Design and Exploratory Neuropharmacological Evaluation of Novel Thyrotropin-Releasing Hormone Analogs and Their Brain-Targeting Bioprecursor Prodrugs

 ,
, 

Abstract
:1. Introduction
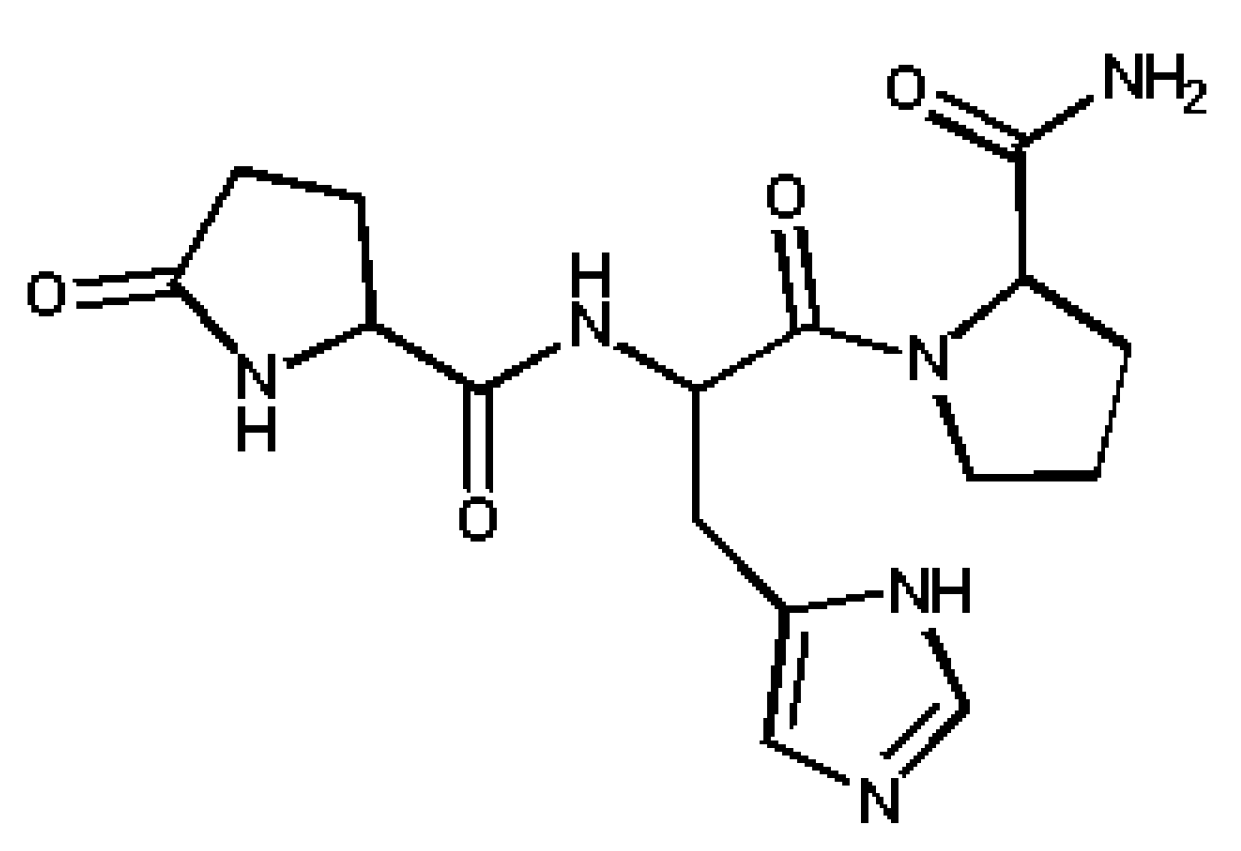
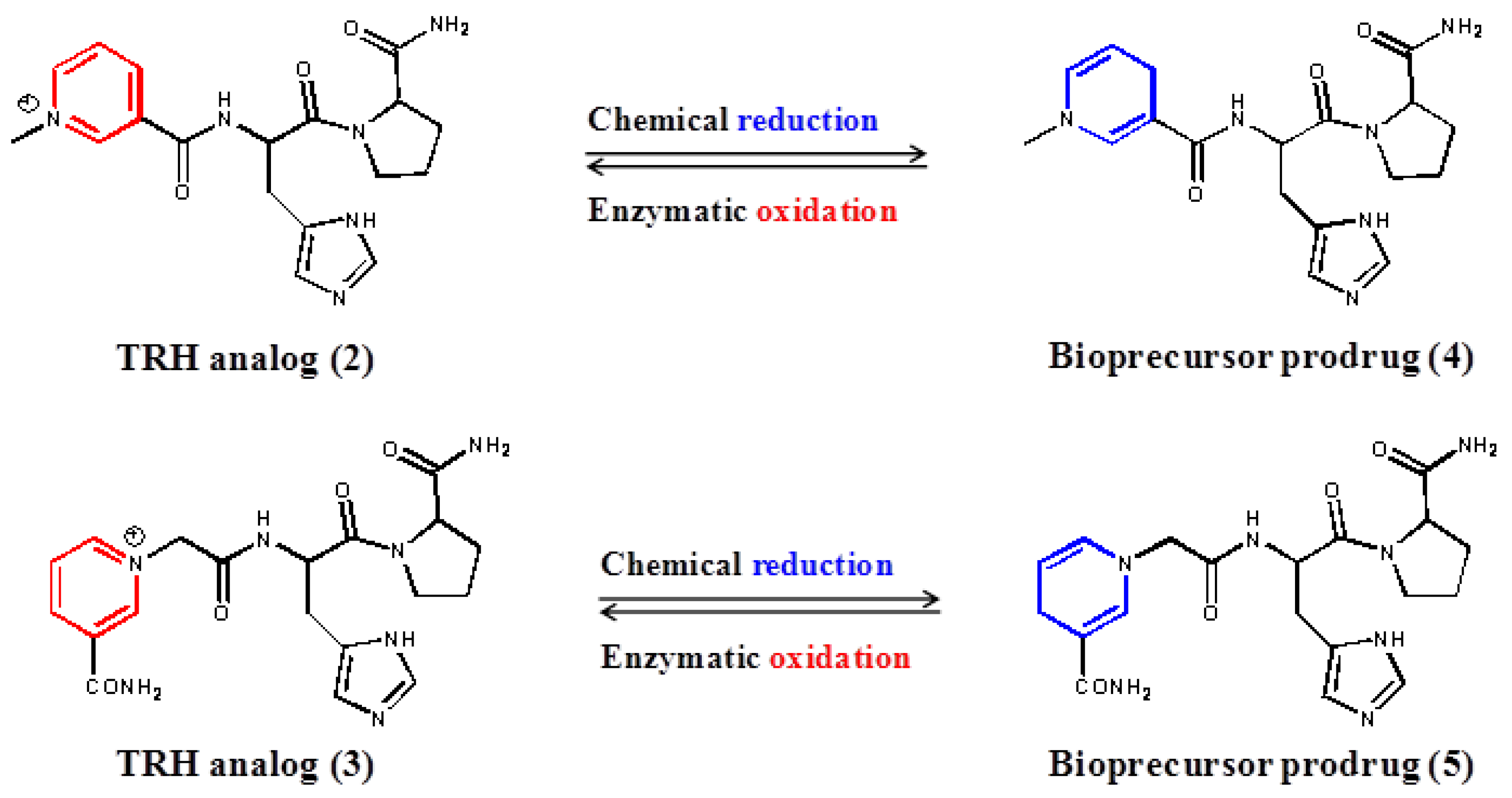
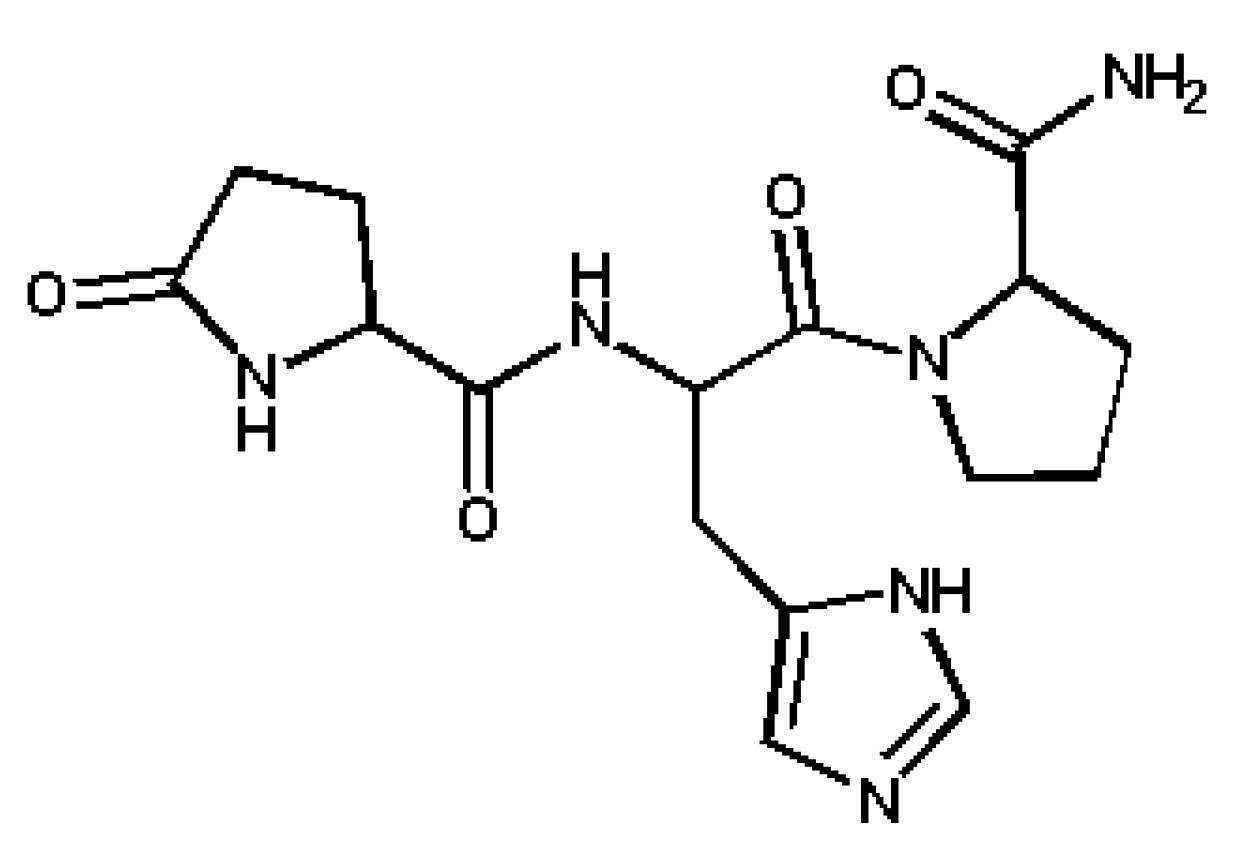
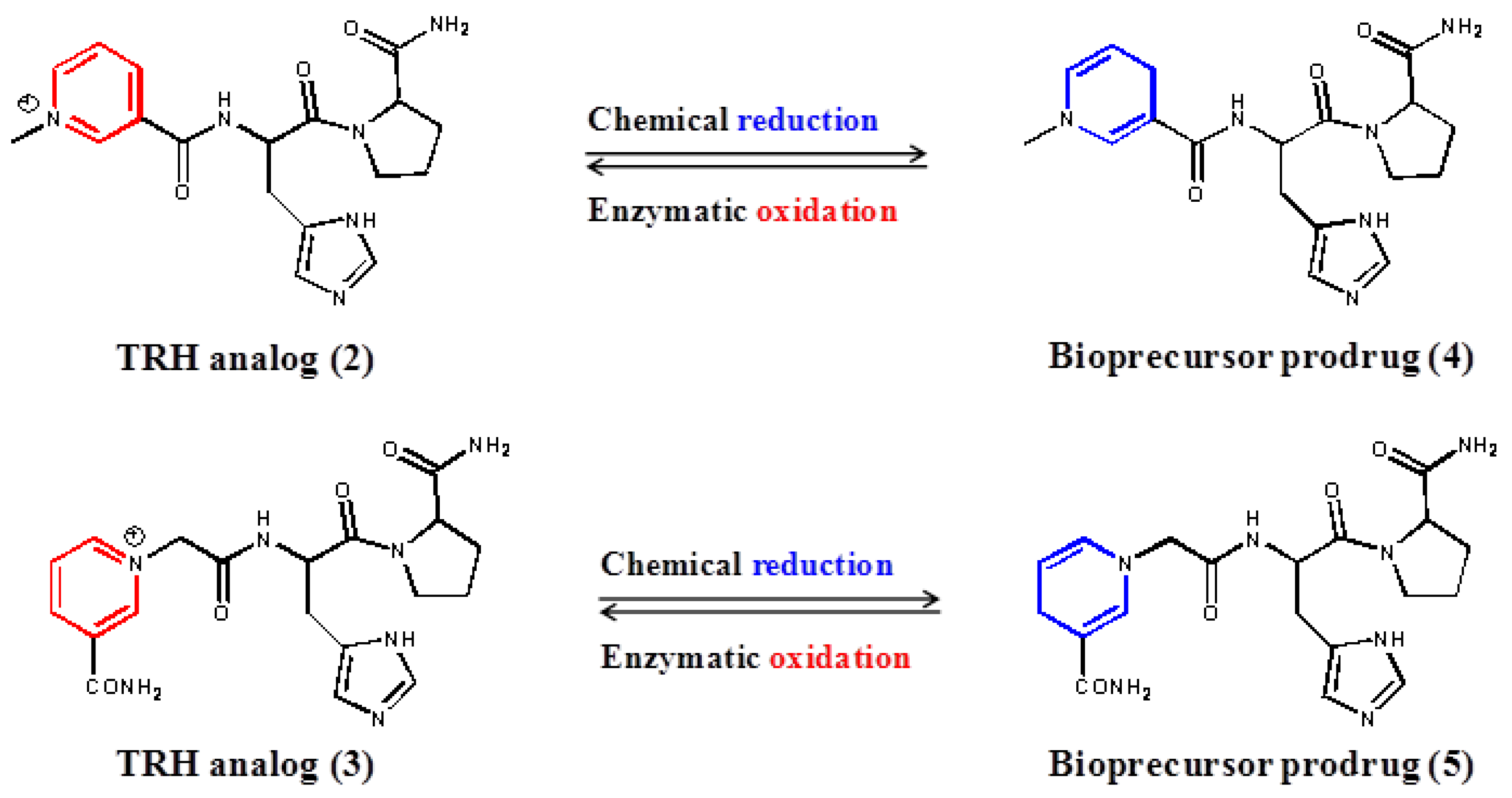
2. Experimental Section
2.1. Materials
2.2. Animals
2.3. Synthesis of Test Compounds
2.4. In Vitro Metabolic Stability
2.5. Analeptic Effect
2.6. Porsolt’s Swim Test (PST) to Assess Antidepressant-Like Activity
2.7. Data Analysis
3. Results and Discussion


{kind=link}
{kind=link}
{kind=link}
| Test agent | Immobility time (s) mean ± SEM | % Change in immobility time compared to control a | % Change in sleeping time compared to control a,b |
|---|---|---|---|
| Saline | 288 ± 6 | 0 | 0 |
| TRH | 184 ± 5 * | −36 | −51 * |
| Analog 2 | 279 ± 8 | ~0 | ~0 |
| Prodrug of 2 (4) | 231 ± 6 * | −19 | −46 * |
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Prokai, L.; Prokai-Tatrai, K.; Ouyang, X.; Kim, H.S.; Wu, W.M.; Zharikova, A.D.; Bodor, N. Metabolism-based brain-targeting system for a thyrotropin-releasing hormone analogue. J. Med. Chem. 1999, 42, 4563–4571. [Google Scholar]
- Prokai-Tatrai, K.; Perjési, P.; Zharikova, A.D.; Li, X.; Prokai, L. Design, synthesis and biological evaluation of novel, centrally-acting thyrotropin-releasing hormone analogues. Bioorg. Med. Chem. Lett. 2002, 12, 2171–2174. [Google Scholar]
- Prokai, L.; Prokai-Tatrai, K.; Zharikova, A.D.; Nguyen, V.; Stevens, S.M., Jr. Centrally acting and metabolically stable thyrotropin-releasing hormone analogues by replacement of histidine with substituted pyridinium. J. Med. Chem. 2004, 47, 6025–6033. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Prokai, L. Prodrugs of thyrotropin-releasing hormone and related peptides as central nervous system agents. Molecules 2009, 14, 633–654. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Prokai, L. Prodrug design for brain delivery of small- and medium-sized neuropeptides. Methods Mol. Biol. 2011, 789, 313–336. [Google Scholar] [CrossRef]
- Boler, J.; Enzmann, F.; Folkers, K.; Bowers, C.Y.; Schally, A.V. The identity of chemical and hormonal properties of the thyrotropin releasing hormone and pyroglutamyl-histidyl-proline amide. Biochem. Biophys. Res. Commun. 1969, 37, 705–710. [Google Scholar] [CrossRef]
- Prokai, L. Central nervous system effects of thyrotropin-releasing hormone and its analogues: Opportunities and perspectives for drug discovery and development. Prog. Drug Res. 2002, 59, 134–169. [Google Scholar]
- Kelly, J.A. Thyrotropin-releasing hormone: Basis and potential for its therapeutic use. Essays Biochem. 1995, 30, 133–149. [Google Scholar]
- Griffiths, E.C. Thyrotrophin releasing hormone: Endocrine and central effect. Psychoneuroendocrinology 1985, 10, 225–235. [Google Scholar]
- Monga, V.; Meena, C.L.; Kaur, N.; Jain, R. Chemistry and biology of thyrotropin-releasing hormone (TRH) and its analogs. Curr. Med. Chem. 2008, 15, 2718–2733. [Google Scholar] [CrossRef]
- Khomane, K.S.; Meena, C.L.; Jain, R.; Bansal, A.K. Novel thyrotropin-releasing hormone analogs: A patent review. Expert Opin. Ther. Pat. 2011, 21, 1673–1691. [Google Scholar]
- Kelly, J.A.; Slator, G.R.; Tipton, K.F.; Williams, C.H.; Bauer, K. Kinetic investigation of the specificity of porcine brain thyrotropin-releasing hormone-degrading ectoenzyme for thyrotropin-releasing hormone-like peptides. J. Biol. Chem. 2000, 275, 16746–16751. [Google Scholar]
- Zloković, B.V.; Lipovac, M.N.; Begley, D.J.; Davson, H.; Rakić, L. Slow penetration of thyrotropin-releasing hormone across the blood-brain barrier of an in situ perfused guinea pig brain. J. Neurochem. 1988, 51, 252–257. [Google Scholar]
- Scalabrino, G.A.; Hogan, N.; O’Boyle, K.M.; Slator, G.R.; Gregg, D.J.; Fitchett, C.M.; Draper, S.M.; Bennett, G.W.; Hinkle, P.M.; Bauer, K.; et al. Discovery of a dual action first-in-class peptide that mimics and enhances CNS-mediated actions of thyrotropin-releasing hormone. Neuropharmacology 2007, 52, 1472–1481. [Google Scholar]
- Szirtes, T.L.; Kisfaludi, L.; Pálosi, E.; Szporny, L. Synthesis of thyrotropin-releasing hormone analogues. 1. Complete dissociation of central nervous system effect from thyrotropin-releasing activity. J. Med. Chem. 1984, 27, 741–745. [Google Scholar] [CrossRef]
- Teixido, M.; Prokai-Tatrai, K.; Wang, X.; Nguyen, V.; Prokai, L. Exploratory neuropharmacological evaluation of a bridged thyrotropin-releasing hormone analogue. Brain Res. Bull. 2007, 73, 103–107. [Google Scholar] [CrossRef]
- Begley, D.J.; Brightman, M.W. Structural and functional aspects of the blood-brain barrier. Prog. Drug Res. 2003, 61, 39–78. [Google Scholar]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Kastin, A.J.; Pan, W. Peptide transport across the blood-brain barrier. Prog. Drug Res. 2003, 61, 79–100. [Google Scholar]
- Patel, M.M.; Goyal, B.R.; Bhadada, S.V.; Bhatt, J.S.; Amin, A.F. Getting into the brain: Approaches to enhance brain drug delivery. CNS Drugs 2009, 23, 35–58. [Google Scholar]
- De Boer, A.G.; Gaillard, P.J. Strategies to improve drug delivery across the blood-brain barrier. Clin. Pharmacokin. 2007, 46, 553–576. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, L. Modern methods for delivery of drugs across the blood-brain barrier. Adv. Drug Deliv. Rev. 2012, 64, 640–665. [Google Scholar] [CrossRef]
- Pavan, B.; Dalpiaz, A.; Ciliberti, N.; Biondi, C.; Manfredini, S.; Vertuani, S. Progress in drug delivery to the central nervous system by the prodrug approach. Molecules 2008, 13, 1035–1065. [Google Scholar] [CrossRef]
- Avdeef, A. Physicochemical profiling (solubility, permeability and charge state). Curr. Top. Med. Chem. 2001, 1, 277–351. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Prokai, L. Modifying peptide properties by prodrug design for enhanced transport into the CNS. Prog. Drug Res. 2003, 61, 155–188. [Google Scholar]
- Kokil, G.R.; Rewatkar, P.V. Bioprecursor prodrugs: Molecular modification of the active principle. Mini Rev. Med. Chem. 2010, 10, 1316–1330. [Google Scholar] [CrossRef]
- Rivier, J.; Vale, W.; Monahan, M.; Ling, N.; Burgus, R. Synthetic thyrotropin-releasing factor analogs. 3. Effect of replacement or modification of histidine residue on biological activity. J. Med. Chem. 1972, 15, 479–482. [Google Scholar] [CrossRef]
- Prokai, L.; Prokai-Tatrai, K.; Bodor, N. Targeting drugs to the brain by redox chemical delivery systems. Med. Res. Rev. 2000, 20, 367–416. [Google Scholar]
- Breese, G.; Cott, J.; Cooper, B.; Prange, A.; Lippton, M.; Plotnikoff, N. Effects of thyrotropin-releasing hormone (TRH) on the actions of pentobarbital and other centrally acting drugs. J. Pharmacol. Exp. Ther. 1975, 193, 11–22. [Google Scholar]
- Hinkle, P.M.; Pekary, A.E.; Senanayaki, S.; Sattin, A. Role of TRH receptors as possible mediators of analeptic actions of TRH-like peptides. Brain Res. 2002, 10, 59–64. [Google Scholar]
- Lloyd, R.L.; Pekary, A.E.; Sattin, A.; Amundson, T. Antidepressant effects of thyrotropin-releasing hormone analogues using a rodent model of depression. Pharmacol. Biochem. Behav. 2001, 70, 15–22. [Google Scholar] [CrossRef]
- Callahan, A.M.; Frye, M.A.; Marangell, L.B.; George, M.S.; Ketter, T.A.; L’Herrou, T.; Post, R.M. Comparative antidepressant effects of intravenous and intrathecal thyrotropin-releasing hormone: Confounding effects of tolerance and implications for therapeutics. Biol. Psychiatry 1997, 41, 264–272. [Google Scholar]
- Porsolt, R.D.; Le Pichon, M.; Jalfre, M. Depression: A new animal model sensitive to antidepressant treatment. Nature 1977, 266, 730–732. [Google Scholar]
- Schmidt, D. Effects of thyrotropin releasing hormone (TRH) on pentobarbital-induced decrease in cholinergic neuronal activity. Commun. Psychopharm. 1977, 1, 469–473. [Google Scholar]
- Pekary, U.A.; Faull, A.; Paulson, K.F.; Lloyd, R.L.; Sattin, A. TRH-like antidepressant peptide, pyroglutamyltyroslyprolineamide, occurs in rat brain. J. Mass Spectrom. 2005, 40, 1232–1236. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Nguyen, V.; Zharikova, A.D.; Braddy, A.C.; Stevens, S.M., Jr.; Prokai, L. Prodrugs to enhance central nervous system effects of the TRH-like peptide pGlu-Glu-Pro-NH2. Bioorg. Med. Chem. Lett. 2003, 13, 1011–1014. [Google Scholar]
- Prokai-Tatrai, K.; Teixido, M.; Nguyen, V.; Zharikova, A.D.; Prokai, L. A pyridinium-substituted analogue of the TRH-like tripeptide pGlu-Glu-Pro-NH2 and its prodrugs as central nervous system agents. Med. Chem. 2005, 2, 141–152. [Google Scholar]
- Brown, W. Taltirelin. Tanabe Seiyaku. IDrugs 1999, 2, 1059–1068. [Google Scholar]
- Prokai-Tatrai, K.; Szarka, S.; Nguyen, V.; Sahyouni, F.; Walker, C.; White, S.; Talamantes, T.; Prokai, L. “All in the mind”? Brain-targeting chemical delivery system of 17β-estradiol (Estredox) produces significant uterotrophic side effect. Pharm. Anal. Acta 2012, S7. [Google Scholar] [CrossRef]
- Ogawa, N.; Mizuno, S.; Mori, A.; Nukina, I.; Ota, Z.; Yamamoto, M. Potential antidepressive effects of thyrotropin releasing hormone (TRH) and its analogues. Peptides 1984, 5, 743–746. [Google Scholar] [CrossRef]
- Keppel, G.; Wickens, T.D. Design and Analysis: A Researcher’s Handbook, 4th ed; Pearson Prentice Hall: Upper Saddle River, NJ, USA, 2004. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Prokai-Tatrai, K.; Nguyen, V.; Szarka, S.; Konya, K.; Prokai, L. Design and Exploratory Neuropharmacological Evaluation of Novel Thyrotropin-Releasing Hormone Analogs and Their Brain-Targeting Bioprecursor Prodrugs. Pharmaceutics 2013, 5, 318-328. https://doi.org/10.3390/pharmaceutics5020318
Prokai-Tatrai K, Nguyen V, Szarka S, Konya K, Prokai L. Design and Exploratory Neuropharmacological Evaluation of Novel Thyrotropin-Releasing Hormone Analogs and Their Brain-Targeting Bioprecursor Prodrugs. Pharmaceutics. 2013; 5(2):318-328. https://doi.org/10.3390/pharmaceutics5020318
Chicago/Turabian StyleProkai-Tatrai, Katalin, Vien Nguyen, Szabolcs Szarka, Krisztina Konya, and Laszlo Prokai. 2013. "Design and Exploratory Neuropharmacological Evaluation of Novel Thyrotropin-Releasing Hormone Analogs and Their Brain-Targeting Bioprecursor Prodrugs" Pharmaceutics 5, no. 2: 318-328. https://doi.org/10.3390/pharmaceutics5020318
APA StyleProkai-Tatrai, K., Nguyen, V., Szarka, S., Konya, K., & Prokai, L. (2013). Design and Exploratory Neuropharmacological Evaluation of Novel Thyrotropin-Releasing Hormone Analogs and Their Brain-Targeting Bioprecursor Prodrugs. Pharmaceutics, 5(2), 318-328. https://doi.org/10.3390/pharmaceutics5020318