The Potential Role of Nanotechnology in Therapeutic Approaches for Triple Negative Breast Cancer

Abstract
:1. Profiling and Current Therapeutic Approaches for Triple Negative Breast Cancer (TNBC)

{kind=link}
{kind=link}
| Classes | ER | PR | HER2 | GRADE | PROGNOSIS |
|---|---|---|---|---|---|
| Luminal A | Pos | Pos | Neg | Low | Good |
| Luminal B | Pos/Neg | Pos/Neg | Pos/Neg | Intermediate/High | Intermediate |
| HER2 | Neg | Neg | Pos | High | Poor |
| Basal-like | Neg | Neg | Neg | High | Poor |
| Subtype | GEP |
|---|---|
| Basal-like 1 (BL1) | expresses cell cycle, DNA repair and proliferating genes |
| Basal-like 2 (BL2) | expresses growth factor signaling genes such as EGFR, MET, Wnt, IGF-1R |
| Immunomodulatory (IM) | expresses genes involved in immune cell processes |
| Mesenchymal (M) | expresses genes involved in cell motility, differentiation and EMT processes |
| Mesenchymal stem-like (MSL) | expresses growth factor signaling genes and low levels of proliferating genes |
| Luminal androgen receptor (LAR) | expresses androgen receptor and downstream genes |
2. Currents Status of TNBC Therapeutics
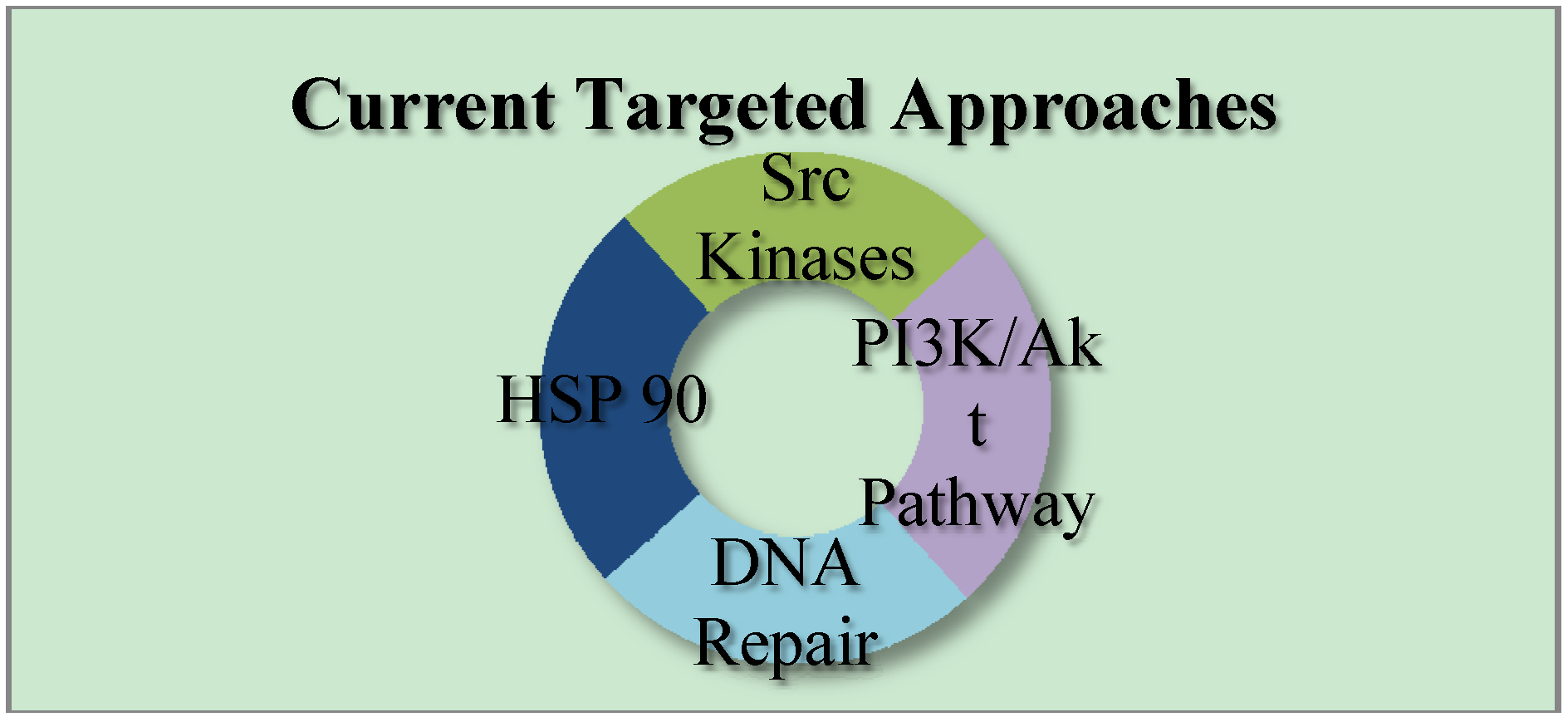
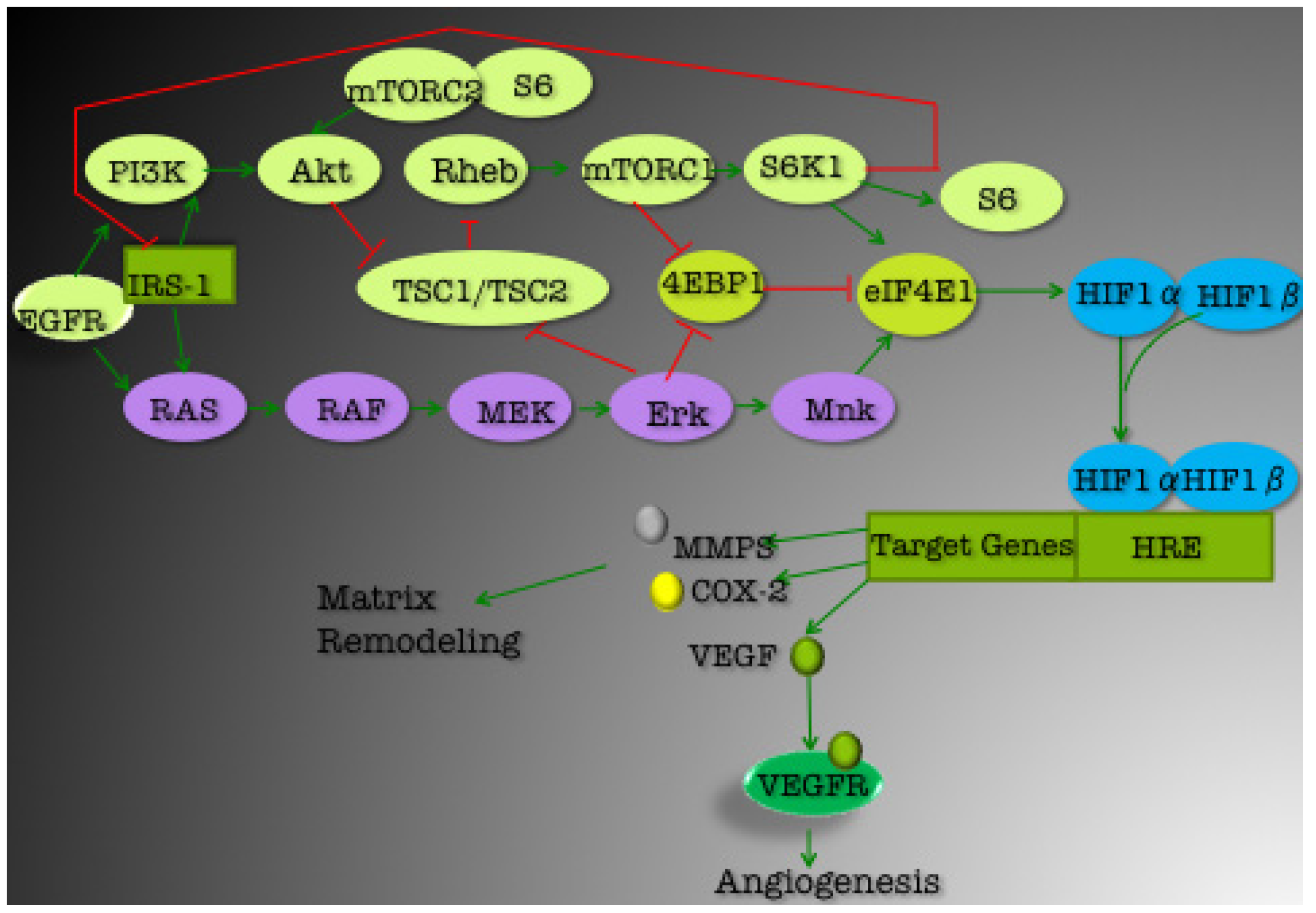
3. Current Targets for TNBC Therapeutics
3.1. PI3K/Akt Pathway
3.1.1. mTOR
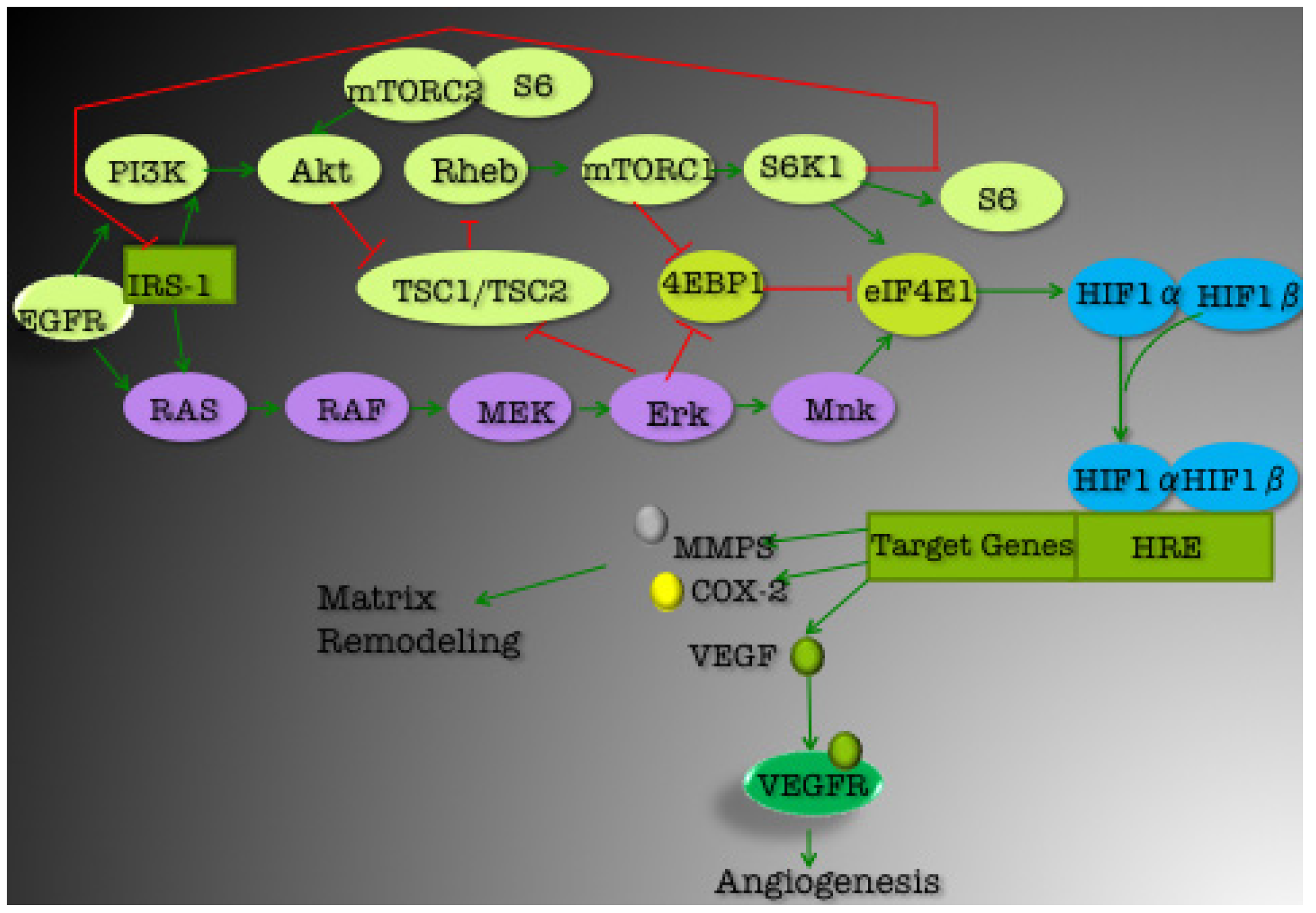
3.1.2. EGFR
3.1.3. IGF1R
3.2. DNA Repair
PARP
3.3. SRC Kinases
3.4. Heat Shock Protein 90
3.5. Combined Targeted Therapy
4. Nanoparticles as Drug Delivery Vehicles to Treat TNBC
Metabolic Profile of TNBC Cells Could Provide New Treatment Opportunities via Biocompatible Nanoparticles
5. Conclusion
Acknowledgement
Conflict of Interest
References
- Department of Health and Human Services, Center for Disease Control and Prevention. United States Cancer Statistics (USCS), 1999–2008 “Incidence and Mortality” web-based report; Centers for Disease Control and Prevention: Atlanta, GA, USA.
- Fornier, M.; Fumoleau, P. The paradox of triple negative breast cancer: Novel approaches to treatment. Breast J. 2012, 18, 41–51. [Google Scholar] [CrossRef]
- Rakha, E.A.; Reis-Filho, J.S.; Ellis, I.O. Basal-like breast cancer: A critical review. J. Clin. Oncol. 2008, 26, 2568–2581. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Tutt, A.N. Triple negative tumors: A critical review. Histopathology 2008, 52, 5846–5853. [Google Scholar]
- Sørlie, T.; Perou, C.M.; Tibshirani, R. Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef]
- Rakha, E.A.; El-Sayed, M.E.; Green, A.R.; Lee, A.H.; Robertson, J.F.; Ellis, I.O. Prognostic markers in triple-negative breast cancer. Cancer 2007, 109, 25–32. [Google Scholar]
- Carey, L.A.; Perou, C.M.; Livasy, C.A.; Dressler, L.G.; Cowan, D.; Conway, K.; Karaca, G.; Troester, M.A.; Tse, C.K.; Edmiston, S.; et al. Race, breast cancer subtypes, and survival in the carolina breast cancer study. JAMA 2006, 295, 2492–2502. [Google Scholar] [CrossRef]
- Thike, A.A.; Cheok, P.Y.; Jara-Lazaro, A.R.; Tan, B.; Tan, P.; Tan, P.H. Triple-negative breast cancer: Clinicopathological characteristics and relationship with basal-like breast cancer. Mod. Pathol. 2010, 23, 123–133. [Google Scholar]
- Banerjee, S.; Reis-Filho, J.S.; Ashley, S.; Steele, D.; Ashworth, A.; Lakhani, S.R.; Smith, I.E. Basal-Like breast carcinomas: Clinical outcome and response to chemotherapy. J. Clin. Pathol. 2006, 59, 729–735. [Google Scholar]
- Jumppanen, M.; Gruvberger-Saal, S.; Kauraniemi, P.; Tanner, M.; Bendahl, P.O.; Lundin, M.; Krogh, M.; Kataja, P.; Borg, A.; Ferno, M.; et al. Basal-like phenotype is not associated with patient survival in estrogen-receptor-negative breast cancers. Breast Cancer Res. 2007, 9, R16. [Google Scholar]
- Shakya, R.; Szabolcs, M.; McCarthy, E.; Ospina, E.; Basso, K.; Nandula, S.; Murty, V.; Baer, R.; Ludwig, T. The basal-like mammary carcinomas induced by Brca1 Or Bard1 inactivation implicate the BRCA1/BARD1 heterodimer in tumor suppression. Proc. Natl. Acad. Sci. USA 2008, 105, 7040–7045. [Google Scholar]
- Langerod, A.; Zhao, H.; Borgan, O.; Nesland, J.M.; Bukholm, I.R.; Ikdahl, T.; Karesen, R.; Borresen-Dale, A.L.; Jeffrey, S.S. TP53 mutation status and gene expression profiles are powerful prognostic markers of breast cancer. Breast Cancer Res. 2007, 9, R30. [Google Scholar] [CrossRef]
- Subhawong, A.P.; Subhawong, T.; Nassar, H.; Kouprina, N.; Begum, S.; Vang, R.; Westra, W.H.; Argani, P. Most basal-like breast carcinomas demonstrate the same Rb−/p16+ immunophenotype as the HPV-related poorly differentiated squamous cell carcinomas which they resemble morphologically. Am. J. Surg. Pathol. 2009, 33, 163–175. [Google Scholar] [CrossRef]
- Gauthier, M.L.; Berman, H.K.; Miller, C.; Kozakeiwicz, K.; Chew, K.; Moore, D.; Rabban, J.; Chen, Y.Y.; Kerlikowske, K.; Tlsty, T.D. Abrogated response to cellular stress identifies DCIS associated with subsequent tumor events and defines basal-like breast tumors. Cancer. Cell 2007, 12, 479–491. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar]
- Cleator, S.; Heller, W.; Coombes, R.C. Triple-negative breast cancer: Therapeutic options. Lancet Oncol. 2007, 8, 235–244. [Google Scholar] [CrossRef]
- Liu, T.; Yacoub, R.; Taliaferro-Smith, L.D. Sun, S.Y.; Graham, T.R.; Dolan, R.; Lobo, C.; Tighiouart, M.; Yang, L.; Adams, A.; et al. Combinatorial effects of lapatinib and rapamycin in triple-negative breast cancer cells. Mol. Cancer Ther. 2011, 10, 1460–1469. [Google Scholar] [CrossRef]
- Banerji, S.; Cibulskis, K.; Rangel-Escareno, C.; Brown, K.K.; Carter, S.L.; Frederick, A.M.; Lawrence, M.S.; Sivachenko, A.Y.; Sougnez, C.; Zou, L.; et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 2012, 486, 405–409. [Google Scholar] [CrossRef]
- Rakha, E.A.; Ellis, I.O. Triple-negative/basal-like breast cancer: Review. Pathology 2009, 41, 40–47. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar]
- Rouzier, R.; Perou, C.M.; Symmans, W.F.; Ibrahim, N.; Cristofanilli, M.; Anderson, K.; Hess, K.R.; Stec, J.; Ayers, M.; Wagner, P.; et al. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin. Cancer Res. 2005, 11, 5678–5685. [Google Scholar] [CrossRef]
- Calza, S.; Hall, P.; Auer, G.; Bjohle, J.; Klaar, S.; Kronenwett, U.; Liu, E.T.; Miller, L.; Ploner, A.; Smeds, J.; et al. Intrinsic molecular signature of breast cancer in a population-based cohort of 412 patients. Breast Cancer Res. 2006, 8, R34. [Google Scholar]
- Bertucci, F.; Finetti, P.; Cervera, N.; Esterni, B.; Hermitte, F.; Viens, P.; Birnbaum, D. How basal are triple-negative breast cancers? Int. J. Cancer 2008, 123, 236–240. [Google Scholar] [CrossRef]
- Tischkowitz, M.; Brunet, J.S.; Begin, L.R.; Huntsman, D.G.; Cheang, M.C.; Akslen, L.A.; Nielsen, T.O.; Foulkes, W.D. Use of immunohistochemical markers can refine prognosis in triple negative breast cancer. BMC Cancer 2007, 7, 134. [Google Scholar] [CrossRef]
- Bidard, F.C.; Conforti, R.; Boulet, T.; Michiels, S.; Delaloge, S.; Andre, F. Does triple-negative phenotype accurately identify basal-like tumour? An immunohistochemical analysis based on 143 “triple-negative” breast cancers. Ann. Oncol. 2007, 18, 1285–1286. [Google Scholar] [CrossRef]
- Tan, D.S.; Marchio, C.; Jones, R.L.; Savage, K.; Smith, I.E.; Dowsett, M.; Reis-Filho, J.S. Triple negative breast cancer: Molecular profiling and prognostic impact in adjuvant anthracycline-treated patients. Breast Cancer Res. Treat. 2008, 111, 27–44. [Google Scholar] [CrossRef]
- Schneider, B.P.; Winer, E.P.; Foulkes, W.D.; Garber, J.; Perou, C.M.; Richardson, A.; Sledge, G.W.; Carey, L.A. Triple-negative breast cancer: Risk factors to potential targets. Clin. Cancer Res. 2008, 14, 8010–8018. [Google Scholar] [CrossRef]
- Lin, N.U.; Vanderplas, A.; Hughes, M.E.; Theriault, R.L.; Edge, S.B.; Wong, Y.N.; Blayney, D.W.; Niland, J.C.; Winer, E.P.; Weeks, J.C. Clinicopathologic features, patterns of recurrence, and survival among women with triple-negative breast cancer in the national comprehensive cancer network. Cancer 2012, 118, 5463–5472. [Google Scholar] [CrossRef]
- Oakman, C.; Viale, G.; di Leo, A. Management of triple negative breast cancer. Breast 2010, 19, 312–321. [Google Scholar] [CrossRef]
- Badve, S.; Dabbs, D.J.; Schnitt, S.J.; Baehner, F.L.; Decker, T.; Eusebi, V.; Fox, S.B.; Ichihara, S.; Jacquemier, J.; Lakhani, S.R.; et al. Basal-like and triple-negative breast cancers: A critical review with an emphasis on the implications for pathologists and oncologists. Mod. Pathol. 2011, 24, 157–167. [Google Scholar]
- Hudis, C.A.; Gianni, L. Triple-negative breast cancer: An unmet medical need. Oncologist 2011, 16, 1–11. [Google Scholar] [CrossRef]
- Gril, B.; Palmieri, D.; Bronder, J.L.; Herring, J.M.; Vega-Valle, E.; Feigenbaum, L.; Liewehr, D.J.; Steinberg, S.M.; Merino, M.J.; Rubin, S.D.; et al. Effect of lapatinib on the outgrowth of metastatic breast cancer cells to the brain. J. Natl. Cancer Inst. 2008, 100, 1092–1103. [Google Scholar] [CrossRef]
- Chitnis, M.M.; Yuen, J.S.P.; Protheroe, A.S.; Pollack, M.; Macaulay, V.M. The type 1 insulin-like growth factor receptor pathway. Mol. Path. 2008, 14, 6364–6370. [Google Scholar]
- Jones, S.E.; Savin, M.A.; Holmes, F.A.; O’Shaughnessy, J.A. Phase III trial comparing doxorubicin plus cyclophosphamide with docetaxel plus cyclophosphamide as adjuvant therapy for operable breast cancer. J. Clin. Oncol. 2007, 24, 5381–5387. [Google Scholar]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; Andre, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar]
- Bos, P.D.; Zhang, X.H.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; van de Vijver, M.J.; Gerald, W.L.; Foekens, J.A.; et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459, 1005–1009. [Google Scholar]
- El Guerrab, A.; Zegrour, R.; Nemlin, C.C.; Vigier, F.; Cayre, A.; Penault-Llorca, F.; Rossignol, F.; Bignon, Y.J. Differential impact of egfr-targeted therapies on hypoxia responses: Implications for treatment sensitivity in triple-negative metastatic breast cancer. PLoS One 2011, 6, e25080. [Google Scholar]
- Fitzgerald, D.P.; Palmieri, D.; Hua, E.; Hargrave, E.; Herring, J.M.; Qian, Y.; Vega-Valle, E.; Weil, R.J.; Stark, A.M.; Vortmeyer, A.O.; et al. Reactive glia are recruited by highly proliferative brain metastases of breast cancer and promote tumor cell colonization. Clin. Exp. Metastasis 2008, 25, 799–810. [Google Scholar] [CrossRef]
- Kassam, F.; Enright, K.; Dent, R.; Dranitsaris, G.; Myers, J.; Flynn, C.; Fralick, M.; Kumar, R.; Clemons, M. Survival outcomes for patients with metastatic triple-negative breast cancer: Implications for clinical practice and trial design. Clin. Breast Cancer 2009, 1, 29–33. [Google Scholar]
- Lin, N.U.; Claus, E.; Sohl, J.; Razzak, A.R.; Arnaout, A.; Winer, E.P. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: High incidence of central nervous system metastases. Cancer 2008, 113, 2638–2645. [Google Scholar]
- Reis-Filho, J.S.; Tutt, A.N. Triple negative tumours: A critical review. Histopathology 2008, 1, 108–118. [Google Scholar]
- Cancello, G.; Maisonneuve, P.; Rotmensz, N.; Viale, G.; Mastropasqua, M.G.; Pruneri, G.; Veronesi, P.; Torrisi, R.; Montagna, E.; Luini, A.; et al. Prognosis and adjuvant treatment effects in selected breast cancer subtypes of very young women (<35 years) with operable breast cancer. Ann. Oncol. 2010, 21, 1974–1981. [Google Scholar]
- Klein, A.; Olendrowitz, C.; Schmutzler, R.; Hampl, J.; Schlag, P.M.; Maass, N.; Arnold, N.; Wessel, R.; Ramser, J.; Meindl, A.; et al. Identification of brain- and bone-specific breast cancer metastasis genes. Cancer Lett. 2009, 276, 212–220. [Google Scholar] [CrossRef]
- Dufour, M.; Dormaond-Meuwly, A.; Demartines, N.; Dormond, O. Targeting the mammalian target of rapamycin (mtor) in cancer therapy: Lessons from past and future perspectives. Cancer 2011, 3, 2478–2500. [Google Scholar] [CrossRef]
- Populo, H.; Lopes, J.M.; Soares, P. The mTOR signaling pathway in human cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef]
- Paranjape, T.; Heneghan, H.; Lindner, R.; Keane, F.K.; Hoffman, A.; Hollestelle, A.; Dorairaj, J.; Geyda, K.; Pelletier, C.; Nallur, S.; et al. A 3'-untranslated region KRAS variant and triple-negative breast cancer: A case-control and genetic analysis. Lancet Oncol. 2011, 12, 377–386. [Google Scholar] [CrossRef]
- Solit, D.B.; Garraway, L.A.; Pratilas, C.A.; Sawai, A.; Getz, G.; Basso, A.; Ye, Q.; Lobo, J.M.; She, Y.; Osman, I.; et al. BRAF mutation predicts sensitivity to mek inhibition. Nature 2006, 439, 358–362. [Google Scholar] [CrossRef]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar]
- Liu, Q.; Chang, J.W.; Wang, J.; Kang, S.A.; Thoreen, C.C.; Markhard, A.; Hur, W.; Zhang, J.; Sim, T.; Sabatini, D.M.; et al. Discovery of 1-(4-(4-Propionylpiperazin-1-Yl)-3-(Trifluoromethyl)Phenyl)-9-(Quinolin-3-Yl)Benz o[h][1,6]Naphthyridin-2(1H)-One as a highly potent, selective mammalian target of rapamycin (mTOR) inhibitor for the treatment of cancer. J. Med. Chem. 2010, 53, 7146–7155. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, J.; Kang, S.A.; Thoreen, C.C.; Hur, W.; Choi, H.G.; Waller, D.L.; Sim, T.; Sabatini, D.M.; Gray, N.S. Discovery and optimization of potent and selective benzonaphthyridinone analogs as small molecule mTOR inhibitors with improved mouse microsome stability. Bioorg. Med. Chem. Lett. 2011, 21, 4036–4040. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Liu, Q.; Thoreen, C.; Wang, J.; Sabatini, D.; Gray, N.S. mTOR mediated anti-cancer drug discovery. Drug Dis. Today Ther. Strat. 2009, 6, 47–55. [Google Scholar] [CrossRef]
- Ozes, O.N.; Akca, H.; Mayo, L.D.; Gustin, J.A.; Maehama, T.; Dixon, J.E.; Donner, D.B. A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proc. Natl. Acad. Sci. USA 2001, 98, 4640–4645. [Google Scholar]
- Tsutsui, S.; Ohno, S.; Murakami, S.; Hachitanda, Y.; Oda, S. Prognostic value of epidermal growth factor receptor (EGFR) and its relationship to the estrogen receptor status in 1029 patients with breast cancer. Breast Cancer Res. Treat. 2002, 71, 67–75. [Google Scholar] [CrossRef]
- Agrawal, A.; Gutteridge, E.; Gee, J.M.; Nicholson, R.I.; Robertson, J.F. Overview of tyrosine kinase inhibitors in clinical breast cancer. Endocr. Relat. Cancer 2005, 12, 135–144. [Google Scholar] [CrossRef]
- Inoue, S.; Patil, R.; Portilla-Arias, J.; Ding, H.; Konda, B.; Espinoza, A.; Mongayt, D.; Markman, J.L.; Elramsisy, A.; Phillips, H.W.; et al. Nanobiopolymer for direct targeting and inhibition of egfr expression in triple negative breast cancer. PLoS One 2012, 7, e31070. [Google Scholar]
- Peddi, P.F.; Ellis, M.J.; Ma, C. Molecular basis of triple negative breast cancer and implications for therapy. Int. J. Breast Cancer 2012, 2012, 217185. [Google Scholar]
- Irvin, W.J., Jr; Carey, L.A. What is triple-negative breast cancer? Eur. J. Cancer 2008, 44, 2799–2805. [Google Scholar] [CrossRef]
- Dogu, G.G.; Ozkan, M.; Ozturk, F.; Dikilitas, M.; Er, O.; Ozturk, A. Triple-negative breast cancer: Immunohistochemical correlation with basaloid markers and prognostic value of survivin. Med. Oncol. 2010, 27, 34–39. [Google Scholar] [CrossRef]
- Ueno, N.T.; Zhang, D. Targeting EGFR in triple negative breast cancer. J. Cancer 2011, 2, 324–328. [Google Scholar] [CrossRef]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 2012, 389, 349–352. [Google Scholar] [CrossRef]
- Baserga, R.; Peruzzi, F.; Reiss, K. The IGF-1 receptor in cancer biology. Int. J. Cancer 2003, 107, 873–877. [Google Scholar] [CrossRef]
- Sell, C.; Dumenil, G.; Deveaud, C.; Miura, M.; Coppola, D.; DeAngelis, T.; Rubin, R.; Efstratiadis, A.; Baserga, R. Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Mol. Cell. Biol. 1994, 14, 3604–3612. [Google Scholar]
- Lopez, T.; Hanahan, D. Elevated levels of IGF-1 receptor convey invasive and metastatic capability in a mouse model of pancreatic Islet tumorigenesis. Cancer Cell. 2002, 1, 339–353. [Google Scholar] [CrossRef]
- Jones, R.A.; Campbell, C.I.; Gunther, E.J.; Chodosh, L.A.; Petrik, J.J.; Khokha, R.; Moorehead, R.A. Transgenic overexpression of IGF-IR disrupts mammary ductal morphogenesis and induces tumor formation. Oncogene 2007, 26, 1636–1644. [Google Scholar] [CrossRef]
- Plymate, S.R.; Haugk, K.; Coleman, I.; Woodke, L.; Vessella, R.; Nelson, P.; Montgomery, R.B.; Ludwig, D.L.; Wu, J.D. An antibody targeting the type I insulin-like growth factor receptor enhances the castration-induced response in androgen-dependent prostate cancer. Clin. Cancer Res. 2007, 13, 6429–6439. [Google Scholar] [CrossRef]
- Natrajan, R.; Lambros, M.B.; Rodriguez-Pinilla, S.M.; Moreno-Bueno, G.; Tan, D.S.; Marchio, C.; Vatcheva, R.; Rayter, S.; Mahler-Araujo, B.; Fulford, L.G.; et al. Tiling path genomic profiling of grade 3 invasive ductal breast cancers. Clin. Cancer Res. 2009, 15, 2711–2722. [Google Scholar] [CrossRef]
- Bergamaschi, A.; Kim, Y.H.; Wang, P.; Sorlie, T.; Hernandez-Boussard, T.; Lonning, P.E.; Tibshirani, R.; Borresen-Dale, A.L.; Pollack, J.R. Distinct patterns of DNA copy number alterationare associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes Chromosomes Cancer 2006, 45, 1033–1040. [Google Scholar] [CrossRef]
- Cheang, M.C.; Voduc, D.; Bajdik, C.; Leung, S.; McKinney, S.; Chia, S.K.; Perou, C.M.; Nielsen, T.O. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin. Cancer Res. 2008, 14, 1368–1376. [Google Scholar] [CrossRef]
- Anderson, W.F.; Chatterjee, N.; Ershler, W.B.; Brawley, O.W. Estrogen receptor breast cancer phenotypes in the surveillance, epidemiology, and end results database. Breast Cancer Res. Treat. 2002, 76, 27–36. [Google Scholar] [CrossRef]
- Rakha, E.A.; El-Sayed, M.E.; Green, A.R.; Paish, E.C.; Powe, D.G.; Gee, J.; Nicholson, R.I.; Lee, A.H.; Robertson, J.F.; Ellis, I.O. Biologic and clinical characteristics of breast cancer with single hormone receptor positive phenotype. J. Clin. Oncol. 2007, 25, 4772–4778. [Google Scholar] [CrossRef]
- Prat, A.; Baselga, J. The role of hormonal therapy in the management of hormonal-receptor-positive breast cancer with co-expression of HER2. Nat. Clin. Pract. Oncol. 2008, 5, 531–542. [Google Scholar] [CrossRef]
- Atchley, D.P.; Albarracin, C.T.; Lopez, A.; Valero, V.; Amos, C.I.; Gonzalez-Angulo, A.M.; Hortobagyi, G.N.; Arun, B.K. Clinical and pathologic characteristics of patients with BRCA-positive and BRCA-negative breast cancer. J. Clin. Oncol. 2008, 26, 4282–4288. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Brunet, J.S.; Stefansson, I.M.; Straume, O.; Chappuis, P.O.; Begin, L.R.; Hamel, N.; Goffin, J.R.; Wong, N.; Trudel, M.; et al. The prognostic implication of the basal-like (Cyclin E high/p27 low/p53+/+glomeruloid-microvascular-proliferation) phenotype of BRCA1-related breast cancer. Cancer Res. 2004, 64, 830–835. [Google Scholar] [CrossRef]
- Gonzalez-Angulo, A.M.; Hortobagyi, G.N. Is there an ideal way to combine trastuzumab with chemotherapy? J. Clin. Oncol. 2011, 29, 4474–4476. [Google Scholar] [CrossRef]
- Sirohi, B.; Arnedos, M.; Popat, S.; Ashley, S.; Nerurkar, A.; Walsh, G.; Johnston, S.; Smith, I.E. Platinum-based chemotherapy in triple-negative breast cancer. Ann. Oncol. 2008, 19, 1847–1852. [Google Scholar] [CrossRef]
- Chalmers, A.J. The potential role and application of PARP inhibitors in cancer treatment. Breast Med. Bull. 2009, 89, 23–40. [Google Scholar] [CrossRef]
- Guo, G.; Zhang, F.; Gao, R.; Delsite, R.; Feng, Z.; Powell, S. DNA repair and synthetic lethality. Int. J. Oral Sci. 2011, 3, 176–179. [Google Scholar] [CrossRef]
- Au-Yong, I.T.; Evans, A.J.; Taneja, S.; Rakha, E.A.; Green, A.R.; Paish, C.; Ellis, I.O. Sonographic correlations with the new molecular classification of invasive breast cancer. Eur. Radiol. 2009, 19, 2342–2348. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Finn, R.S.; Bengala, C.; Ibrahim, N.; Roche, H.; Sparano, J.; Strauss, L.; Fairchild, J.; Sy, O.; Goldstein, L.J. Dasatinib as a single agent in triple negative breast cancer: Results of an open-labeled phase 2 study. Clin. Cancer Res. 2011, 21, 6905–6913. [Google Scholar]
- Gnoni, A.; Marech, I.; Silvestris, N.; Vacca, A.; Lorusso, V. Dasatinib: An anti-tumour agent via Src inhibition. Curr. Drug Targets 2011, 4, 563–578. [Google Scholar]
- Tryfonopoulos, D.; Walsh, S.; Collins, D.M.; Flanagan, L.; Quinn, C.; Corkery, B.; McDermott, E.W.; Evoy, D.; Pierce, A.; O’Donovan, N.; et al. Src: A potential target for the treatment of triple-negative breast cancer. Ann. Oncol. 2011, 10, 2234–2240. [Google Scholar]
- Chiosis, G.; Caldas-Lopes, E.; Solit, D. Heat shock protein-90 inhibitors: A chronicle from geldanamycin to today’s agents. Curr. Opin. Inv. Drugs 2006, 6, 534–541. [Google Scholar]
- Caldas-lopes, E.; Cerchietti, L.; Ahn, J.; Clement, C.; Robles, A.; Rodina, A.I.; Rodina, A.; Moylick, K.; Taldone, T.; Gozman, A.; et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc. Natl. Acad. Sci. USA 2009, 106, 8368–8373. [Google Scholar] [CrossRef]
- Patel, H.J.; Modi, S.; Chiosis, G.; Taldone, T. Advances in the discovery and development of heat-shock protein 90 inhibitors for cancer treatment. Expert Opin. Drug Discov. 2011, 6, 559–587. [Google Scholar] [CrossRef]
- Modi, S.; Stopeck, A.; Linden, H.; Solit, D.; Chandarlapaty, S.; Rosen, N.; D’Andrea, G.; Dickler, M.; Moynahan, M.; Sugarman, S.; et al. HSP90 inhibition is effective in breast cancer: A phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin. Cancer Res. 2011, 17, 5132–5139. [Google Scholar] [CrossRef]
- Haluska, P.; Shaw, H.M.; Batzel, G.N.; Yin, D.; Molina, J.R.; Molife, L.R.; Yap, T.A.; Roberts, M.L.; Sharma, A.; Gualberto, A.; et al. Phase I dose escalation study of the anti insulin-like growth factor-I receptor monoclonal antibody CP-751,871 in patients with refractory solid tumors. Clin. Cancer Res. 2007, 13, 5834–5840. [Google Scholar] [CrossRef]
- Ryan, C.J.; Harzstark, A.H.; Rosenberg, J.; Lin, A.; Claros, C.; Goldfine, I.D.; Kerner, J.F.; Small, E.J.; Youngren, J.F. A Pilot Dose-escalation study of the effects of nordihydroguareacetic acid on hormone and prostate specific antigen levels in patients with relapsed prostate cancer. BJU Int. 2008, 101, 436–439. [Google Scholar] [CrossRef]
- Rochester, M.A.; Riedemann, J.; Hellawell, G.O.; Brewster, S.F.; Macaulay, V.M. Silencing of the IGF1R gene enhances sensitivity to DNA-damaging agents in both PTEN wild-type and mutant human prostate cancer. Cancer Gene Ther. 2005, 12, 90–100. [Google Scholar] [CrossRef]
- Yeh, A.H.; Bohula, E.A.; Macaulay, V.M. Human melanoma cells expressing V600E B-RAF are susceptible to IGF1R targeting by small interfering RNAs. Oncogene 2006, 25, 6574–6581. [Google Scholar] [CrossRef]
- Haluska, P.; Carboni, J.M.; TenEyck, C.; Attar, R.M.; Hou, X.; Yu, C.; Sagar, M.; Wong, T.W.; Gottardis, M.M.; Erlichman, C. HER receptor signaling confers resistance to the insulin-like growth factor-I receptor inhibitor, BMS-536924. Mol. Cancer Ther. 2008, 7, 2589–2598. [Google Scholar] [CrossRef]
- Jerome, L.; Alami, N.; Belanger, S.; Page, V.; Yu, Q.; Paterson, J.; Shiry, L.; Pegram, M.; Leyland-Jones, B. Recombinant human insulin-like growth factor binding protein 3 inhibits growth of human epidermal growth factor receptor-2-overexpressing breast tumors and potentiates herceptin activity in vivo. Cancer Res. 2006, 66, 7245–7252. [Google Scholar] [CrossRef]
- Lu, D.; Zhang, H.; Koo, H. Tonra, J.; Balderes, P.; Prewett, M.; Corcoran, E.; Mangalampalli, V.; Bassi, R.; Anselma, D.; et al. A fully human recombinant IgG-like bispecific antibody to both the epidermal growth factor receptor and the insulin-like growth factor receptor for enhanced antitumor activity. J. Biol. Chem. 2005, 280, 19665–19672. [Google Scholar] [CrossRef]
- Oliveras-Ferraros, C.; Vazquez-Martin, A.; Lopez-Bonet, E.; Martin-Castillo, B.; del Barco, S.; Brunet, J.; Menendez, J.A. Growth and molecular interactions of the anti-EGFR antibody cetuximab and the DNA cross-linking agent cisplatin in gefitinib-resistant MDA-MB-468 cells: New prospects in the treatment of triple-negative/basal-like breast cancer. Int. J. Oncol. 2008, 33, 1165–1176. [Google Scholar]
- Nowsheen, S.; Cooper, T.; Stanley, J.A.; Yang, E.S. Synthetic lethal interactions between EGFR and PARP inhibition in human triple negative breast cancer cells. PLoS One 2012, 7, e46614. [Google Scholar]
- Lee, E.S.; Na, K.; Bae, Y.H. Doxorubicin loaded pH-sensitive polymeric micelles for reversal of resistant MCF-7 tumor. J. Contr. Release 2005, 103, 405–418. [Google Scholar] [CrossRef]
- Patel, N.R.; Rathi, A.; Mongayt, D.; Torchilin, V.P. Reversal of multidrug resistance by Co-delivery of tariquidar (XR9576) and paclitaxel using long-circulating liposomes. Int. J. Pharm. 2011, 416, 296–299. [Google Scholar] [CrossRef]
- Li, B.; Xu, H.; Li, Z.; Yao, M.; Xie, M.; Shen, H.; Shen, S.; Wang, X.; Jin, Y. Bypassing multidrug resistance in human breast cancer cells with lipid/polymer particle assemblies. Int. J. Nanomed. 2012, 7, 187–197. [Google Scholar]
- Lee, S.M.; Ahn, R.W.; Chen, F.; Fought, A.J.; O’Halloran, T.V.; Cryns, V.L.; Nguyen, S.T. Biological evaluation of pH-responsive polymer-caged nanobins for breast cancer therapy. ACS Nano 2010, 4, 4971–4978. [Google Scholar] [CrossRef]
- Ahn, R.W.; Chen, F.; Chen, H.; Stern, S.T.; Clogston, J.D.; Patri, A.K.; Raja, M.R.; Swindell, E.P.; Parimi, V.; Cryns, V.L.; et al. A novel nanoparticulate formulation of arsenic trioxide with enhanced therapeutic efficacy in a murine model of breast cancer. Clin. Cancer Res. 2010, 16, 3607–3617. [Google Scholar] [CrossRef]
- Shao, W.; Paul, A.; Abbasi, S.; Chahal, P.S.; Mena, J.A.; Montes, J.; Kamen, A.; Prakesh, S. A novelpolyethyleneimine-coatedadeno-associated virus-like particle formulation for efficient siRNAdelivery in breast cancer therapy: Preparation and in vitro analysis. Int. J. Nanomed. 2012, 7, 1575–1586. [Google Scholar]
- Hussein, Y.R.; Sood, A.K.; Bandyopadhyay, S.; Albashiti, B.; Semaan, A.; Nahleh, Z.; Roh, J.; Han, H.D.; Lopez-Berestein, G.; Ali-Fehmi, R. Clinical and biological relevance of enhancer of zeste homolog 2 in triple-negative breast cancer. Hum. Pathol. 2012, 43, 1638–1644. [Google Scholar] [CrossRef]
- Tekedereli, I.; Alpay, S.N.; Tavares, C.D.; Cobanoglu, Z.E.; Kaoud, T.S.; Sahin, I.; Sood, A.K.; Lopez-Berestein, G.; Dalby, K.N.; Ozpolat, B. Targeted silencing of elongation factor 2 kinase suppresses growth and sensitizes tumors to doxorubicin in an orthotopic model of breast cancer. PLoS One 2012, 7, e41171. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Sabnis, N.; Lacko, A.G. Drug delivery via lipoprotein-based carriers: Answering the challenges in systemic therapeutics. Ther. Deliv. 2012, 3, 599–608. [Google Scholar] [CrossRef]
- Lacko, A.G.; Nair, M.; Paranjape, S.; Mooberry, L.; McConathy, W.J. Trojan horse meets magic bullet to spawn a novel, highly effective drug delivery model. Chemotherapy 2006, 52, 171–173. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Johnson, R.; Sabnis, N.; McConathy, W.J.; Lacko, A.G. The Potential Role of Nanotechnology in Therapeutic Approaches for Triple Negative Breast Cancer. Pharmaceutics 2013, 5, 353-370. https://doi.org/10.3390/pharmaceutics5020353
Johnson R, Sabnis N, McConathy WJ, Lacko AG. The Potential Role of Nanotechnology in Therapeutic Approaches for Triple Negative Breast Cancer. Pharmaceutics. 2013; 5(2):353-370. https://doi.org/10.3390/pharmaceutics5020353
Chicago/Turabian StyleJohnson, Rebecca, Nirupama Sabnis, Walter J. McConathy, and Andras G. Lacko. 2013. "The Potential Role of Nanotechnology in Therapeutic Approaches for Triple Negative Breast Cancer" Pharmaceutics 5, no. 2: 353-370. https://doi.org/10.3390/pharmaceutics5020353
APA StyleJohnson, R., Sabnis, N., McConathy, W. J., & Lacko, A. G. (2013). The Potential Role of Nanotechnology in Therapeutic Approaches for Triple Negative Breast Cancer. Pharmaceutics, 5(2), 353-370. https://doi.org/10.3390/pharmaceutics5020353