Timing and Duration of Drug Exposure Affects Outcomes of a Drug-Nutrient Interaction During Ontogeny

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals, Diet, and Chemicals
2.2. Chronic Cefepime Administration in Rat Pups
2.3. Free L-Carnitine Analysis in Serum and Heart
2.4. Total mRNA Isolation and Quantitative RT-PCR Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Accession Number | Primers | |
|---|---|---|---|
| Forward | Reverse | ||
| β-actin | NM_031144 | agcgtggctacagcttcacc | tgccacaggattccataccc |
| Octn1 | NM_022270 | catggctgtgcagactgg | gcaccatgtagccgatgg |
| Octn2 | NM_019269 | ggcgcaaccacagtatcc | ggggctttccagtcatcc |
| Octn3 | NM_019723 | gacaccgtgaacctgagc | ccatccaggcagttctcc |
| Cpt1b | NM_013200 | cagccatgccaccaagatc | aagggccgcacagaatcc |
| Cpt2 | NM_012930 | gctccgaggcgtttctca | tggccgttgccagatagc |
| Bbh | NM_022629 | acgatggggcagagtcc | ctggcctcctgagaaaagc |
| Tmlh | NM_133387 | aatgtccctcccactcagg | tcggtatggcgatctaggg |
2.5. Validation of the 2-ΔΔCT Method
2.6. Liver γ-Butyrobetaine Hydroxylase (Bbh) Enzyme Activity
2.7. Heart Carnitine Palmitoyltransferase (Cpt) Enzyme Activities
2.8. Heart High Energy Phosphate Substrate Determination
2.9. Histopathology of the Heart
2.10. Statistical Analysis
3. Results and Discussion
3.1. Body Weight Gain
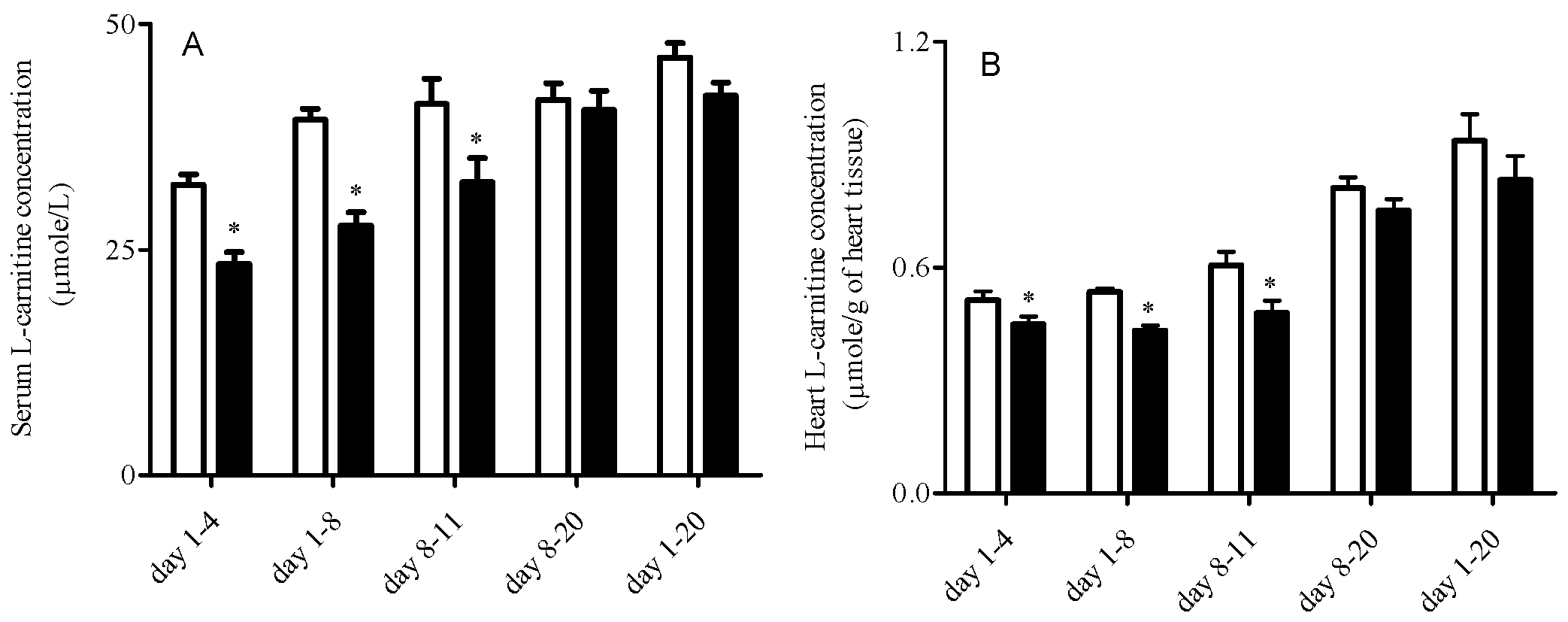
3.2. L-Carnitine Levels
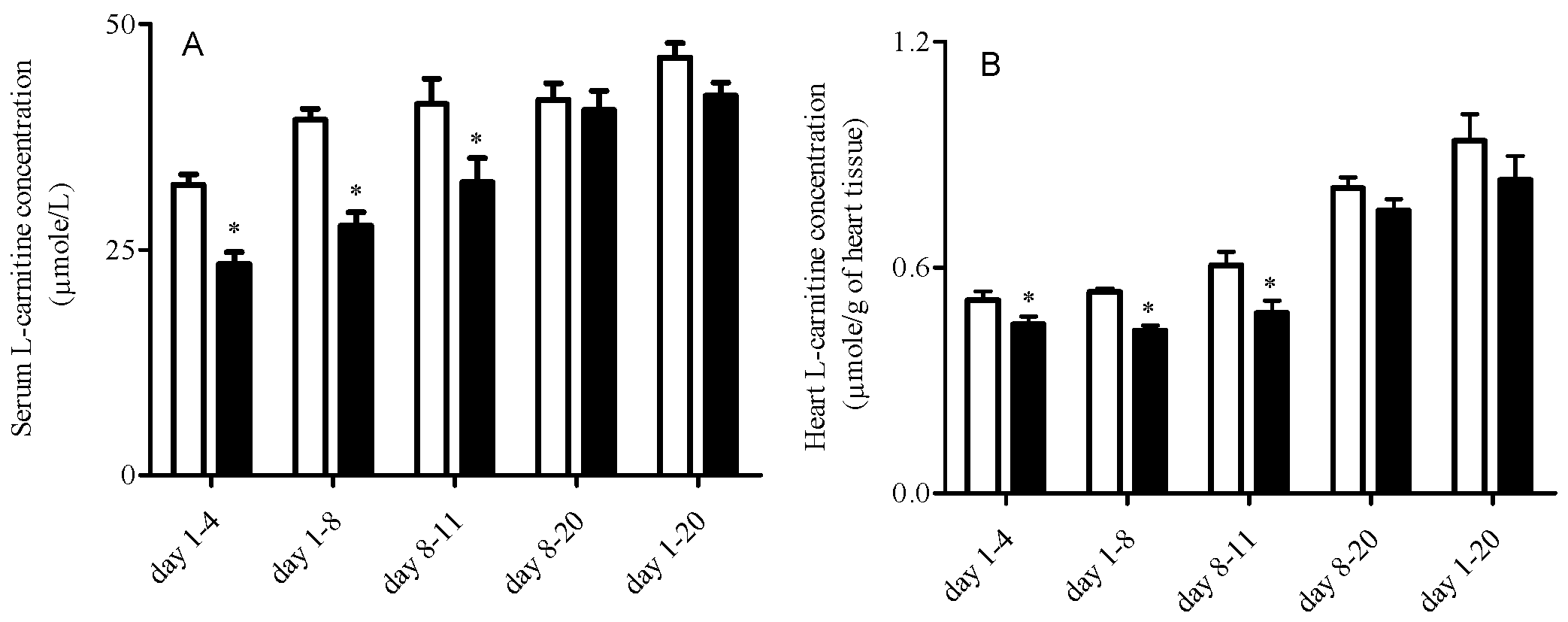
| Day 1-4 | Day 1-8 | Day 8-11 | Day 8-20 | Day 1-20 | |
|---|---|---|---|---|---|
| Kidney Octn2 | 1.27 ± 0.10* | 1.17 ± 0.15 | 0.98 ± 0.14 | 2.24 ± 0.37* | 1.24 ± 0.05* |
| Intestinal Octn1 | 0.99 ± 0.09 | 0.90 ± 0.09 | 1.89 ± 0.33* | 0.84 ± 0.08 | 1.80 ± 0.16* |
| Intestinal Octn2 | 1.18 ± 0.28 | 1.06 ± 0.16 | 1.22 ± 0.10* | 1.09 ± 0.17 | 0.76 ± 0.21 |
| Intestinal Octn3 | 1.39 ± 0.50 | 1.84 ± 0.26* | 1.02 ± 0.17 | 2.21 ± 0.89 | 1.51 ± 0.12* |
| Heart Octn2 | 0.99 ± 0.03 | 0.88 ± 0.13 | 0.95 ± 0.14 | 1.09 ± 0.12 | 0.92 ± 0.10 |
| Liver Bbh | 1.15 ± 0.56 | 0.90 ± 0.26 | 0.89 ± 0.20 | 0.73 ± 0.22 | 0.90 ± 0.33 |
| Liver Tmlh | 0.82 ± 0.18 | 1.58 ± 0.28* | 1.12 ± 0.12 | 1.28 ± 0.09 | 1.18 ± 0.21 |
| Heart Cpt1b | 1.02 ± 0.17 | 1.03 ± 0.15 | 1.34 ± 0.22 | 1.00 ± 0.13 | 0.83 ± 0.09 |
| Heart Cpt2 | 1.19 ± 0.12 | 0.97 ± 0.07 | 1.32 ± 0.16 | 1.15 ± 0.18 | 1.10 ± 0.09 |
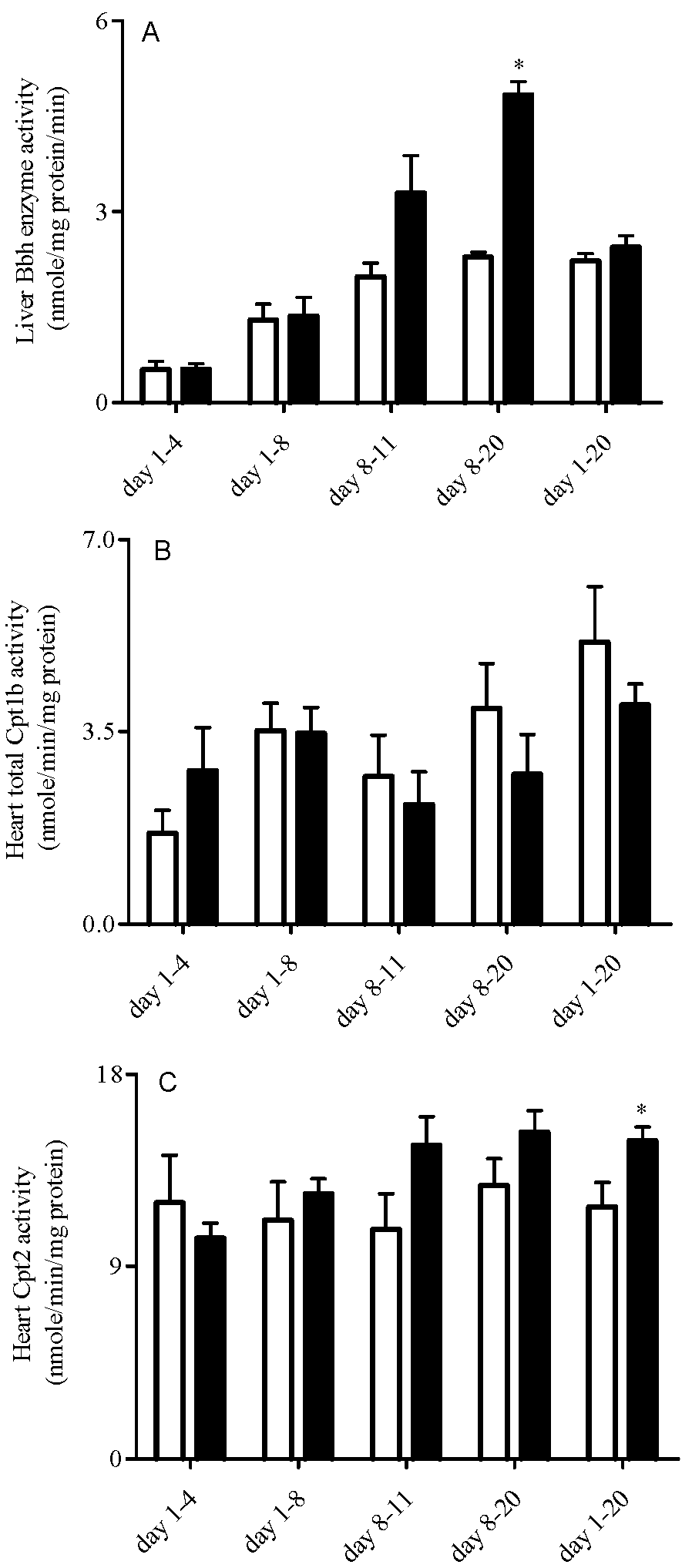
3.4. Liver Bbh and Tmlh and Heart Cpt Enzyme Expression and Activity
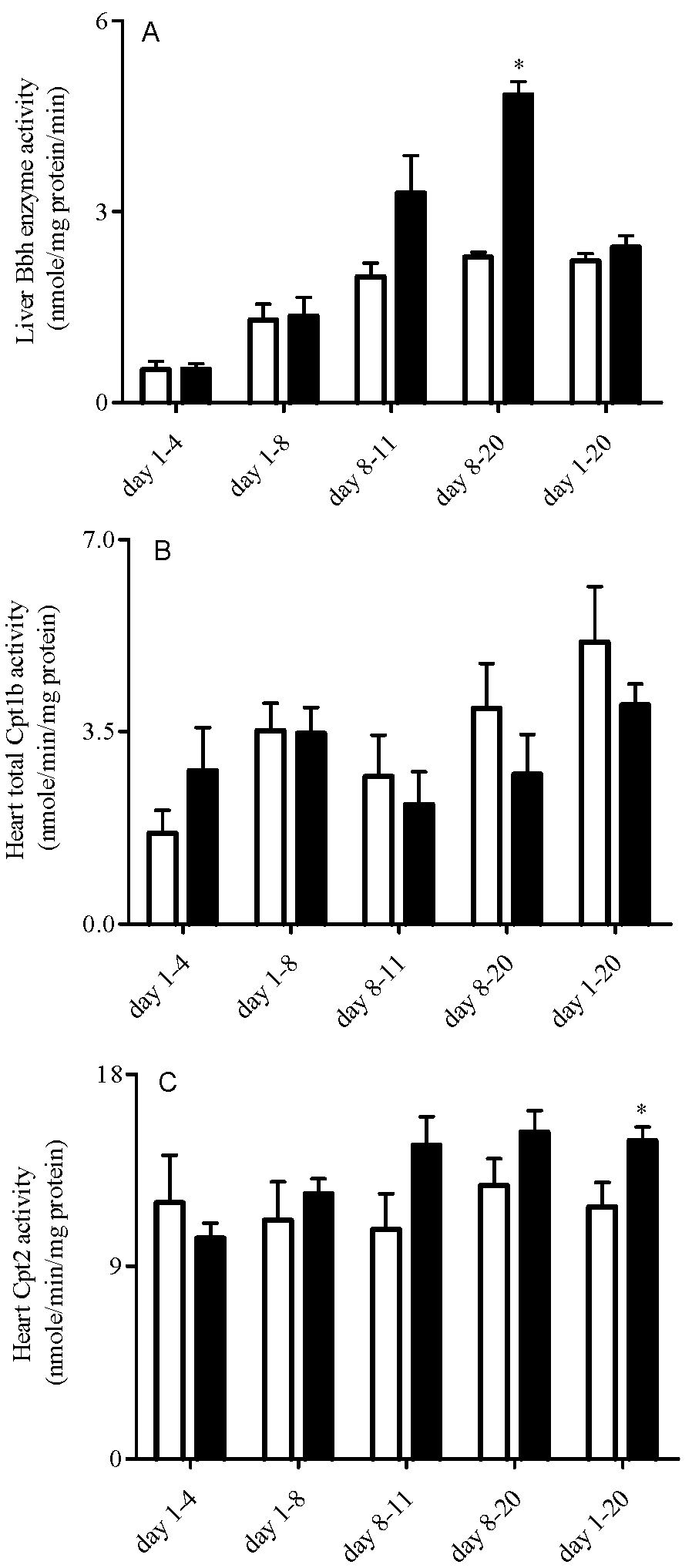
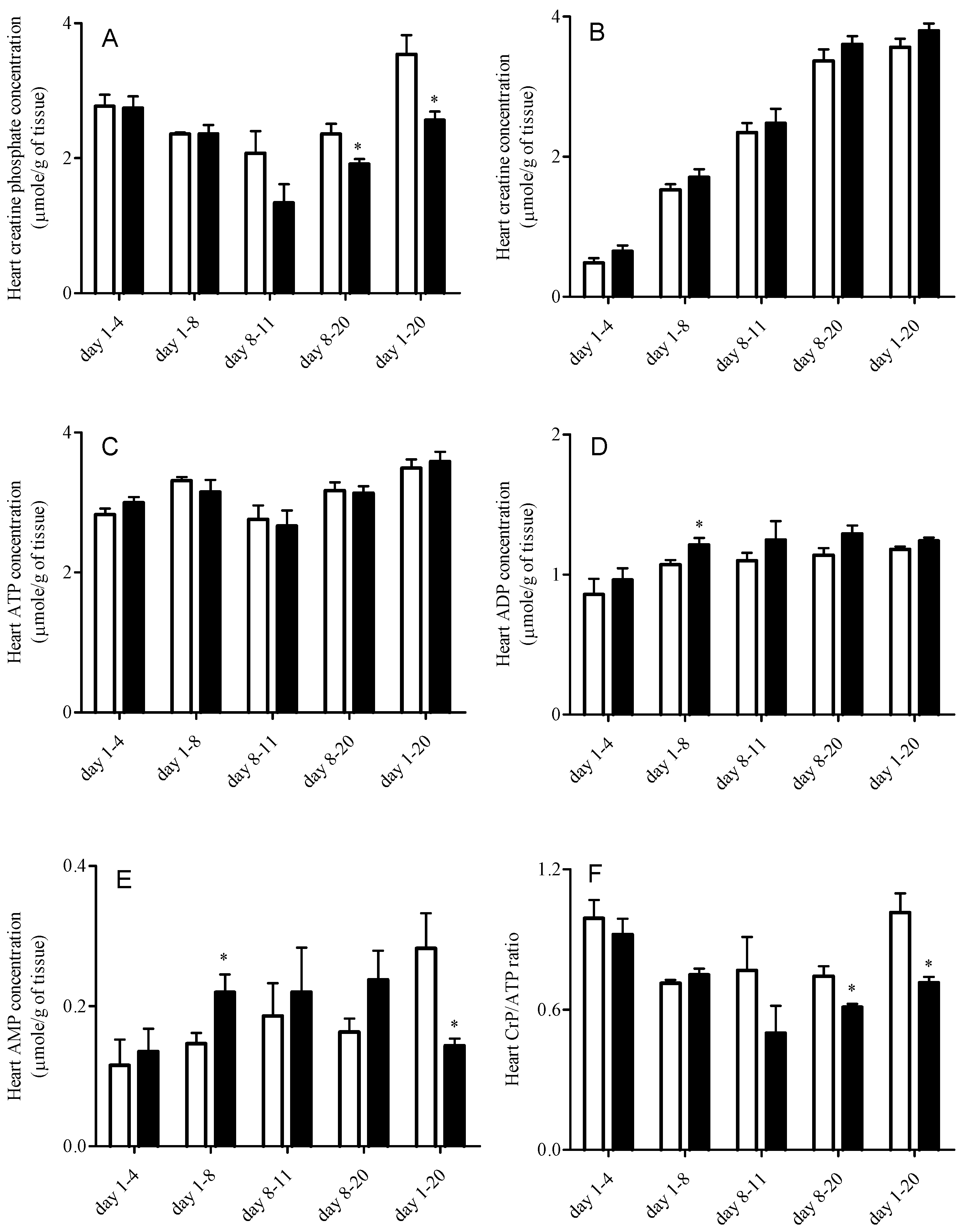
3.5. Heart High Energy Phosphate Substrate Levels

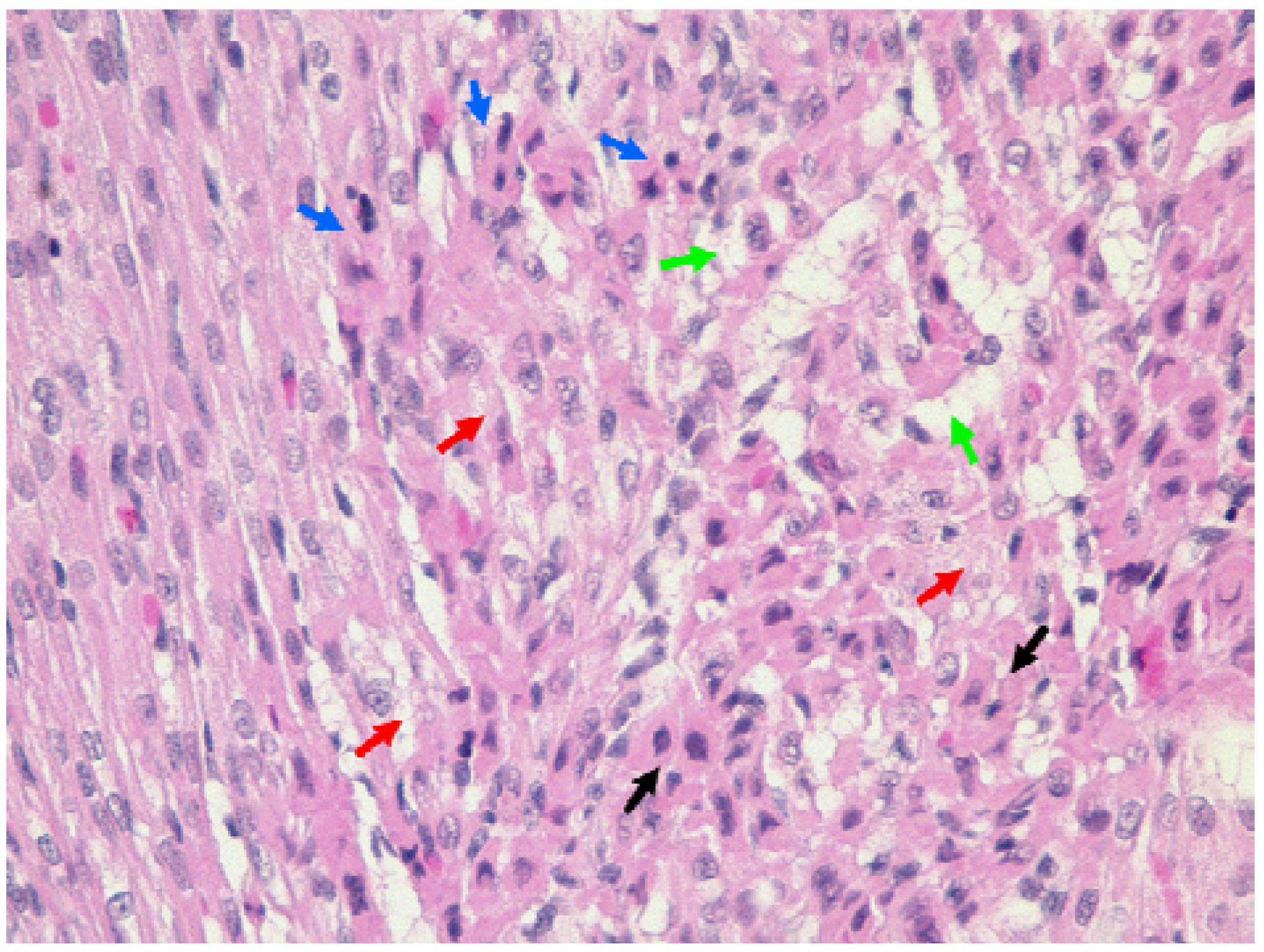
3.6. Histological Analysis
| Day 1-4 | Day 1-8 | Day 8-11 | Day 8-20 | Day 1-20 | |
|---|---|---|---|---|---|
| Control | 1.67 ± 0.49 | 2.17 ± 0.31 | 1.17 ± 0.40 | 1.33 ± 0.42 | 1.00 ± 0.52 |
| Treated | 2.50 ± 0.34 | 2.83 ± 0.17 | 2.17 ± 0.31 | 1.17 ± 0.40 | 0.33 ± 0.33 |
| P value | 0.196 | 0.086 | 0.076 | 0.781 | 0.304 |

3.7. Correlation Analysis
| Parameters | Control Animals | Treated Animals | ||
|---|---|---|---|---|
| Pearson Correlation Coefficient | P value | Pearson Correlation Coefficient | P value | |
| Serum L-carnitine | 0.642 | 0.000 | 0.861 | P < 0.0001 |
| Heart L-carnitine | 0.840 | P < 0.0001 | 0.846 | P < 0.0001 |
| Kidney Octn2 mRNA | 0.369 | 0.045 | 0.665 | P < 0.0001 |
| Heart Octn2 mRNA | 0.827 | P < 0.0001 | 0.879 | P < 0.0001 |
| Intestinal Octn1 mRNA | 0.305 | 0.101 | 0.370 | 0.044 |
| Intestinal Octn2 mRNA | -0.465 | 0.010 | 0.083 | 0.663 |
| Intestinal Octn3 mRNA | 0.803 | P < 0.0001 | 0.098 | 0.608 |
| Liver Bbh activity | 0.417 | 0.022 | 0.707 | P < 0.0001 |
| Liver Bbh mRNA | 0.034 | 0.857 | 0.387 | 0.035 |
| Liver Tmlh mRNA | 0.047 | 0.807 | -0.037 | 0.848 |
| CrP | 0.248 | 0.186 | -0.168 | 0.375 |
| Cr | 0.956 | P < 0.0001 | 0.956 | P < 0.0001 |
| ATP | 0.421 | 0.021 | 0.358 | 0.052 |
| ADP | 0.533 | 0.002 | 0.419 | 0.021 |
| AMP | 0.415 | 0.023 | 0.093 | 0.627 |
| Cpt1 activity | 0.564 | 0.001 | 0.268 | 0.152 |
| Cpt2 activity | 0.086 | 0.653 | 0.603 | 0.000 |
| Heart Cpt1b mRNA | 0.692 | P < 0.0001 | 0.382 | 0.037 |
| Heart Cpt2 mRNA | 0.838 | P < 0.0001 | 0.901 | P < 0.0001 |
3.8. Discussion
4. Conclusion
References
- Gluckman, P.D.; Hanson, M.A.; Spencer, H.G.; Bateson, P. Environmental influences during development and their later consequences for health and disease: implications for the interpretation of empirical studies. Proc. Roy. Soc. Biol. Sci. 2005, 272, 671–677. [Google Scholar] [CrossRef]
- Patel, M.S.; Srinivasan, M. Metabolic programming: causes and consequences. J. Biol. Chem. 2002, 277, 1629–1632. [Google Scholar] [CrossRef]
- Gurr, M.I. Individuals can or do adapt their metabolism to changes in diet. Br. J. Nutr. 1989, 62, 1–3. [Google Scholar] [CrossRef]
- Waterlow, J.C. Nutritional adaptation in man: general introduction and concepts. Am. J. Clin. Nutr. 1990, 51, 259–263. [Google Scholar]
- Symonds, M.E.; Pearce, S.; Bispham, J.; Gardner, D.S.; Stephenson, T. Timing of nutrient restriction and programming of fetal adipose tissue development. Proc. Nutr. Soc. 2004, 63, 397–403. [Google Scholar] [CrossRef]
- Waterlow, J.C. The nature and significance of nutritional adaptation. Eur. J. Clin. Nutr. 1999, 53, S2–S5. [Google Scholar]
- Kimmel, G.L. An overview of children as a special population--relevance to predictive biomarkers. Toxicol. Appl. Pharmacol. 2005, 206, 215–218. [Google Scholar] [CrossRef]
- Blake, M.J.; Abdel-Rahman, S.M.; Pearce, R.E.; Leeder, J.S.; Kearns, G.L. Effect of diet on the development of drug metabolism by cytochrome P-450 enzymes in healthy infants. Pediatr. Res. 2006, 60, 717–723. [Google Scholar] [CrossRef]
- Genser, D. Food and drug interaction: consequences for the nutrition/health status. Ann. Nutr. Metab. 2008, 52, 29–32. [Google Scholar] [CrossRef]
- Hines, R.N. The ontogeny of drug metabolism enzymes and implications for adverse drug events. Pharmacol. Ther. 2008, 118, 250–267. [Google Scholar] [CrossRef]
- Karasov, W.H.; Solberg, D.H.; Chang, S.D.; Hughes, M.; Stein, E.D.; Diamond, J.M. Is intestinal transport of sugars and amino acids subject to critical-period programming? Amer. J. Physiol. 1985, 249, G770–G785. [Google Scholar]
- Patel, M.S.; Srinivasan, M.; Laychock, S.G. Metabolic programming: Role of nutrition in the immediate postnatal life. J. Inherit. Metab. Dis. 2009, 32, 218–228. [Google Scholar] [CrossRef]
- Godarova, A.; Litzlbauer, E.; Brunner, S.; Agu, A.C.; Lohninger, A.; Hofbauer, R. L-Carnitine Regulates mRNA ExpressionLevels of the Carnitine Acyltransferases –CPT1A, CPT2, and CRAT. Chem. Monthly 2005, 136, 1349–1363. [Google Scholar] [CrossRef]
- Santos, C.A.; Boullata, J.I. An approach to evaluating drug-nutrient interactions. Pharmacotherapy 2005, 25, 1789–1800. [Google Scholar] [CrossRef]
- Chan, L.N. Drug-Nutrient Interactions. In Modern Nutrition in Health and Disease; Shils, M.E., Shike, M., Ross, A.C., Caballero, B., Cousins, R.J., Eds.; Lippincott Williams & Wilkins.: Baltimore, MD, USA, 2006; pp. 1540–1553. [Google Scholar]
- Bailey, D.G. Grapefruit and Other Fruit Juices Interactions with Medicines. In Handbook of Drug-Nutrient Interactions, 2nd ed.; Boullata, J.I., Armenti, V.A., Eds.; Humana Press: Totowa, NJ, USA, 2010; pp. 267–302. [Google Scholar]
- Moretti, M.E.; Caprara, D.L. Drug–Nutrient Interaction Considerations in Pregnancy and Lactation. In Handbook of Drug-Nutrient Interactions, 2nd ed.; Boullata, J.I., Armenti, V.A., Eds.; Humana Press: Totowa, NJ, USA, 2010; pp. 593–616. [Google Scholar]
- Felipez, L.; Sentongo, T.A. Drug-induced nutrient deficiencies. Pediatr. Clin. North Am. 2009, 56, 1211–1224. [Google Scholar] [CrossRef]
- Yan, Q.; Sadee, W. Human membrane transporter database: a Web-accessible relational database for drug transport studies and pharmacogenomics. AAPS Pharm. Sci. 2000, 2, E20. [Google Scholar]
- Hahn, P. The development of carnitine synthesis from gamma-butyrobetaine in the rat. Life Sci. 1981, 28, 1057–1060. [Google Scholar] [CrossRef]
- Sharma, S.; Black, S.M. Carnitine homeostasis, mitochondrial function and cardiovascular disease. Drug Discov. Today: Dis. Mech. 2009, in press. [Google Scholar]
- Alnouti, Y.; Petrick, J.S.; Klaassen, C.D. Tissue distribution and ontogeny of organic cation transporters in mice. Drug Metab. Disposition 2006, 34, 477–482. [Google Scholar]
- Bressler, R.; Brendel, K. The role of carnitine and carnitine acyltransferase in biological acetylations and fatty acid synthesis. J. Biol. Chem. 1966, 241, 4092–4097. [Google Scholar]
- Arenas, J.; Rubio, J.C.; Martin, M.A.; Campos, Y. Biological roles of L-carnitine in perinatal metabolism. Early Hum. Dev. 1998, 53, S43–S50. [Google Scholar] [CrossRef]
- Onay-Besikci, A.; Campbell, F.M.; Hopkins, T.A.; Dyck, J.R.; Lopaschuk, G.D. Relative importance of malonyl CoA and carnitine in maturation of fatty acid oxidation in newborn rabbit heart. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H283–H289. [Google Scholar]
- Ganapathy, M.E.; Huang, W.; Rajan, D.P.; Carter, A.L.; Sugawara, M.; Iseki, K.; Leibach, F.H.; Ganapathy, V. beta-lactam antibiotics as substrates for OCTN2, an organic cation/carnitine transporter. J. Biol. Chem. 2000, 275, 1699–1707. [Google Scholar] [CrossRef]
- Kano, T.; Kato, Y.; Ito, K.; Ogihara, T.; Kubo, Y.; Tsuji, A. Carnitine/organic cation transporter OCTN2 (Slc22a5) is responsible for renal secretion of cephaloridine in mice. Drug Metab. Disposition 2009, 37, 1009–1016. [Google Scholar] [CrossRef]
- Ling, B.; Alcorn, J. Acute administration of cefepime lowers L-carnitine concentrations in early lactation stage rat milk. J. Nutr. 2008, 138, 1317–1322. [Google Scholar]
- Galland, S.; Le Borgne, F.; Bouchard, F.; Georges, B.; Clouet, P.; Grand-Jean, F.; Demarquoy, J. Molecular cloning and characterization of the cDNA encoding the rat liver gamma-butyrobetaine hydroxylase. Biochim. Biophys. Acta 1999, 1441, 85–92. [Google Scholar]
- Feng, Y.; Zhang, S.; Xu, Y.; Zhoum, J.; Tao, Y.; Cai, W. Study on determination of carnitine in serum by HPLC. Acta Nutrimenta Sin. 2006, 28, 177–179. [Google Scholar]
- Bieber, L.L.; Abraham, T.; Helmrath, T. A rapid spectrophotometric assay for carnitine palmitoyltransferase. Anal. Biochem. 1972, 50, 509–518. [Google Scholar] [CrossRef]
- Alhomida, A.S. Evaluation of theophylline-stimulated changes in carnitine palmitoyltransferase activity in skeletal muscle and liver of rats. J. Enzym. Inhib. 2001, 16, 177–183. [Google Scholar] [CrossRef]
- Yu, X. X.; Odle, J.; Drackley, J.K. Differential induction of peroxisomal beta-oxidation enzymes by clofibric acid and aspirin in piglet tissues. Amer. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R1553–R1561. [Google Scholar]
- Olkowski, A.A.; Nain, S.; Wojnarowicz, C.; Laarveld, B.; Alcorn, J.; Ling, B.B. Comparative study of myocardial high energy phosphate substrate content in slow and fast growing chicken and in chickens with heart failure and ascites. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2007, 148, 230–238. [Google Scholar] [CrossRef]
- Bartelink, I.H.; Rademaker, C.M.; Schobben, A.F.; van den Anker, J.N. Guidelines on paediatric dosing on the basis of developmental physiology and pharmacokinetic considerations. Clin. Pharmacokinet. 2006, 45, 1077–1097. [Google Scholar] [CrossRef]
- Brass, E.P.; Hoppel, C.L.; Hiatt, W.R. Effect of intravenous L-carnitine on carnitine homeostasis and fuel metabolism during exercise in humans. Clin. Pharmacol. Ther. 1994, 55, 681–692. [Google Scholar] [CrossRef]
- Knapp, A.C.; Todesco, L.; Torok, M.; Beier, K.; Krahenbuhl, S. Effect of carnitine deprivation on carnitine homeostasis and energy metabolism in mice with systemic carnitine deficiency. Ann. Nutr. Metab. 2008, 52, 136–144. [Google Scholar] [CrossRef]
- Srinivasan, M.; Laychock, S.G.; Hill, D.J.; Patel, M.S. Neonatal nutrition: metabolic programming of pancreatic islets and obesity. Exp. Biol. Med. (Maywood) 2003, 228, 15–23. [Google Scholar]
- Amat di San Filippo, C.; Taylor, M.R.; Mestroni, L.; Botto, L.D.; Longo, N. Cardiomyopathy and carnitine deficiency. Mol. Genet. Metab. 2008, 94, 162–166. [Google Scholar] [CrossRef]
- Rebouche, C.J. Kinetics, pharmacokinetics, and regulation of L-carnitine and acetyl-L-carnitine metabolism. Ann. N. Y. Acad. Sci. 2004, 1033, 30–41. [Google Scholar] [CrossRef]
- Flores, C.A.; Hu, C.; Edmond, J.; Koldovsky, O. Milk carnitine affects organ carnitine concentration in newborn rats. J. Nutr. 1996, 126, 1673–1682. [Google Scholar]
- Rebouche, C.J.; Lombard, K.A.; Chenard, C.A. Renal adaptation to dietary carnitine in humans. Am. J. Clin. Nutr. 1993, 58, 660–665. [Google Scholar]
- Tamai, I.; China, K.; Sai, Y.; Kobayashi, D.; Nezu, J.; Kawahara, E.; Tsuji, A. Na(+)-coupled transport of L-carnitine via high-affinity carnitine transporter OCTN2 and its subcellular localization in kidney. Biochim. Biophys. Acta 2001, 1512, 273–284. [Google Scholar]
- Dyer, J.; Barker, P.J.; Shirazi-Beechey, S.P. Nutrient regulation of the intestinal Na+/glucose co-transporter (SGLT1) gene expression. Biochem. Biophys. Res. Commun. 1997, 230, 624–629. [Google Scholar] [CrossRef]
- Dyer, J.; Vayro, S.; King, T.P.; Shirazi-Beechey, S.P. Glucose sensing in the intestinal epithelium. Eur. J. Biochem. 2003, 270, 3377–3388. [Google Scholar] [CrossRef]
- Hyatt, S.L.; Aulak, K.S.; Malandro, M.; Kilberg, M.S.; Hatzoglou, M. Adaptive regulation of the cationic amino acid transporter-1 (Cat-1) in Fao cells. J. Biol. Chem. 1997, 272, 19951–19957. [Google Scholar]
- Vega, Y.M.; Puchal, A.A.; Buddington, R.K. Intestinal amino acid and monosaccharide transport in suckling pigs fed milk replacers with different sources of carbohydrate. J. Nutr. 1992, 122, 2430–2439. [Google Scholar]
- Stefanovic-Racic, M.; Perdomo, G.; Mantell, B.S.; Sipula, I.J.; Brown, N.F.; O'Doherty, R.M. A moderate increase in carnitine palmitoyltransferase 1a activity is sufficient to substantially reduce hepatic triglyceride levels. Amer. J. Physiol. Endocrinol. Metab. 2008, 294, E969–E977. [Google Scholar] [CrossRef]
- Paulson, D.J. Carnitine deficiency-induced cardiomyopathy. Mol. Cell. Biochem. 1998, 180, 33–41. [Google Scholar] [CrossRef]
- Weiss, R.G.; Bottomley, P.A.; Hardy, C.J.; Gerstenblith, G. Regional myocardial metabolism of high-energy phosphates during isometric exercise in patients with coronary artery disease. N. Engl. J. Med. 1990, 323, 1593–1600. [Google Scholar] [CrossRef]
- Neubauer, S.; Horn, M.; Cramer, M.; Harre, K.; Newell, J.B.; Peters, W.; Pabst, T.; Ertl, G.; Hahn, D.; Ingwall, J.S.; Kochsiek, K. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation 1997, 96, 2190–2196. [Google Scholar] [CrossRef]
- Cederbaum, S.D.; Koo-McCoy, S.; Tein, I.; Hsu, B.Y.; Ganguly, A.; Vilain, E.; Dipple, K.; Cvitanovic-Sojat, L.; Stanley, C. Carnitine membrane transporter deficiency: a long-term follow up and OCTN2 mutation in the first documented case of primary carnitine deficiency. Mol. Genet. Metab. 2002, 77, 195–201. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ling, B.; Aziz, C.; Wojnarowicz, C.; Olkowski, A.; Alcorn, J. Timing and Duration of Drug Exposure Affects Outcomes of a Drug-Nutrient Interaction During Ontogeny. Pharmaceutics 2010, 2, 321-338. https://doi.org/10.3390/pharmaceutics2040321
Ling B, Aziz C, Wojnarowicz C, Olkowski A, Alcorn J. Timing and Duration of Drug Exposure Affects Outcomes of a Drug-Nutrient Interaction During Ontogeny. Pharmaceutics. 2010; 2(4):321-338. https://doi.org/10.3390/pharmaceutics2040321
Chicago/Turabian StyleLing, Binbing, Caroline Aziz, Chris Wojnarowicz, Andrew Olkowski, and Jane Alcorn. 2010. "Timing and Duration of Drug Exposure Affects Outcomes of a Drug-Nutrient Interaction During Ontogeny" Pharmaceutics 2, no. 4: 321-338. https://doi.org/10.3390/pharmaceutics2040321
APA StyleLing, B., Aziz, C., Wojnarowicz, C., Olkowski, A., & Alcorn, J. (2010). Timing and Duration of Drug Exposure Affects Outcomes of a Drug-Nutrient Interaction During Ontogeny. Pharmaceutics, 2(4), 321-338. https://doi.org/10.3390/pharmaceutics2040321