRef]
- Brasel, K.; McKenna, H.J.; Morrissey, P.J.; Charrier, K.; Morris, A.E.; Lee, C.C.; Williams, D.E.; Lyman, S.D. Hematologic effects of flt3 ligand in vivo in mice. Blood 1996, 88, 2004–2012. [Google Scholar]
- Maraskovsky, E.; Brasel, K.; Teepe, M.; Roux, E.R.; Lyman, S.D.; Shortman, K.; McKenna, H.J. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand-treated mice: multiple dendritic cell subpopulations identified. J. Exp. Med. 1996, 184, 1953–1962. [Google Scholar] [CrossRef]
- Williamson, E.; Westrich, G.M.; Viney, J.L. Modulating dendritic cells to optimize mucosal immunization protocols. J. Immunol. 1999, 163, 3668–3675. [Google Scholar]
- Hung, C.F.; Hsu, K.F.; Cheng, W.F.; Chai, C.Y.; He, L.; Ling, M.; Wu, T.C. Enhancement of DNA vaccine potency by linkage of antigen gene to a gene encoding the extracellular domain of Fms-like tyrosine kinase 3-ligand. Cancer Res. 2001, 61, 1080–1088. [Google Scholar]
- Kataoka, K.; McGhee, J.R.; Kobayashi, R.; Fujihashi, K.; Shizukuishi, S.; Fujihashi, K. Nasal Flt3 ligand cDNA elicits CD11c+CD8+ dendritic cells for enhanced mucosal immunity. J. Immunol. 2004, 172, 3612–3619. [Google Scholar] [CrossRef]
- Kataoka, K.; Fujihashi, K.; Oma, K.; Fukuyama, Y.; Hollingshead, S.K.; Sekine, S.; Kawabata, S.; Ito, H.O.; Briles, D.E.; Oishi, K. The nasal dendritic cell-targeting Flt3 ligand as a safe adjuvant elicits effective protection against fatal pneumococcal pneumonia. Infect. Immun. 2011, 79, 2819–2828. [Google Scholar] [CrossRef]
- Ermak, T.H.; Dougherty, E.P.; Bhagat, H.R.; Kabok, Z.; Pappo, J. Uptake and transport of copolymer biodegradable microspheres by rabbit Peyer’s patch M cells. Cell Tissue Res. 1995, 279, 433–436. [Google Scholar] [CrossRef]
- Shakweh, M.; Besnard, M.; Nicolas, V.; Fattal, E. Poly (lactide-co-glycolide) particles of different physicochemical properties and their uptake by Peyer’s patches in mice. Eur. J. Pharm. Biopharm. 2005, 61, 1–13. [Google Scholar] [CrossRef]
- Gupta, P.N.; Khatri, K.; Goyal, A.K.; Mishra, N.; Vyas, S.P. M-cell targeted biodegradable PLGA nanoparticles for oral immunization against hepatitis B. J. Drug Target. 2007, 15, 701–713. [Google Scholar] [CrossRef]
- Rajapaksa, T.E.; Stover-Hamer, M.; Fernandez, X.; Eckelhoefer, H.A.; Lo, D.D. Claudin 4-targeted protein incorporated into PLGA nanoparticles can mediate M cell targeted delivery. J. Control. Release 2010, 142, 196–205. [Google Scholar]
- Sharma, S.; Mukkur, T.K.; Benson, H.A.; Chen, Y. Pharmaceutical aspects of intranasal delivery of vaccines using particulate systems. J. Pharm. Sci. 2009, 98, 812–843. [Google Scholar] [CrossRef]
- Reddy, S.T.; van der Vlies, A.J.; Simeoni, E.; Angeli, V.; Randolph, G.J.; O’Neil, C.P.; Lee, L.K.; Swartz, M.A.; Hubbell, J.A. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol. 2007, 25, 1159–1164. [Google Scholar] [CrossRef]
- Manocha, M.; Pal, P.C.; Chitralekha, K.T.; Thomas, B.E.; Tripathi, V.; Gupta, S.D.; Paranjape, R.; Kulkarni, S.; Rao, D.N. Enhanced mucosal and systemic immune response with intranasal immunization of mice with HIV peptides entrapped in PLG microparticles in combination with Ulex Europaeus-I lectin as M cell target. Vaccine 2005, 23, 5599–5617. [Google Scholar] [CrossRef]
- Wolf, J.L.; Rubin, D.H.; Finberg, R.; Kauffman, R.S.; Sharpe, A.H.; Trier, J.S.; Fields, B.N. Intestinal M cells: A pathway for entry of reovirus into the host. Science 1981, 212, 471–472. [Google Scholar]
- Jones, B.D.; Ghori, N.; Falkow, S. Salmonella typhimurium initiates murine infection by penetrating and destroying the specialized epithelial M cells of the Peyer’s patches. J. Exp. Med. 1994, 180, 15–23. [Google Scholar]
- Teitelbaum, R.; Schubert, W.; Gunther, L.; Kress, Y.; Macaluso, F.; Pollard, J.W.; McMurray, D.N.; Bloom, B.R. The M cell as a portal of entry to the lung for the bacterial pathogen Mycobacterium tuberculosis. Immunity 1999, 10, 641–650. [Google Scholar] [CrossRef]
- Fujkuyama, Y.; Tokuhara, D.; Kataoka, K.; Gilbert, R.S.; McGhee, J.R.; Yuki, Y.; Kiyono, H.; Fujihashi, K. Novel vaccine development strategies for inducing mucosal immunity. Expert Rev. Vaccines 2012, 11, 367–379. [Google Scholar]
- Kim, S.H.; Seo, K.W.; Kim, J.; Lee, K.Y.; Jang, Y.S. The M cell-targeting ligand promotes antigen delivery and induces antigen-specific immune responses in mucosal vaccination. J. Immunol. 2010, 185, 5787–5795. [Google Scholar] [CrossRef]
- Lo, D.D.; Ling, J.; Eckelhoefer, A.H. M cell targeting by a Claudin 4-targeting peptide can enhance mucosal IgA responses. BMC Biotechnol. 2012, 12, 7. [Google Scholar] [CrossRef]
- Nochi, T.; Yuki, Y.; Matsumura, A.; Mejima, M.; Terahara, K.; Kim, D.Y.; Fukuyama, S.; Iwatsuki-Horimoto, K.; Kawaoka, Y.; Kohda, T.; et al. A novel M cell-specific carbohydrate-targeted mucosal vaccine effectively induces antigen-specific immune responses. J. Exp. Med. 2007, 204, 2789–2796. [Google Scholar] [CrossRef]
- Rynda, A.; Maddaloni, M.; Mierzejewska, D.; Ochoa-Reparaz, J.; Maslanka, T.; Crist, K.; Riccardi, C.; Barszczewska, B.; Fujihashi, K.; McGhee, J.R.; et al. Low-dose tolerance is mediated by the microfold cell ligand, reovirus protein sigma1. J. Immunol. 2008, 180, 5187–5200. [Google Scholar] [CrossRef]
- Boussif, O.; Lezoualc’h, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar]
- Shim, B.S.; Park, S.M.; Quan, J.S.; Jere, D.; Chu, H.; Song, M.K.; Kim, D.W.; Jang, Y.S.; Yang, M.S.; Han, S.H.; et al. Intranasal immunization with plasmid DNA encoding spike protein of SARS-coronavirus/polyethylenimine nanoparticles elicits antigen-specific humoral and cellular immune responses. BMC Immunol. 2010, 11, 65. [Google Scholar] [CrossRef]
- Torrieri-Dramard, L.; Lambrecht, B.; Ferreira, H.L.; van den Berg, T.; Klatzmann, D.; Bellier, B. Intranasal DNA vaccination induces potent mucosal and systemic immune responses and cross-protective immunity against influenza viruses. Mol. Ther. 2011, 19, 602–611. [Google Scholar] [CrossRef]
- Mann, J.F.; McKay, P.F.; Arokiasamy, S.; Patel, R.K.; Klein, K.; Shattock, R.J. Pulmonary delivery of DNA vaccine constructs using deacylated PEI elicits immune responses and protects against viral challenge infection. J. Control. Release 2013, 170, 452–459. [Google Scholar] [CrossRef]
- Kang, M.L.; Cho, C.S.; Yoo, H.S. Application of chitosan microspheres for nasal delivery of vaccines. Biotechnol. Adv. 2009, 27, 857–865. [Google Scholar] [CrossRef]
- Mao, S.; Sun, W.; Kissel, T. Chitosan-based formulations for delivery of DNA and siRNA. Adv. Drug Deliv. Rev. 2010, 62, 12–27. [Google Scholar] [CrossRef]
- Illum, L.; Davis, S.S. Nasal vaccination: A non-invasive vaccine delivery method that holds great promise for the future. Adv. Drug Deliv. Rev. 2001, 51, 81. [Google Scholar] [CrossRef]
- Issa, M.M.; Köping-Höggård, M.; Artursson, P. Chitosan and the mucosal delivery of biotechnology drugs. Drug Discov. Today 2005, 2, 1–6. [Google Scholar]
- Casettari, L.; Vllasaliu, D.; Lam, J.K.; Soliman, M.; Illum, L. Biomedical applications of amino acid-modified chitosans: A review. Biomaterials 2012, 33, 7565–7583. [Google Scholar] [CrossRef]
- Seferian, P.G.; Martinez, M.L. Immune stimulating activity of two new chitosan containing adjuvant formulations. Vaccine 2000, 19, 661–668. [Google Scholar] [CrossRef]
- Kumar, M.; Behera, A.K.; Lockey, R.F.; Zhang, J.; Bhullar, G.; de La Cruz, C.P.; Chen, L.C.; Leong, K.W.; Huang, S.K.; Mohapatra, S.S. Intranasal gene transfer by chitosan-DNA nanospheres protects BALB/c mice against acute respiratory syncytial virus infection. Hum. Gene Ther. 2002, 13, 1415–1425. [Google Scholar] [CrossRef]
- Iqbal, M.; Lin, W.; Jabbal-Gill, I.; Davis, S.; Steward, M.; Illum, L. Nasal delivery of chitosan–DNA plasmid expressing epitopes of respiratory syncytial virus (RSV) induces protective CTL responses in BALB/c mice. Vaccine 2003, 21, 1478–1485. [Google Scholar] [CrossRef]
- Bernkop-Schnurch, A.; Hornof, M.; Guggi, D. Thiolated chitosans. Eur. J. Pharm. Biopharm. 2004, 57, 9–17. [Google Scholar] [CrossRef]
- Lee, D.; Zhang, W.; Shirley, S.A.; Kong, X.; Hellermann, G.R.; Lockey, R.F.; Mohapatra, S.S. Thiolated chitosan/DNA nanocomplexes exhibit enhanced and sustained gene delivery. Pharm. Res. 2007, 24, 157–167. [Google Scholar]
- Panyam, J.; Labhasetwar, V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv. Drug Deliv. Rev. 2003, 55, 329–347. [Google Scholar] [CrossRef]
- Bivas-Benita, M.; Romeijn, S.; Junginger, H.E.; Borchard, G. PLGA–PEI nanoparticles for gene delivery to pulmonary epithelium. Eur. J. Pharm. Biopharm. 2004, 58, 1–6. [Google Scholar] [CrossRef]
- Nafee, N.; Taetz, S.; Schneider, M.; Schaefer, U.F.; Lehr, C.M. Chitosan-coated PLGA nanoparticles for DNA/RNA delivery: Effect of the formulation parameters on complexation and transfection of antisense oligonucleotides. Nanomedicine 2007, 3, 173–183. [Google Scholar] [CrossRef]
- Bordelon, H.; Biris, A.S.; Sabliov, C.M.; Todd Monroe, W. Characterization of plasmid DNA location within Chitosan/PLGA/pDNA nanoparticle complexes designed for gene delivery. J. Nanomater. 2011, 2011, 1–9. [Google Scholar]
- Tahara, K.; Sakai, T.; Yamamoto, H.; Takeuchi, H.; Kawashima, Y. Establishing chitosan coated PLGA nanosphere platform loaded with wide variety of nucleic acid by complexation with cationic compound for gene delivery. Int. J. Pharm. 2008, 354, 210–216. [Google Scholar] [CrossRef]
- Oster, C.G.; Kim, N.; Grode, L.; Barbu-Tudoran, L.; Schaper, A.K.; Kaufmann, S.H.; Kissel, T. Cationic microparticles consisting of poly(lactide-co-glycolide) and polyethylenimine as carriers systems for parental DNA vaccination. J. Control. Release 2005, 104, 359–377. [Google Scholar] [CrossRef]
- Wang, G.; Pan, L.; Zhang, Y.; Wang, Y.; Zhang, Z.; Lü, J.; Zhou, P.; Fang, Y.; Jiang, S. Intranasal delivery of cationic PLGA nano/microparticles-loaded FMDV DNA vaccine encoding IL-6 elicited protective immunity against FMDV challenge. PLoS One 2011, 6, e27605. [Google Scholar]
- Ravi Kumar, M.N.; Bakowsky, U.; Lehr, C.M. Preparation and characterization of cationic PLGA nanospheres as DNA carriers. Biomaterials 2004, 25, 1771–1777. [Google Scholar] [CrossRef]
- Bu, J.; Song, Y.; Rompato, G.; Burgess, D.J.; Garmendia, A.E. Co-delivery of IL-2 or liposomes augment the responses of mice to a DNA vaccine for pseudorabies virus IE180. Comp. Immunol. Microbiol. Infect. Dis. 2003, 26, 175–187. [Google Scholar] [CrossRef]
- Jiao, X.; Wang, R.Y.; Feng, Z.; Alter, H.J.; Shih, J.W. Modulation of cellular immune response against hepatitis C virus nonstructural protein 3 by cationic liposome encapsulated DNA immunization. Hepatology 2003, 37, 452–460. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Imazu, S.; Gao, J.Q.; Hayashi, K.; Tsuda, Y.; Shimokawa, M.; Sugita, T.; Niwa, T.; Oda, A.; Akashi, M.; et al. Augmentation of antigen-specific immune responses using DNA-fusogenic liposome vaccine. Biochem. Biophys. Res. Commun. 2004, 325, 500–505. [Google Scholar] [CrossRef]
- Brito, L.A.; Malyala, P.; O’Hagan, D.T. Vaccine adjuvant formulations: A pharmaceutical perspective. Semin. Immunol. 2013, 25, 130–145. [Google Scholar] [CrossRef]
- Legendre, J.Y.; Szoka, F.C., Jr. Delivery of plasmid DNA into mammalian cell lines using pH-sensitive liposomes: Comparison with cationic liposomes. Pharm. Res. 1992, 9, 1235–1242. [Google Scholar] [CrossRef]
- Khatri, K.; Goyal, A.K.; Gupta, P.N.; Mishra, N.; Mehta, A.; Vyas, S.P. Surface modified liposomes for nasal delivery of DNA vaccine. Vaccine 2008, 26, 2225–2233. [Google Scholar] [CrossRef]
- Lay, M.; Callejo, B.; Chang, S.; Hong, D.K.; Lewis, D.B.; Carroll, T.D.; Matzinger, S.; Fritts, L.; Miller, C.J.; Warner, J.F.; et al. Cationic lipid/DNA complexes (JVRS-100) combined with influenza vaccine (Fluzone) increases antibody response, cellular immunity, and antigenically drifted protection. Vaccine 2009, 27, 3811–3820. [Google Scholar] [CrossRef]
- D’Souza, S.; Rosseels, V.; Denis, O.; Tanghe, A.; de Smet, N.; Jurion, F.; Palfliet, K.; Castiglioni, N.; Vanonckelen, A.; Wheeler, C.; et al. Improved tuberculosis DNA vaccines by formulation in cationic lipids. Infect. Immun. 2002, 70, 3681–3688. [Google Scholar] [CrossRef]
- Rosada, R.S.; de la Torre, L.G.; Frantz, F.G.; Trombone, A.P.; Zarate-Blades, C.R.; Fonseca, D.M.; Souza, P.R.; Brandao, I.T.; Masson, A.P.; Soares, E.G.; et al. Protection against tuberculosis by a single intranasal administration of DNA-hsp65 vaccine complexed with cationic liposomes. BMC Immunol. 2008, 9, 38. [Google Scholar] [CrossRef]
- Wong, J.P.; Zabielski, M.A.; Schmaltz, F.L.; Brownlee, G.G.; Bussey, L.A.; Marshall, K.; Borralho, T.; Nagata, L.P. DNA vaccination against respiratory influenza virus infection. Vaccine 2001, 19, 2461–2467. [Google Scholar] [CrossRef]
- Wang, D.; Christopher, M.E.; Nagata, L.P.; Zabielski, M.A.; Li, H.; Wong, J.P.; Samuel, J. Intranasal immunization with liposome-encapsulated plasmid DNA encoding influenza virus hemagglutinin elicits mucosal, cellular and humoral immune responses. J. Clin. Virol. 2004, 31S, 99–106. [Google Scholar]
- Gogev, S.; de Fays, K.; Versali, M.F.; Gautier, S.; Thiry, E. Glycol chitosan improves the efficacy of intranasally administrated replication defective human adenovirus type 5 expressing glycoprotein D of bovine herpesvirus 1. Vaccine 2004, 22, 1946–1953. [Google Scholar] [CrossRef]
- Kataoka, K.; Fujihashi, K. Dendritic cell-targeting DNA-based mucosal adjuvants for the development of mucosal vaccines. Expert Rev. Vaccines 2009, 8, 1183–1193. [Google Scholar] [CrossRef]
- Fraser, C.K.; Diener, K.R.; Brown, M.P.; Hayball, J.D. Improving vaccines by incorporating immunological coadjuvants. Expert Rev. Vaccines 2007, 6, 559–578. [Google Scholar] [CrossRef]
- Vajdy, M.; Srivastava, I.; Polo, J.; Donnelly, J.; O’Hagan, D.; Singh, M. Mucosal adjuvants and delivery systems for protein-, DNA- and RNA-based vaccines. Immunol. Cell. Biol. 2004, 82, 617–627. [Google Scholar] [CrossRef]
- Ryan, E.J.; McNeela, E.; Murphy, G.A.; Stewart, H.; O’Hagan, D.; Pizza, M.; Rappuoli, R.; Mills, K.H. Mutants of Escherichia coli heat-labile toxin act as effective mucosal adjuvants for nasal delivery of an acellular pertussis vaccine: Differential effects of the nontoxic AB complex and enzyme activity on Th1 and Th2 cells. Infect. Immun. 1999, 67, 6270–6280. [Google Scholar]
- Jakobsen, H.; Schulz, D.; Pizza, M.; Rappuoli, R.; Jonsdottir, I. Intranasal immunization with pneumococcal polysaccharide conjugate vaccines with nontoxic mutants of Escherichia coli heat-labile enterotoxins as adjuvants protects mice against invasive pneumococcal infections. Infect. Immun. 1999, 67, 5892–5897. [Google Scholar]
- Neidleman, J.A.; Vajdy, M.; Ugozzoli, M.; Ott, G.; O’Hagan, D. Genetically detoxified mutants of heat-labile enterotoxin from Escherichia coli are effective adjuvants for induction of cytotoxic T-cell responses against HIV-1 gag-p55. Immunology 2000, 101, 154–160. [Google Scholar] [CrossRef]
- Fujihashi, K.; Koga, T.; van Ginkel, F.W.; Hagiwara, Y.; McGhee, J.R. A dilemma for mucosal vaccination: Efficacy versus toxicity using enterotoxin-based adjuvants. Vaccine 2002, 20, 2431–2438. [Google Scholar] [CrossRef]
- Pizza, M.; Giuliani, M.; Fontana, M.; Monaci, E.; Douce, G.; Dougan, G.; Mills, K.; Rappuoli, R.; Del Giudice, G. Mucosal vaccines: Non toxic derivatives of LT and CT as mucosal adjuvants. Vaccine 2001, 19, 2534–2541. [Google Scholar] [CrossRef]
- Mutsch, M.; Zhou, W.; Rhodes, P.; Bopp, M.; Chen, R.T.; Linder, T.; Spyr, C.; Steffen, R. Use of the inactivated intranasal influenza vaccine and the risk of Bell’s palsy in Switzerland. N. Engl. J. Med. 2004, 350, 896–903. [Google Scholar] [CrossRef]
- Liang, S.; Hajishengallis, G. Heat-labile enterotoxins as adjuvants or anti-inflammatory agents. Immunol. Investig. 2010, 39, 449–467. [Google Scholar] [CrossRef]
- Spangler, B.D. Structure and function of cholera toxin and the related Escherichia coli heat-labile enterotoxin. Microbiol. Rev. 1992, 56, 622–647. [Google Scholar]
- Mata-Haro, V.; Cekic, C.; Martin, M.; Chilton, P.M.; Casella, C.R.; Mitchell, T.C. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science Signal. 2007, 316, 1628. [Google Scholar]
- Smith, K.A. T-cell growth factor. Immunol. Rev. 1980, 51, 337–357. [Google Scholar] [CrossRef]
- Kim, J.J.; Trivedi, N.N.; Nottingham, L.K.; Morrison, L.; Tsai, A.; Hu, Y.; Mahalingam, S.; Dang, K.; Ahn, L.; Doyle, N.K.; et al. Modulation of amplitude and direction of in vivo immune responses by co-administration of cytokine gene expression cassettes with DNA immunogens. Eur J. Immunol. 1998, 28, 1089–1103. [Google Scholar] [CrossRef]
- Xin, K.-Q.; Hamajima, K.; Sasaki, S.; Tsuji, T.; Watabe, S.; Okada, E.; Okuda, K. IL-15 expression plasmid enhances cell-mediated immunity induced by an HIV-1 DNA vaccine. Vaccine 1999, 17, 858–866. [Google Scholar] [CrossRef]
- Barouch, D.H.; Santra, S.; Schmitz, J.E.; Kuroda, M.J.; Fu, T.-M.; Wagner, W.; Bilska, M.; Craiu, A.; Zheng, X.X.; Krivulka, G.R. Control of viremia and prevention of clinical AIDS in rhesus monkeys by cytokine-augmented DNA vaccination. Science 2000, 290, 486–492. [Google Scholar] [CrossRef]
- Barouch, D.H.; Letvin, N.L.; Seder, R.A. The role of cytokine DNAs as vaccine adjuvants for optimizing cellular immune responses. Immunol. Rev. 2004, 202, 266–274. [Google Scholar] [CrossRef]
- Seaman, M.S.; Peyerl, F.W.; Jackson, S.S.; Lifton, M.A.; Gorgone, D.A.; Schmitz, J.E.; Letvin, N.L. Subsets of memory cytotoxic T lymphocytes elicited by vaccination influence the efficiency of secondary expansion in vivo. J. Virol. 2004, 78, 206–215. [Google Scholar] [CrossRef]
- Jeon, B.Y.; Eoh, H.; Ha, S.J.; Bang, H.; Kim, S.C.; Sung, Y.C.; Cho, S.N. Co-immunization of plasmid DNA encoding IL-12 and IL-18 with Bacillus Calmette-Guerin vaccine against progressive tuberculosis. Yonsei Med. J. 2011, 52, 1008–1015. [Google Scholar] [CrossRef]
- Kim, J.J.; Nottingham, L.K.; Sin, J.I.; Tsai, A.; Morrison, L.; Oh, J.; Dang, K.; Hu, Y.; Kazahaya, K.; Bennett, M.; et al. CD8 positive T cells influence antigen-specific immune responses through the expression of chemokines. J. Clin. Investig. 1998, 102, 1112–1124. [Google Scholar] [CrossRef]
- Sin, J.; Kim, J.J.; Pachuk, C.; Satishchandran, C.; Weiner, D.B. DNA vaccines encoding interleukin-8 and RANTES enhance antigen-specific Th1-type CD4+ T-cell-mediated protective immunity against herpes simplex virus type 2 in vivo. J. Virol. 2000, 74, 11173–11180. [Google Scholar] [CrossRef]
- Barouch, D.H.; McKay, P.F.; Sumida, S.M.; Santra, S.; Jackson, S.S.; Gorgone, D.A.; Lifton, M.A.; Chakrabarti, B.K.; Xu, L.; Nabel, G.J.; et al. Plasmid chemokines and colony-stimulating factors enhance the immunogenicity of DNA priming-viral vector boosting human immunodeficiency virus type 1 vaccines. J. Virol. 2003, 77, 8729–8735. [Google Scholar] [CrossRef]
- Sumida, S.M.; McKay, P.F.; Truitt, D.M.; Kishko, M.G.; Arthur, J.C.; Seaman, M.S.; Jackson, S.S.; Gorgone, D.A.; Lifton, M.A.; Letvin, N.L.; et al. Recruitment and expansion of dendritic cells in vivo potentiate the immunogenicity of plasmid DNA vaccines. J. Clin. Investig. 2004, 114, 1334–1342. [Google Scholar] [CrossRef]
- Chang, D.Z.; Lomazow, W.; Joy Somberg, C.; Stan, R.; Perales, M.A. Granulocyte-macrophage colony stimulating factor: An adjuvant for cancer vaccines. Hematology 2004, 9, 207–215. [Google Scholar] [CrossRef]
- McKay, P.F.; Barouch, D.H.; Santra, S.; Sumida, S.M.; Jackson, S.S.; Gorgone, D.A.; Lifton, M.A.; Letvin, N.L. Recruitment of different subsets of antigen-presenting cells selectively modulates DNA vaccine-elicited CD4+ and CD8+ T lymphocyte responses. Eur. J. Immunol. 2004, 34, 1011–1020. [Google Scholar] [CrossRef]
- Pavlenko, M.; Roos, A.K.; Lundqvist, A.; Palmborg, A.; Miller, A.M.; Ozenci, V.; Bergman, B.; Egevad, L.; Hellstrom, M.; Kiessling, R.; et al. A Phase I trial of DNA vaccination with a plasmid expressing prostate-specific antigen in patients with hormone-refractory prostate cancer. Br. J. Cancer 2004, 91, 688–694. [Google Scholar]
- Brave, A.; Ljungberg, K.; Boberg, A.; Rollman, E.; Isaguliants, M.; Lundgren, B.; Blomberg, P.; Hinkula, J.; Wahren, B. Multigene/multisubtype HIV-1 vaccine induces potent cellular and humoral immune responses by needle-free intradermal delivery. Mol. Ther. 2005, 12, 1197–1205. [Google Scholar] [CrossRef]
- Drexler, H.G.; Quentmeier, H. FLT3: Receptor and ligand. Growth Factors 2004, 22, 71–73. [Google Scholar] [CrossRef]
- Sekine, S.; Kataoka, K.; Fukuyama, Y.; Adachi, Y.; Davydova, J.; Yamamoto, M.; Kobayashi, R.; Fujihashi, K.; Suzuki, H.; Curiel, D.T.; et al. A novel adenovirus expressing Flt3 ligand enhances mucosal immunity by inducing mature nasopharyngeal-associated lymphoreticular tissue dendritic cell migration. J. Immunol. 2008, 180, 8126–8134. [Google Scholar] [CrossRef]
- Grant, E.V.; Thomas, M.; Fortune, J.; Klibanov, A.M.; Letvin, N.L. Enhancement of plasmid DNA immunogenicity with linear polyethylenimine. Eur. J. Immunol. 2012, 42, 2937–2948. [Google Scholar] [CrossRef]
- Waeckerle-Men, Y.; Groettrup, M. PLGA microspheres for improved antigen delivery to dendritic cells as cellular vaccines. Adv. Drug Deliv. Rev. 2005, 57, 475–482. [Google Scholar] [CrossRef]
- Aspden, T.J.; Mason, J.D.; Jones, N.S.; Lowe, J.; Skaugrud, Ø.; Illum, L. Chitosan as a nasal delivery system: The effect of chitosan solutions on in vitro and in vivo mucociliary transport rates in human turbinates and volunteers. J. Pharm. Sci. 1997, 86, 509–513. [Google Scholar] [CrossRef]
- Phase I Study of IV DOTAP: Cholesterol-Fus1 in Non-Small-Cell Lung Cancer. Available online: http://clinicaltrials.gov/ct2/show/NCT00059605?term=NCT00059605&rank=1 (accessed on 20 June 2014).
- Cheng, J.Y.; Huang, H.N.; Tseng, W.C.; Li, T.L.; Chan, Y.L.; Cheng, K.C.; Wu, C.J. Transcutaneous immunization by lipoplex-patch based DNA vaccines is effective vaccination against Japanese encephalitis virus infection. J. Control. Release 2009, 135, 242–249. [Google Scholar] [CrossRef]
- Chen, R.; Lu, S.H.; Tong, Q.B.; Lou, D.; Shi, D.Y.; Jia, B.B.; Huang, G.P.; Wang, J.F. Protective effect of DNA-mediated immunization with liposome-encapsulated GRA4 against infection of Toxoplasma gondii. J. Zhejiang Univ. Sci. B 2009, 10, 512–521. [Google Scholar]
- Korsholm, K.S.; Andersen, P.L.; Christensen, D. Cationic liposomal vaccine adjuvants in animal challenge models: Overview and current clinical status. Expert Rev. Vaccines 2012, 11, 561–577. [Google Scholar] [CrossRef]
- Gustafson, G.L.; Rhodes, M.J. Bacterial cell wall products as adjuvants: Early interferon γ as a marker for adjuvants that enhance protective immunity. Res. Immunol. 1992, 143, 483–488. [Google Scholar] [CrossRef]
- Ulrich, J.T.; Myers, K.R. Monophosphoryl lipid A as an adjuvant. Past experiences and new directions. Pharm. Biotechnol. 1995, 6, 495–524. [Google Scholar] [CrossRef]
- Morrison, D.C.; Silverstein, R.; Luchi, M.; Shnyra, A. Structure-function relationships of bacterial endotoxins. Contribution to microbial sepsis. Infect. Dis. Clin. N. Am. 1999, 13, 313–340. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Kool, M.; Willart, M.A.; Hammad, H. Mechanism of action of clinically approved adjuvants. Curr. Opin. Immunol. 2009, 21, 23–29. [Google Scholar] [CrossRef]
- Manicassamy, S.; Pulendran, B. Modulation of adaptive immunity with Toll-like receptors. Semin. Immunol. 2009, 21, 185–193. [Google Scholar] [CrossRef]
- Evans, J.T.; Cluff, C.W.; Johnson, D.A.; Lacy, M.J.; Persing, D.H.; Baldridge, J.R. Enhancement of antigen-specific immunity via the TLR4 ligands MPL adjuvant and Ribi. 529. Expert Rev. Vaccines 2003, 2, 219–229. [Google Scholar] [CrossRef]
- Childers, N.K.; Miller, K.L.; Tong, G.; Llarena, J.C.; Greenway, T.; Ulrich, J.T.; Michalek, S.M. Adjuvant activity of monophosphoryl lipid A for nasal and oral immunization with soluble or liposome-associated antigen. Infect. Immun. 2000, 68, 5509–5516. [Google Scholar] [CrossRef]
- Hall, M.A.; Stroop, S.D.; Hu, M.C.; Walls, M.A.; Reddish, M.A.; Burt, D.S.; Lowell, G.H.; Dale, J.B. Intranasal immunization with multivalent group A streptococcal vaccines protects mice against intranasal challenge infections. Infect. Immun. 2004, 72, 2507–2512. [Google Scholar] [CrossRef]
- Kamphuis, T.; Shafique, M.; Meijerhof, T.; Stegmann, T.; Wilschut, J.; de Haan, A. Efficacy and safety of an intranasal virosomal respiratory syncytial virus vaccine adjuvanted with monophosphoryl lipid A in mice and cotton rats. Vaccine 2013, 31, 2169–2176. [Google Scholar] [CrossRef]
- Greenland, J.R.; Letvin, N.L. Chemical adjuvants for plasmid DNA vaccines. Vaccine 2007, 25, 3731–3741. [Google Scholar] [CrossRef]
- Heath, A.W. Cytokines as immunological adjuvants. Pharm. Biotechnol. 1995, 6, 645–658. [Google Scholar] [CrossRef]
- Thompson, A.L.; Staats, H.F. Cytokines: The future of intranasal vaccine adjuvants. Clin. Dev. Immunol. 2011, 2011, 289597. [Google Scholar]
- Lynch, J.M.; Briles, D.E.; Metzger, D.W. Increased protection against pneumococcal disease by mucosal administration of conjugate vaccine plus interleukin-12. Infect. Immun. 2003, 71, 4780–4788. [Google Scholar] [CrossRef]
- Staats, H.F.; Bradney, C.P.; Gwinn, W.M.; Jackson, S.S.; Sempowski, G.D.; Liao, H.-X.; Letvin, N.L.; Haynes, B.F. Cytokine requirements for induction of systemic and mucosal CTL after nasal immunization. J. Immunol. 2001, 167, 5386–5394. [Google Scholar] [CrossRef]
- Huber, V.C.; Arulanandam, B.P.; Arnaboldi, P.M.; Elmore, M.K.; Sheehan, C.E.; Kallakury, B.V.; Metzger, D.W. Delivery of IL-12 intranasally leads to reduced IL-12-mediated toxicity. Int. Immunopharmacol. 2003, 3, 801–809. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xu, Y.; Yuen, P.-W.; Lam, J.K.-W. Intranasal DNA Vaccine for Protection against Respiratory Infectious Diseases: The Delivery Perspectives. Pharmaceutics 2014, 6, 378-415. https://doi.org/10.3390/pharmaceutics6030378
Xu Y, Yuen P-W, Lam JK-W. Intranasal DNA Vaccine for Protection against Respiratory Infectious Diseases: The Delivery Perspectives. Pharmaceutics. 2014; 6(3):378-415. https://doi.org/10.3390/pharmaceutics6030378
Chicago/Turabian StyleXu, Yingying, Pak-Wai Yuen, and Jenny Ka-Wing Lam. 2014. "Intranasal DNA Vaccine for Protection against Respiratory Infectious Diseases: The Delivery Perspectives" Pharmaceutics 6, no. 3: 378-415. https://doi.org/10.3390/pharmaceutics6030378
APA StyleXu, Y., Yuen, P.-W., & Lam, J. K.-W. (2014). Intranasal DNA Vaccine for Protection against Respiratory Infectious Diseases: The Delivery Perspectives. Pharmaceutics, 6(3), 378-415. https://doi.org/10.3390/pharmaceutics6030378