1. Introduction
Skin is the largest organ of the body, its primary function being to act as a permeability barrier to the surrounding environment. The outer layer of the skin, the stratum corneum, has an essential role as a barrier to the transport of water and xenobiotics. However, any xenobiotics that do penetrate the skin are biotransformed into harmless or less harmful compounds by various enzymes in the epidermis and dermis. Cutaneous metabolism introduces new chemicals into the systemic circulation and is an important determinant in the penetration of many compounds through the skin [
1]. Polar metabolites are more able to, than non-polar compounds, penetrate an aqueous environment such as the dermis and viable epidermis. A wide range of Phase I and Phase II metabolic biotransformations are able to be carried out in the skin, and most dermal metabolism occurs in basal keratinocytes in the epidermis [
2]. Constitutive expression of xenobiotic-metabolizing enzymes has been detected in normal human keratinocytes [
3].
Prodrugs, which are designed to improve the bioavailability and/or toxicity of parent drugs, are generally modified by ester linkage. Prodrugs are more easily partitioned into the stratum corneum by diffusion due to their high lipophilicity. Some prodrugs are transported into the blood as original prodrugs, but others are subcutaneously hydrolyzed to polar parent drugs by esterases in the viable epidermis underlying the stratum corneum. Polar compounds permeate the dermis and viable epidermis more easily than non-polar compounds, and the penetration rate of prodrugs is controlled by cutaneous hydrolysis. Therefore, both the partition of a prodrug into the stratum corneum and its cutaneous hydrolysis are key factors in the overall permeation of a prodrug through the skin.
The specific activity of dermal enzymes is much lower than their hepatic counterparts. However, because of the retention of lipophylic prodrugs in viable epidermis, they are extensively metabolized in the skin. Hewitt
et al. [
1] showed that esters applied on the surface of the skin were completely hydrolyzed during dermal absorption using an
in vitro diffusion cell. We have previously reported that the ester derivatives of propranolol (PL) are hydrolyzed during permeation through hairless mouse skin, and their hydrolysis rates are positively related with their hydrophobicities [
4].
It has been demonstrated that several esterases such as carboxylesterase (CES; EC 3.1.1.1) and arylesterase, present in human and rat skin, are responsible for the biotransformation of prodrugs [
5]. In addition, we have previously reported that the hydrolysis of PL ester derivatives is catalyzed by CES and butyrylcholinesterase in hairless mouse skin [
6].
CESs are member of the α/β hydrolase fold family and show ubiquitous tissue expression profiles with high levels in liver, small intestine, and lung [
7,
8,
9]. The mammalian CESs comprise a multigene family, and isozymes are classified into five main CES families (CES1–CES5), and several subfamilies, according to the homology of their amino acid sequences [
10]. The majority of CESs involved in detoxification of xenobiotics are from the CES1 and CES2 families. Mammalian CES1 isozymes are highly expressed in most organs, while CES2 isozymes are expressed in a limited number of organs, including intestine, liver, and kidney [
7,
11]. It has been reported that human skin expresses mainly CES1 isozyme (hCE1), and to a lesser extent CES2 isozyme (hCE2) [
5], while Zhu
et al. [
12], reported a high expression of hCE2 and a weak expression of hCE1 in human HaCaT keratinocytes. Unfortunately, esterases, including CESs, are rarely investigated in animal skin.
In the present study, we have focused on the identification of rat dermal esterases, especially CES isozymes. The hairless mouse and minipig are commonly used for percutaneous experiments, but their CES isozymes have hardly been investigated. In comparison, rat CES isozymes have been more extensively analyzed, so we investigated the relation between dermal hydrolysis and CES expression in rat skin. We selected caproyl-PL as a model hydrophobic prodrug, and demonstrated the effect of hydrolysis during prodrug permeation across rat skin.
2. Materials and Methods
2.1. Materials
Racemic
O-caproyl-PL hydrochloride was synthesized from PL hydrochloride (Wako Pure Chemical Industries, Ltd., Osaka, Japan) and caproyl chlorides (Tokyo Kasei, Tokyo, Japan) according to a previously described method [
13].
p-Nitrophenol,
p-nitrophenylacetate (PNPA),
p-nitrophenylbutyrate (PNPB), and bis-
p-nitrophenyl phosphate (BNPP) were purchased from Nacalai Tesque, Inc. (Kyoto, Japan). All other chemicals and reagents were of analytical or biochemical grade.
2.2. Animals
Male Wistar rats (8 weeks, 240–260 g) were used after overnight fasting with free access to water. All animal experimental protocols were approved by the Ethics Review Committee for Animal Experimentation of Kumamoto University.
2.3. Preparation of Skin Homogenate
After hair was carefully clipped and shaved, rats were sacrificed by exsanguination under ether anesthesia. Abdominal and dorsal skin was removed using a scalpel and dissection scissors. A few pieces of skin were stored in liquid nitrogen for extraction of total RNA. The skin was minced, mixed with five volumes of ice-cold 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (50 mM, pH 7.4) containing 0.15 M KCl, and homogenized using a Polytron (Kinematica, Lucerne, Switzerland) on ice. Whole homogenate was filtered through a funnel containing buffer-soaked cotton wool. After centrifugation of the skin homogenate at 9000
g for 20 min at 4 °C, the supernatant fraction (S9) was obtained. Protein content was determined by the method described by Bradford [
14] using bovine serum albumin (BSA) as standard. The skin S9 was stored at −80 °C until use.
2.4. Preparation of Homogenates of Liver, Kidney, and Testis
After rats were sacrificed by exsanguination under ether anesthesia, a cannula was placed in the inferior vena cava, and the liver and kidney were perfused with ice-cold 0.15 M KCl to remove blood. The excised liver, kidney, and testis were washed with ice-cold 0.15 M KCl, and finely minced using scissors. Minced tissues were homogenized with 3 volumes of 50 mM HEPES buffer (pH 7.4) containing 0.15 M KCl in a Potter–Elvehjem glass homogenizer equipped with a Teflon pestle under ice-cold conditions. Homogenates (25% wet
w/
v) were centrifuged at 9000
g for 20 min at 4 °C, to give the S9 fraction. S9 fraction was further centrifuged at 100,000
g for 1 h at 4 °C. The resulting pellets were resuspended in 50 mM HEPES buffer (pH 7.4) containing 0.15 M KCl to prepare the microsomal fraction. Protein concentrations were determined by the method of Bradford [
14] using BSA as standard. S9 and the microsomal fraction were stored at −80 °C until use.
2.5. Native Polyacrylamide Gel Electrophoresis (PAGE)
Polyacrylamide gel electrophoresis (PAGE) was performed as described by Mentlein
et al. [
15]. Polyacrylamide gels (7.5%
w/
w), containing 1% (
w/
v) nonidet P-40 for solubilization of proteins, were used for the separation of native enzymes. After electrophoresis of tissue samples (10–40 μg protein), the gels were stained for esterase activity with 1-naphthylacetate through coupling to liberated 1-naphthol with Fast Red TR-salt.
2.6. Total RNA Preparation for Tissue and Reverse Transcription (RT)-Polymerase Chain Reaction (PCR)
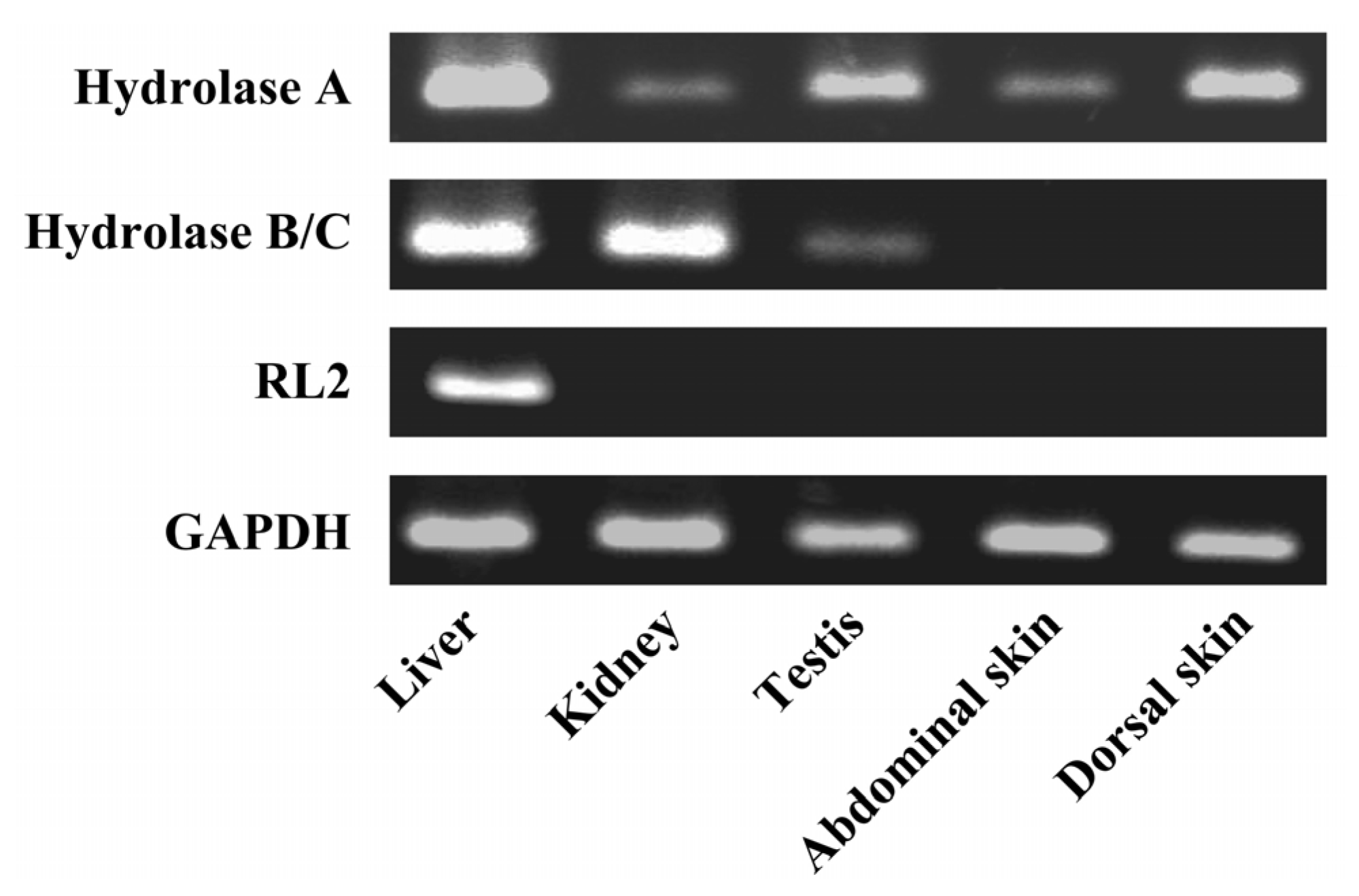
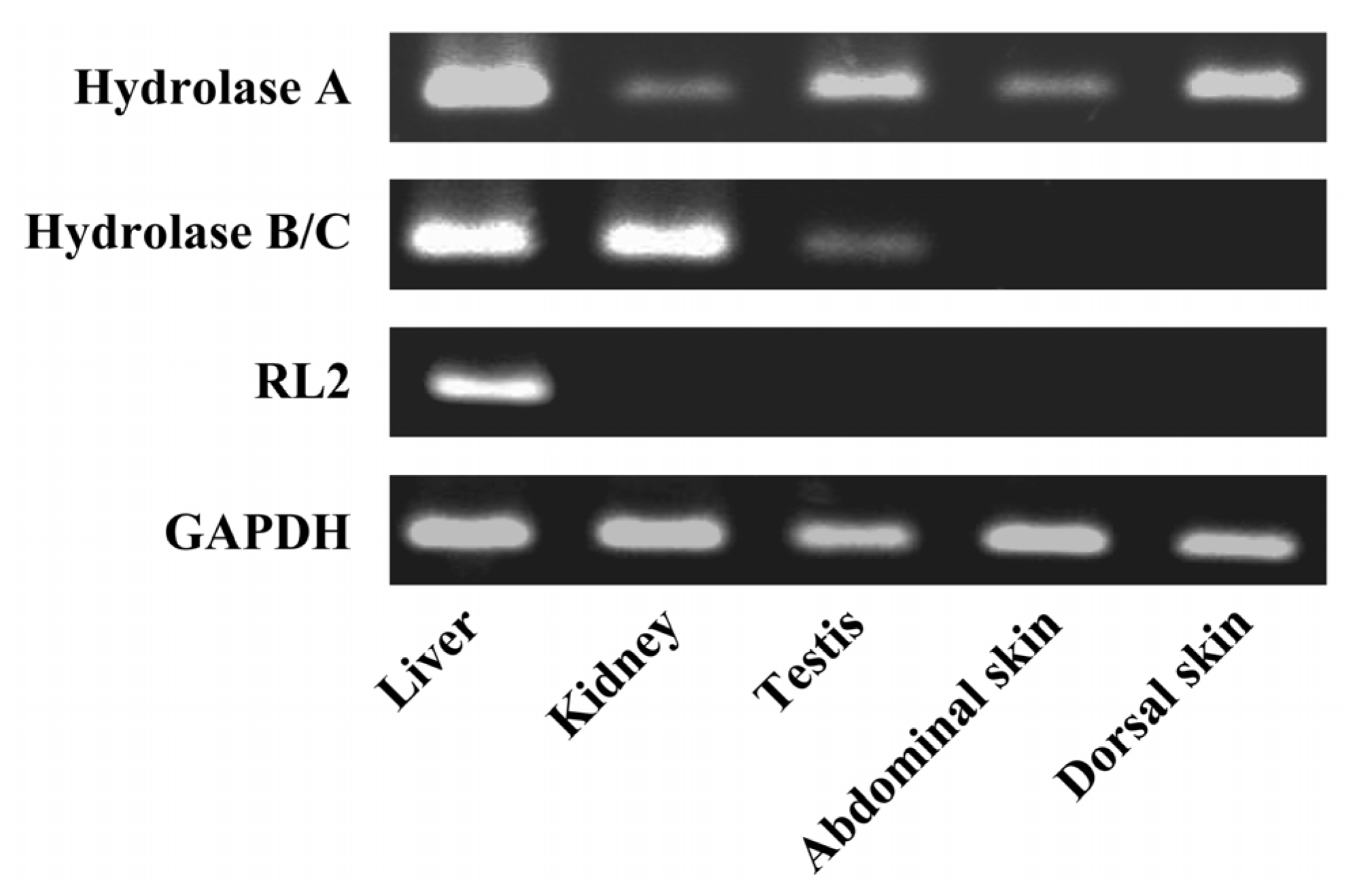
Total RNA was extracted from skin, liver, kidney, and testis using ISOGEN (Nippon Gene Co. Ltd., Toyama, Japan). To prevent contamination with genomic DNA, the extracts were treated with DNase I (Invitrogen, Carlsbad, CA, USA). RNA concentration and purity were determined spectrophotometrically. Total RNA (2 μg) was reverse-transcribed using 100 pmol Oligo (dT) prime, 1 mM dNTP, and RNase H free ReverTra Ace (Toyobo, Osaka, Japan) with one cycle of the RT reaction (42 °C for 60 min). After digestion of the remaining RNA with RNase H, the reverse transcription samples were subjected to subsequence PCR using platinum TaqDNA polymerase (Invitrogen, Carlsbad, CA, USA). Specific primers for rat CES isozymes were as follows: Hydrolase A (X51974) sense: 5'-CTGGACTTACTTGGAAACCC-3' and antisense: 5'-TGCAACCAAGTCCTGGAACA-3'; Hydrolase B/C (X81825/U10697) sense: 5'-CCAAAGACCCAAGGATGTAG-3' and antisense: 5'-TGAGGTTGTCTCTTAGCCAG-3'; RL2 (X81395) sense: 5'-ACACAGATGACCCAGACAGA-3' and antisense: 5'-CAGTGGCTTCATAGCCAGAA-3'; GAPDH (NM_017008) sense: 5'-ACCACAGTCCATGCCATCAC-3' and antisense: 5'-TCCACCACCCTGTTGCTGTA-3'.
2.7. Hydrolysis Experiments
Hydrolysis of PNPA, PNPB, and caproyl-PL were performed in S9 and microsomal fractions of rat liver, testis, kidney and skin. Tissue samples were diluted with 50 mM HEPES (pH 7.4) buffer at appropriate protein concentrations and preincubated for 5 min at 37 °C. The hydrolysis reaction was initiated by the addition of test compounds dissolved in dimethyl sulfoxide (DMSO). The final concentration of DMSO was less than 0.5%, which has no effect on hydrolytic activity. The rate of hydrolysis of PNPA and PNPB (final concentrations 15–500 μM) was determined by the initial linear increase in absorbance of p-nitrophenol at 405 nm (V-530, JASCO Co., Tokyo, Japan).
For the hydrolysis of caproyl-PL, the reaction was terminated by addition of 5 mL ethylacetate. After extraction of PL, as previously described [
16], stereoselective determination was carried out using high-performance liquid chromatography (HPLC).
2.8. In Vitro Skin Permeation Study
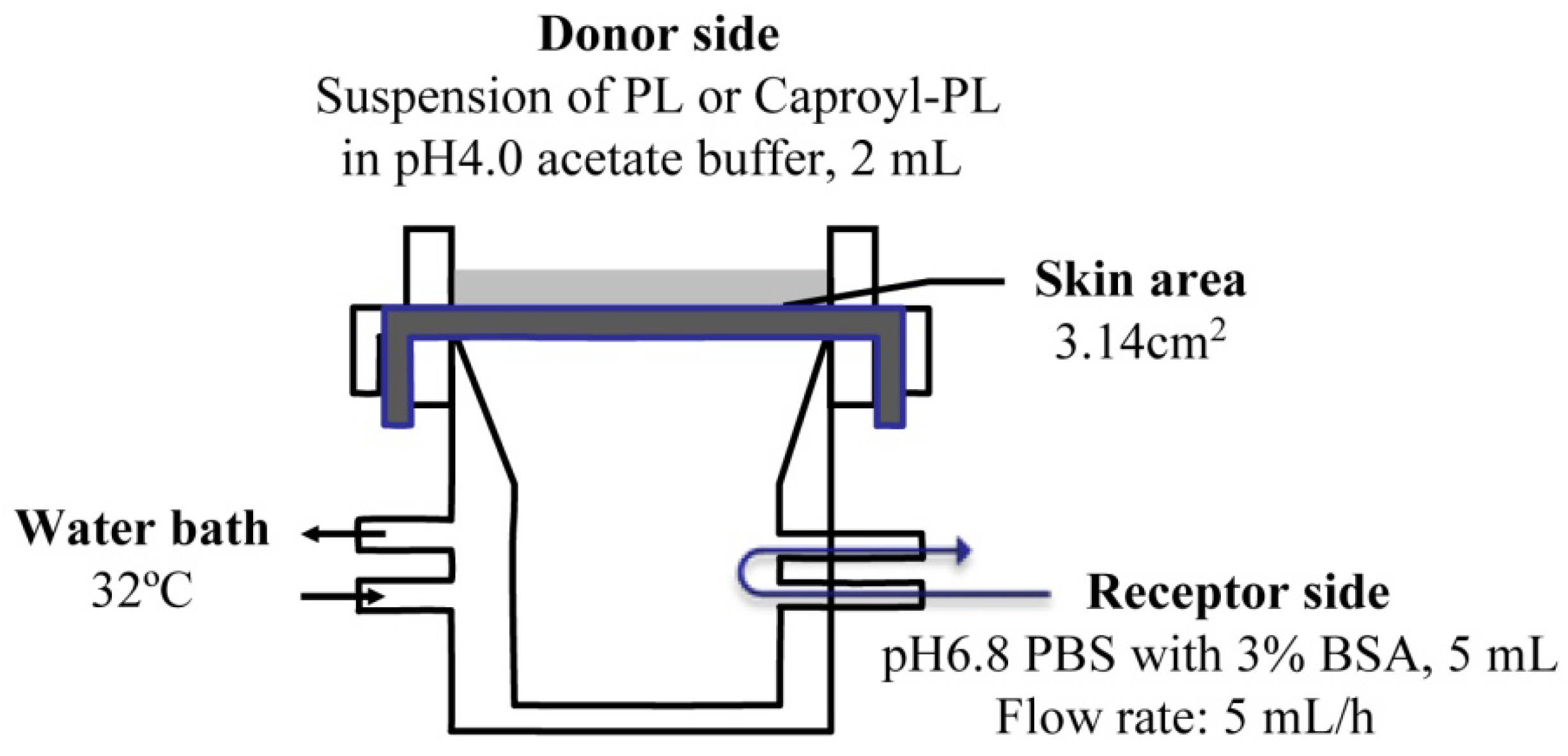
The
in vitro percutaneous penetration study was performed using a flow-through diffusion cell at 32 °C, as shown in
Figure 1. The receiving chamber had a volume of 5 mL and the skin area available for diffusion was 3.14 cm
2. Fresh excised abdominal skin was used for these experiments. To strip skin, stratum corneum was removed by 20 successive strippings using cellophane adhesive tape (Nichiban K.K., Japan) according to conventional methods. The excised skin was mounted between half-cells with the dermal side in contact with receptor fluid (pH 6.8 phosphate-buffered saline (PBS) with 3% BSA). Receptor fluid flow rate was 5 mL/h. Two milliliters of drug suspension (drug amount = twice the amount required for saturation) in pH 4.0 acetate buffer was added to the donor chamber.
Figure 1.
Scheme of transdermal permeation experiment using a flow-through diffusion cell.
Figure 1.
Scheme of transdermal permeation experiment using a flow-through diffusion cell.
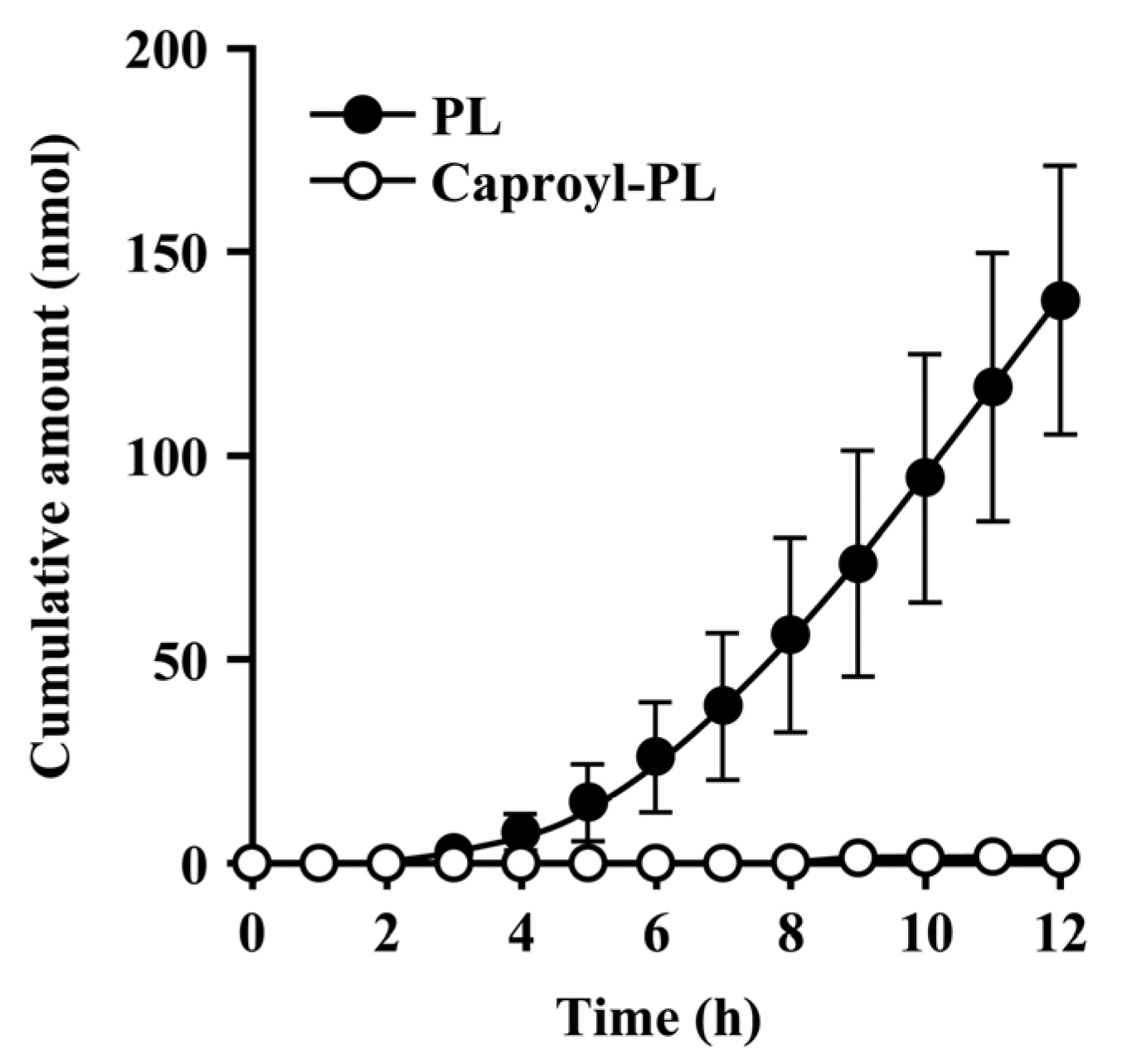
Nonenzymatic hydrolysis of caproyl-PL to PL was observed in buffer. This hydrolysis was <0.1% in 12 h under the most stable conditions of caproyl-PL (pH 4.0 acetate buffer). Hydrolysis was greater under basic buffer conditions (pH 6.8). However, the addition of BSA to the receptor fluid effectively prevented the hydrolysis of caproyl-PL. The hydrolysis of caproyl-PL was <0.05% during the sampling period (1 h) at pH 6.8 in PBS containing 3% BSA.
In an inhibition study for CES, skin mounted between the half-cells was preincubated with the dermal side in contact with 2 mM BNPP (dissolved in pH 6.8 PBS containing 3% BSA) for 1 h.
2.9. HPLC Assay
The HPLC system comprised a JASCO PU-980 pump, a JASCO 980-UV detector, a JASCO AS950 autosampler, a JASCO CO-965 column oven and a JASCO 1520S fluorescence detector (JASCO Co., Tokyo, Japan), and a Shimadzu chromatopac C-R7A plus (Shimadzu Co., Ltd., Kyoto, Japan). PL formation in the hydrolysis experiment was stereoselectively determined using Chiralcell OD column (10 μm, 250 × 4 mm inner diameter (i.d.); Daicel Chemical Industries, Ltd., Tokyo, Japan) with a mobile phase of hexane/2-propanol (9:1, v/v) at a flow rate of 1.0 mL/min. For the skin penetration experiment, LiChrosorb RP-select B column (7 μm, 250 × 4 mm i.d.; Merck Ltd., Tokyo, Japan) was used with a mobile phase of acetonitril/20 mM KH2PO4 (1:1 v/v) at a flow rate of 1.0 mL/min. PL and caproyl-PL were detected with excitation and emission wavelengths of 285 and 340 nm, respectively. The quantitative limitation was 30 pmol for PL and 60 pmol for caproyl-PL as injected amounts.
2.10. Data Analysis
The in vitro permeation parameters were calculated from the penetration data using the following equations: Js = (KmDC)/δ = KpC and τ = δ2/6D, where Js is steady state flux, Km is the solvent membrane partition coefficient of drug, D is diffusion coefficient, C is drug concentration in donor chamber, δ is thickness of rat skin, Kp is the permeability coefficient of drug, and τ represents lag time.
4. Discussion
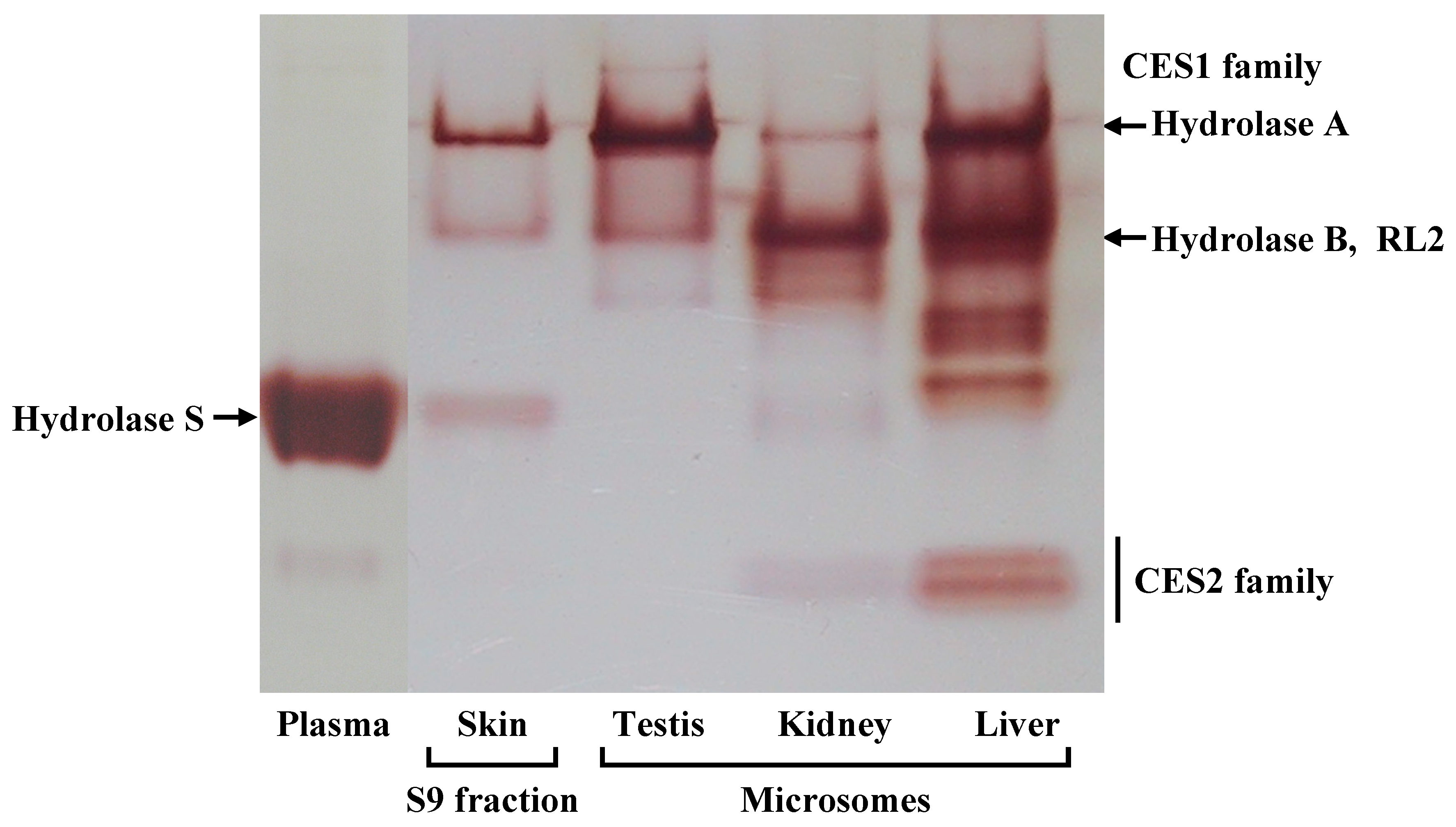
The barrier function of the skin to the absorption of active drugs resides in the outer-most layer, the stratum corneum. The use of a prodrug, a conjugate of active drug with a lipophilic molecule, is a way to achieve or increase cutaneous absorption. Conjugation via an ester bond is a conventional molecular modification. However, lipophilic drugs are retained in the skin. Therefore the conversion of a lipophilic prodrug to the active drug should occur at an appropriate rate in viable epidermis in order to obtain cutaneous activity and/or increase absorption of active drug into the bloodstream. CES is an abundant hydrolase in most organs [
10]. There are at least four CES1 isozymes in the rat CES1 family, Hydrolase A, Hydrolase B, Hydrolase C, and RL2 [
17,
18,
22]. In this study, we found Hydrolase A to be the predominant esterase in rat skin. In native PAGE, although a secondary band migrated to the same position as Hydrolase B, this band probably represents a monomer of Hydrolase A. The result of the RT-PCR also supports this explanation. The dissociation of trimeric CES1 isozyme has occasionally been found even in human liver samples [
23]. Of these four CES1 isozymes, Hydrolase A shows the closest homology to hCE1 (78% homology) [
10]. It has been reported that human skin expresses
hCE1 [
5]. hCE1 hydrolyzes a wide variety of substrates, and its expression is utilized in the hydrolysis of various prodrugs to the active hydrophilic parent drugs [
8,
21].
For the hydrolysis of PNPA and PNPB, microsomes from testis, which expressed Hydrolase A, showed lower Km values for PNPA than PNPB, while in kidney microsomes, which are abundant in Hydrolase B, the values for PNPB were lower than those for PNPA. Skin S9 showed similar properties to testis microsomes in its expression of Hydrolase A. The hydrolysis of PNPA and PNPB in skin S9 was around 20-fold slower than in liver microsomes. Caproyl-PL, a specific CES substrate, was also 20-fold more slowly hydrolyzed in skin S9 than in liver microsomes. As the CES content is higher in microsomes than in the S9 fraction, due to the localization of CES in the ER, these results indicate that the expression level of CES is sufficient to allow the hydrolysis of hydrophobic prodrugs during permeation through skin.
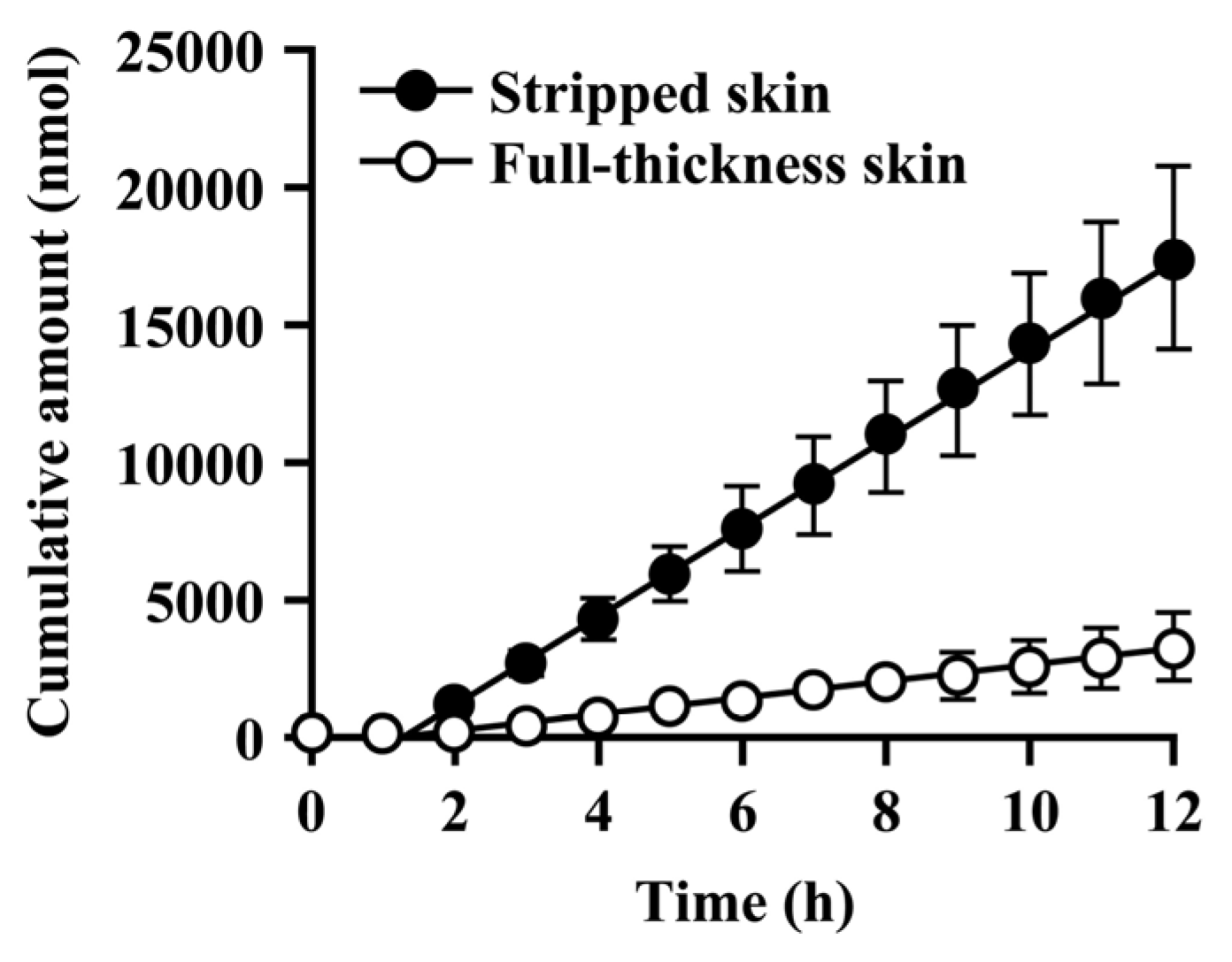
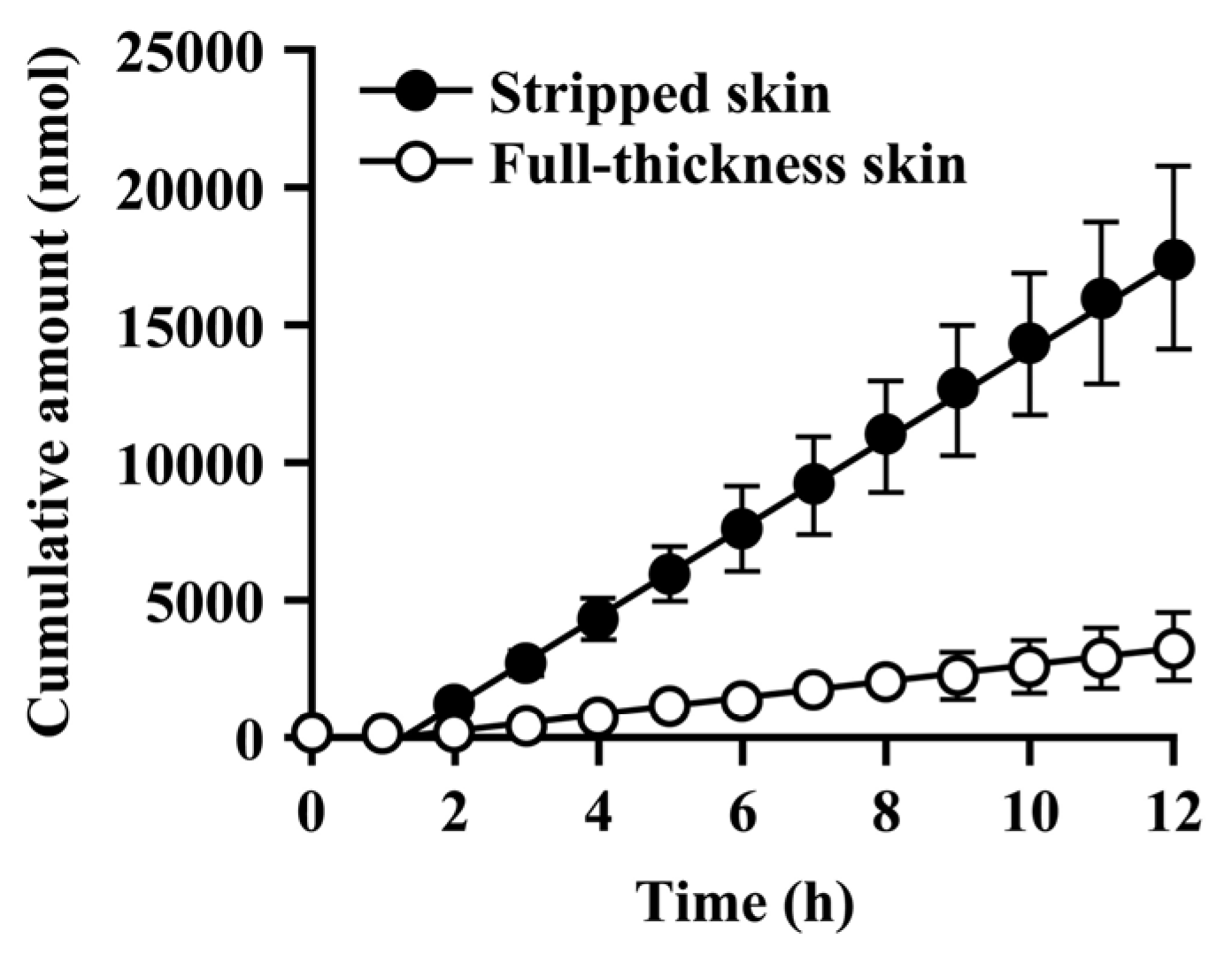
Since the hydrolysis of caproyl-PL by CES proceeds in the ER of viable epidermis and dermis, its rate of hydrolysis during cutaneous permeation depends on its dermal concentration. However, caproyl-PL was hydrolyzed to PL at nearly the same rate in both full-thickness and stripped skin. The concentration of caproyl-PL was 3200 ± 580 nmol/g tissue in stripped skin; this concentration is 160-fold higher than the Km value (19.7 ± 5.36 μM), suggesting that saturation of hydrolysis occurs in stripped skin. These data led us to expect that hydrolysis would also be saturated in full-thickness skin. We therefore estimated the concentration of caproyl-PL in viable epidermis and dermis after its application to full-thickness skin. We assumed that the distribution of caproyl-PL in viable epidermis and dermis would be homogeneous, and that it would move from viable skin to receptor fluid by simple diffusion. From the steady-state flux of caproyl-PL (2.4 ± 0.98 nmol/h) and its cutaneous concentration in stripped skin, the permeation clearance from viable skin to receptor fluid was calculated as 0.75 mg tissue/h. Since steady-state flux was 0.37 ± 0.15 nmol/h in full-thickness skin, the concentration of caproyl-PL was predicted to be about 500 nmol/g tissue in viable epidermis and dermis. This concentration is 25-fold higher than the Km value, suggesting saturation of cutaneous hydrolysis of caproyl-PL. The concentration of caproyl-PL in the stratum corneum can be calculated by subtraction from the 1900 ± 630 nmol/g tissue concentration of caproyl-PL in the skin. We therefore concluded that caproyl-PL applied to full-thickness skin is partitioned into the stratum corneum, where 75% of the dose is retained, only 25% of the dose being transferred into viable skin; this percentage is then hydrolyzed at maximum rate.
When CES-mediated hydrolysis was inhibited by BNPP, conversion to PL was markedly slower and there was a significant decrease in the net transport of caproyl-PL into receptor fluid. This clearly demonstrates that the net flux of prodrug is dependent on its conversion rate to the hydrophilic active drug.
In this study, caproyl-PL was retained not only in the stratum corneum but also in viable epidermis and dermis. Caproyl-PL has a molecular weight of 358, a pKa of 9.1, and a log PC between 1-octanol and pH 4.0 phosphate buffer of 2.3, while PL has a molecular weight of 259, pKa of 9.44 and log PC of 0.38. Molecular size and basicity are important factors in determining skin permeability, in addition to hydrophobicity. The maximum size of molecule able to diffuse through the epidermis and dermis is smaller for human skin than rat skin due to differences in their relative thickness. Therefore, caproyl-PL is retained for longer in human skin than in rat skin. As caproyl-PL is more rapidly hydrolyzed by hCE1 than Hydrolase A [
21], the steady-state flux of PL may be greater in human skin than in rat skin.
In the development of dermally applied prodrugs, there are two different aims, to obtain dermal activity and to obtain systemic activity. For dermal activity, prodrugs should be retained in whole skin where they are hydrolyzed to active drugs in a sustained manner. In this respect, caproyl-PL showed desirable properties as a prodrug. In order to obtain a systemic pharmacological activity, the intact prodrug should either rapidly diffuse into the bloodstream or be rapidly hydrolyzed in the skin. It is difficult to obtain rapid diffusion of hydrophobic prodrugs. Therefore, rapid conversion to active drug during penetration of a prodrug is obtained by selecting the optimum structure for recognition by hCE1.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}