
Enhanced Anticancer Potential of Pd(II)-Thiosemicarbazone Complexes: Selectivity, Mechanisms, and 3D Models

 , , , , , , and
, , , , , , and
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Physical Measurements
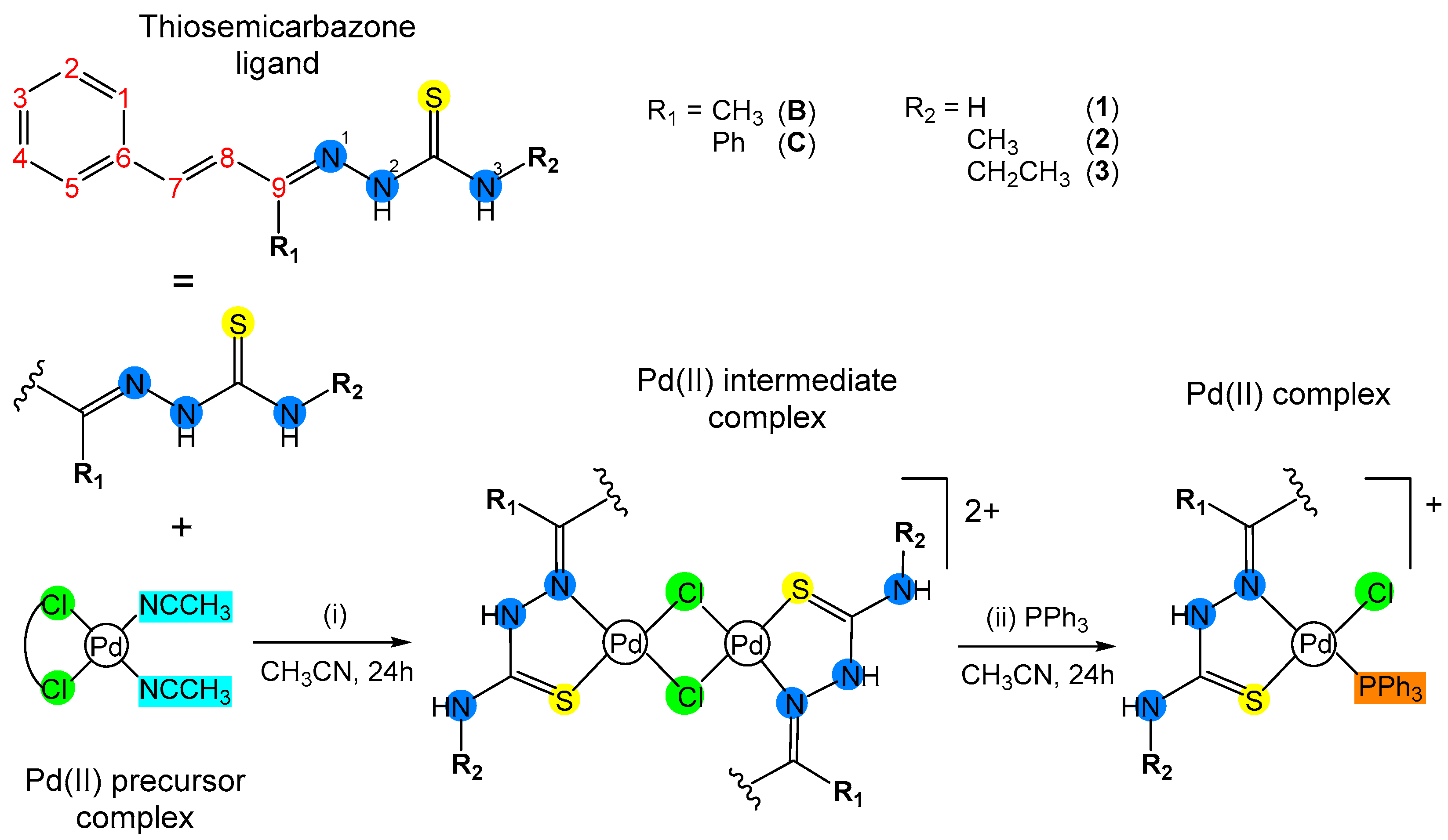
2.3. Synthesis of the Complexes
2.4. Cell Culture Tests (In Vitro)
2.5. Cell Viability (IC50)
2.6. Clonogenic Assay
2.7. Cell Morphology Assay
2.8. Cell Viability (Kit: LIVE/DEAD®)
2.9. Cell Migration Assay—“Wound Healing”
2.10. Cell Cycle Analysis
2.11. Apoptosis Assay
2.12. Three-Dimensional Assay
3. Results and Discussion
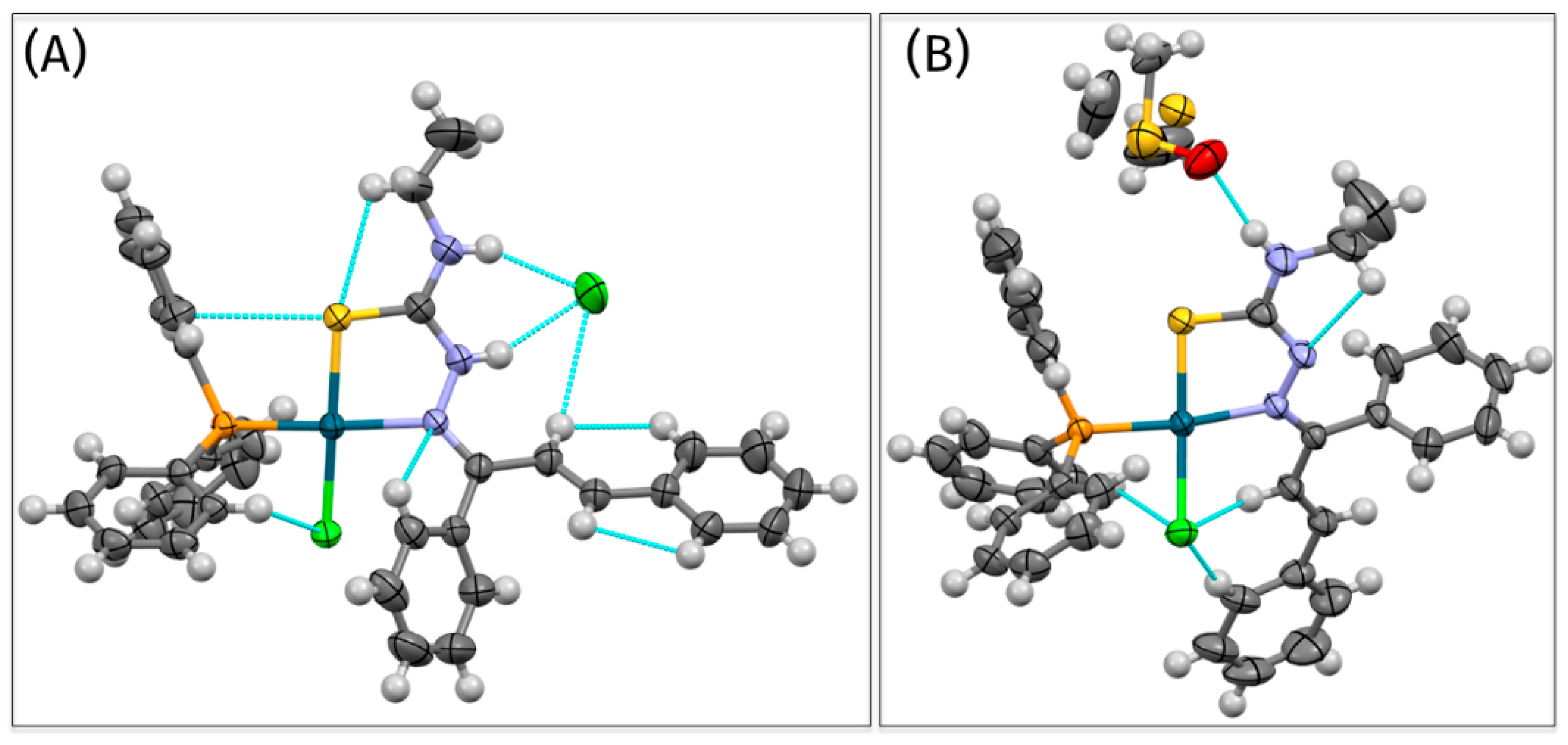
3.1. Synthesis and Characterization
3.2. Cell Tests
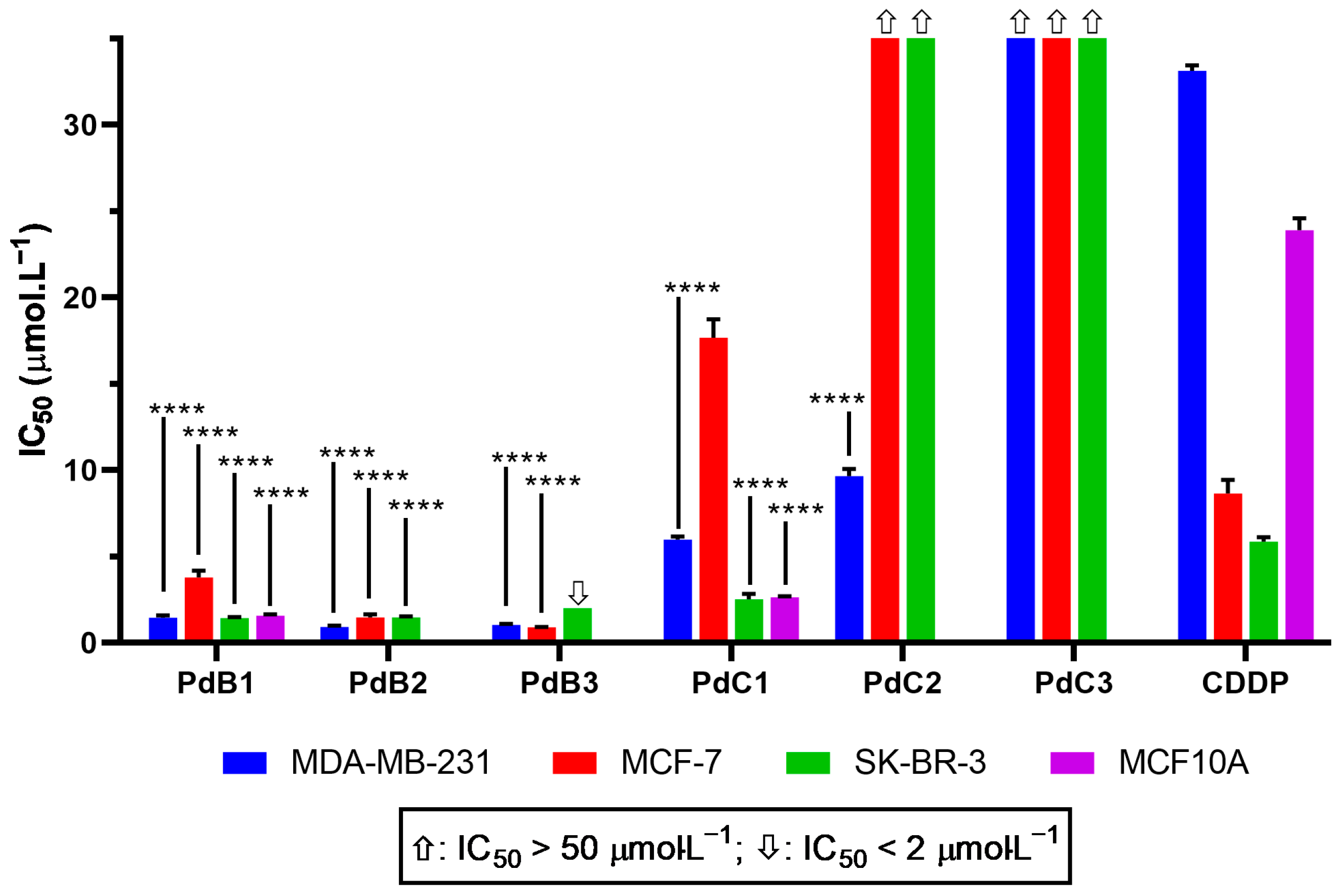
3.2.1. Cell Viability (IC50)
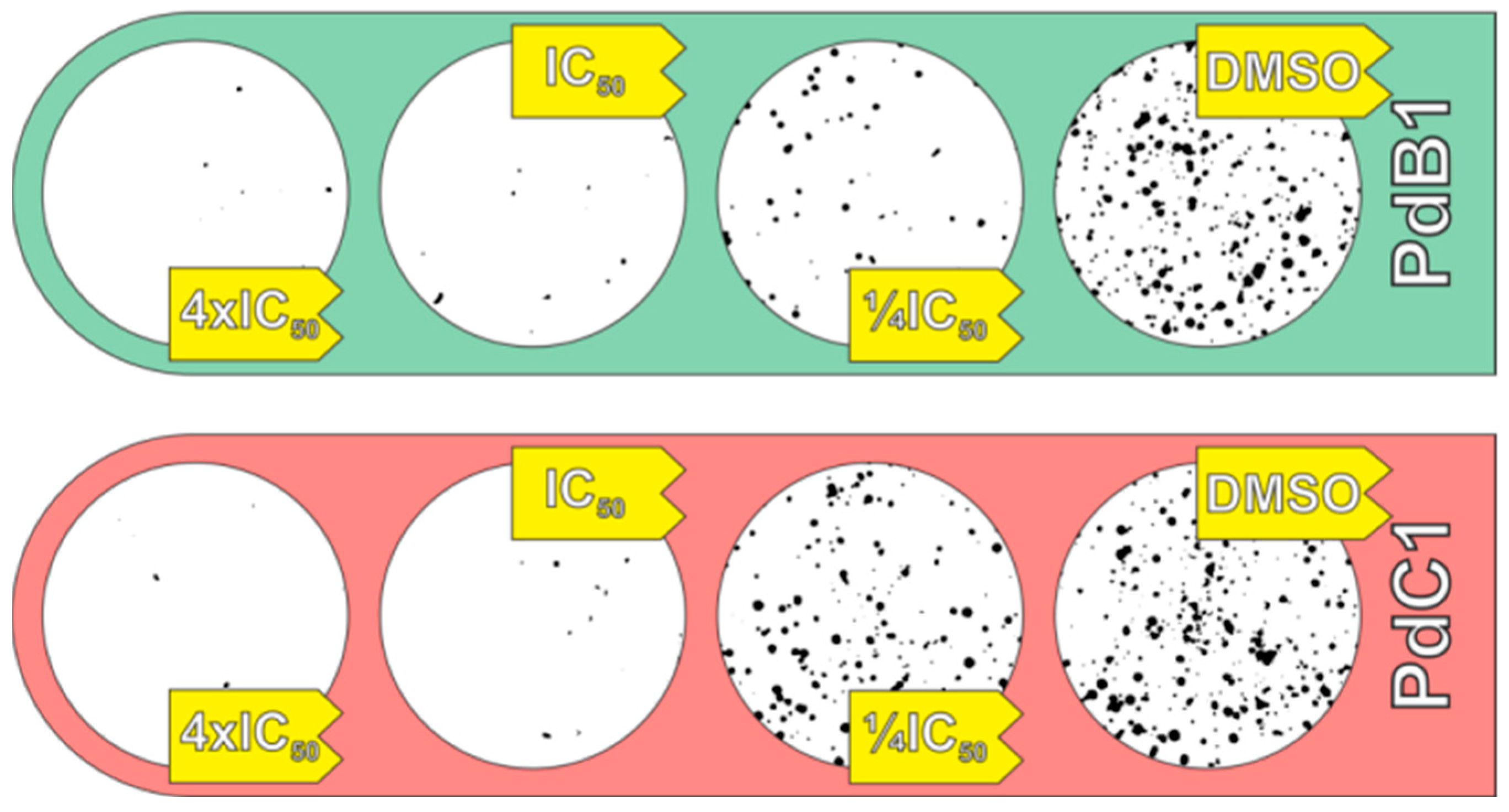
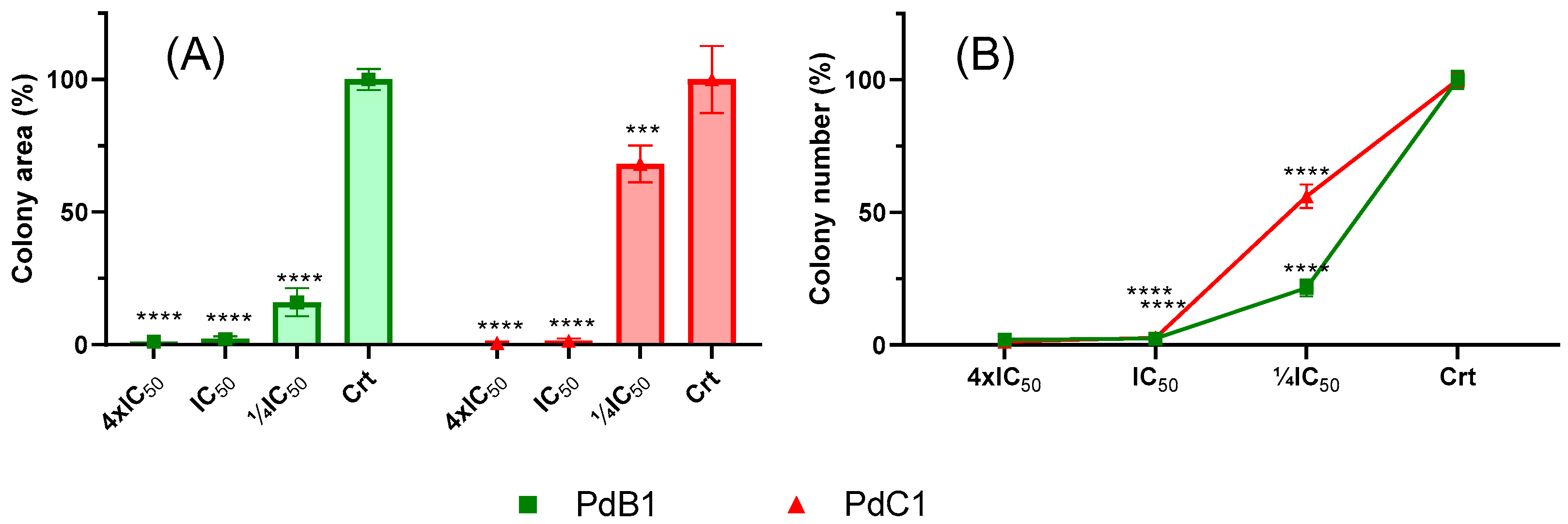
3.2.2. Clonogenic Assay
3.2.3. Cell Morphology Assay
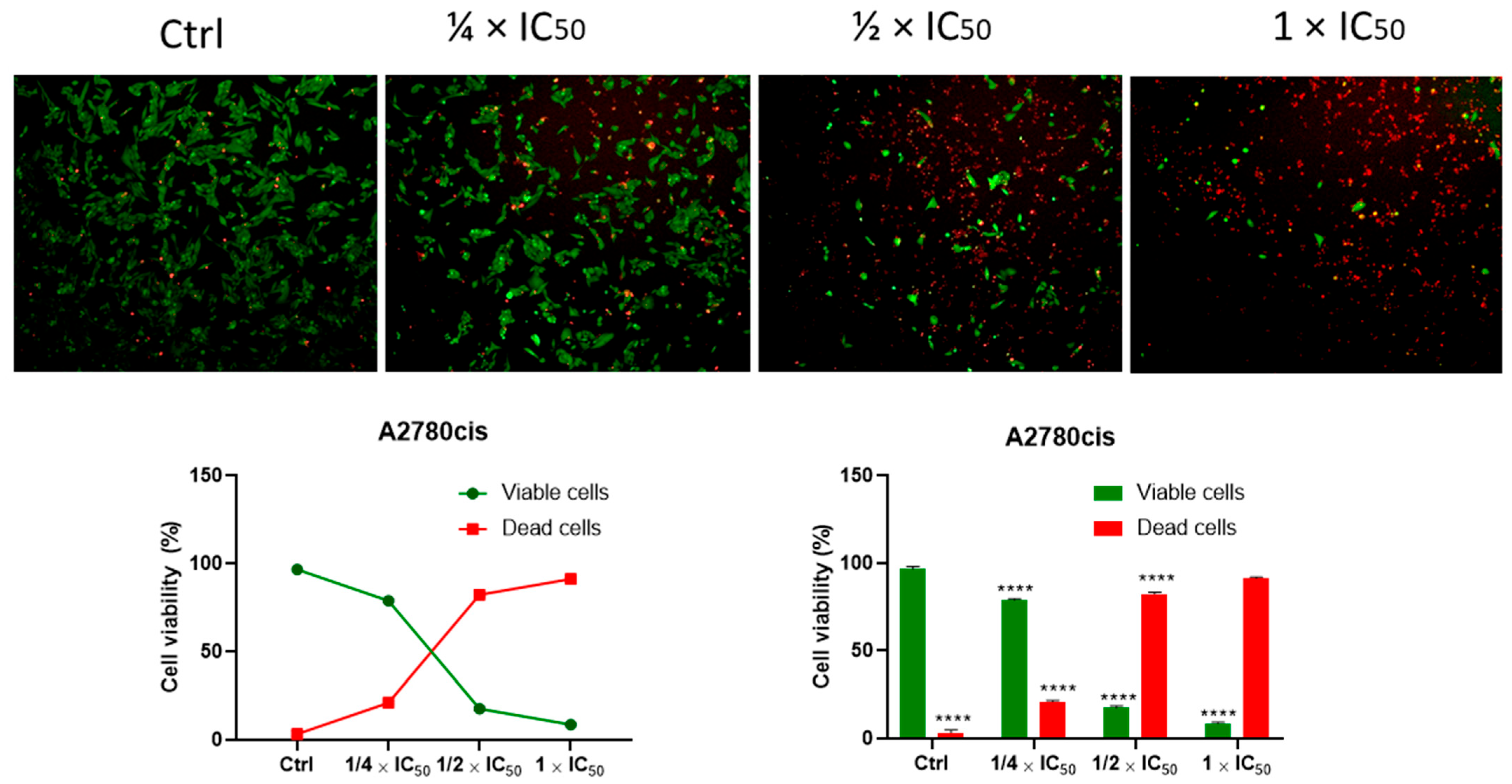
3.2.4. Cell Viability (Kit: LIVE/DEAD®)
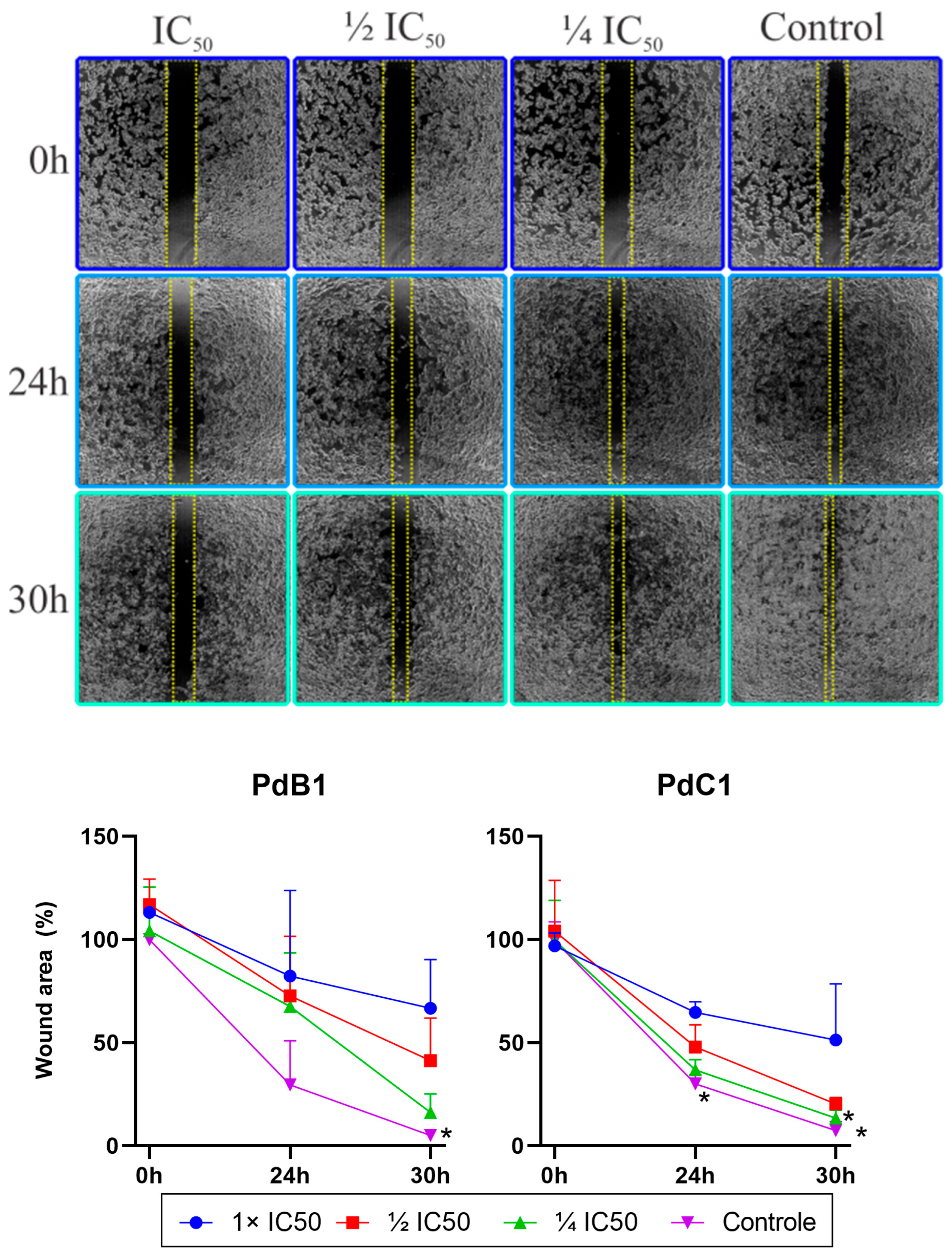
3.2.5. Cell Migration Assay (Wound Healing)
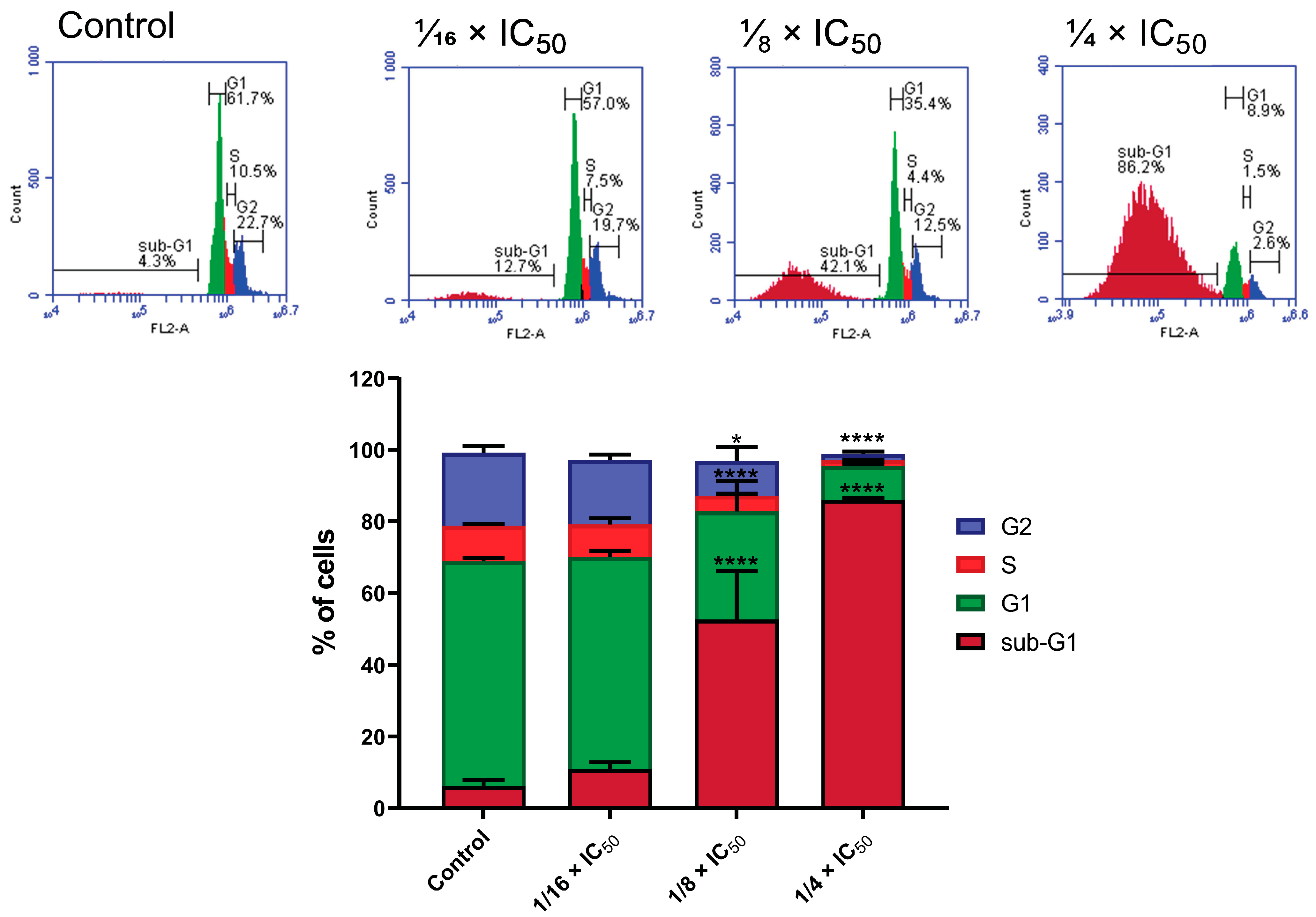
3.2.6. Cell Cycle Analysis
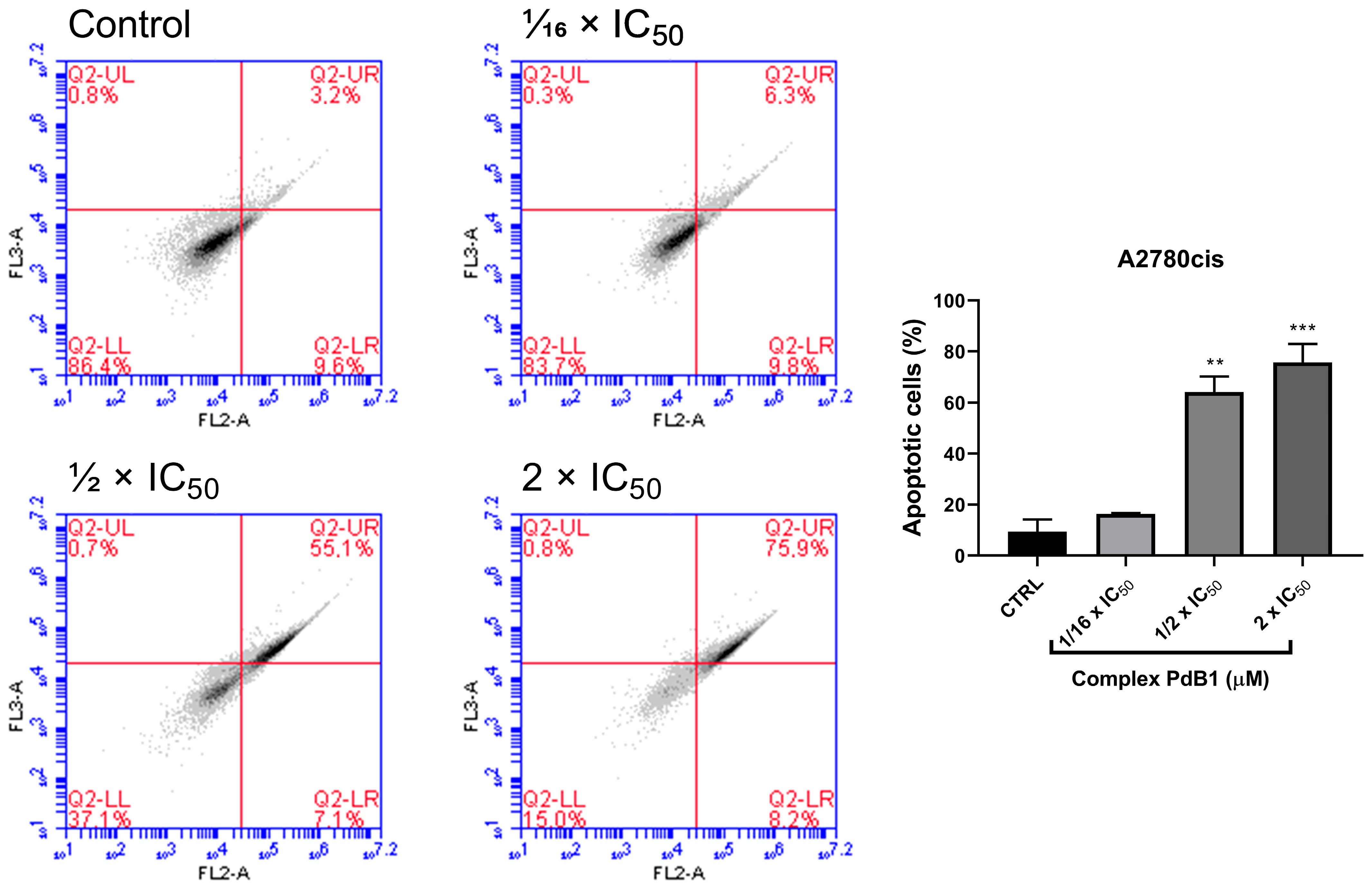
3.2.7. Apoptosis Assay by Anexin V
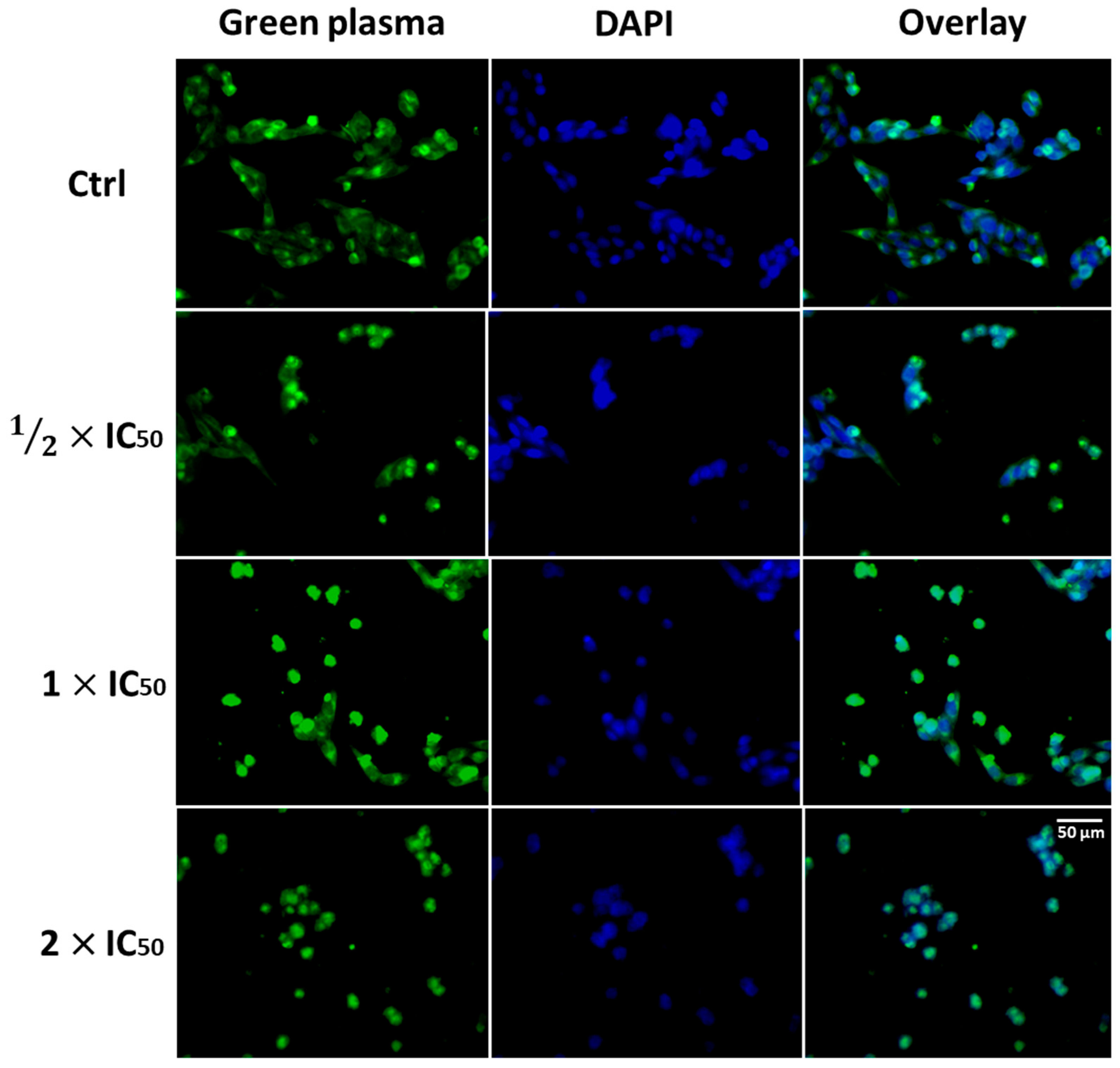
3.2.8. Three-Dimensional Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nenclares, P.; Harrington, K.J. The biology of cancer. Medicine 2020, 48, 67–72. [Google Scholar] [CrossRef]
- Sarkar, S.; Horn, G.; Moulton, K.; Oza, A.; Byler, S.; Kokolus, S.; Longacre, M. Cancer development, progression, and therapy: An epigenetic overview. Int. J. Mol. Sci. 2013, 14, 21087–21113. [Google Scholar] [CrossRef]
- Kennedy, S.R.; Zhang, Y.; Risques, R.A. Cancer-Associated Mutations but No Cancer: Insights into the Early Steps of Carcinogenesis and Implications for Early Cancer Detection. Trends Cancer 2019, 5, 531–540. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Debela, D.T.; Muzazu, S.G.Y.; Heraro, K.D.; Ndalama, M.T.; Mesele, B.W.; Haile, D.C.; Kitui, S.K.; Manyazewal, T. New Approaches and Procedures for Cancer Treatment: Current Perspectives. SAGE Open Med. 2021, 9, 20503121211034370. [Google Scholar] [CrossRef]
- Chabner, B.A.; Roberts, T.G., Jr. Chemotherapy and the War on Cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef]
- Kelland, L. The Resurgence of Platinum-Based Cancer Chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Ghosh, S. Cisplatin: The First Metal Based Anticancer Drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
- Barabas, K.; Milner, R.; Lurie, D.; Adin, C. Cisplatin: A Review of Toxicities and Therapeutic Applications. Vet. Comp. Oncol. 2008, 6, 1–18. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA Repair Pathways and Cisplatin Resistance: An Intimate Relationship. Clinics 2018, 73 (Suppl. 1), e478s. [Google Scholar] [CrossRef]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The Side Effects of Platinum-Based Chemotherapy Drugs: A Review for Chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef] [PubMed]
- Lucaciu, R.L.; Hangan, A.C.; Sevastre, B.; Oprean, L.S. Metallo-Drugs in Cancer Therapy: Past, Present and Future. Molecules 2022, 27, 6485. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Pöthig, A. Metals in Cancer Research: Beyond Platinum Metallodrugs. ACS Cent. Sci. 2024, 10, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.; Arjmand, F.; Tabassum, S. Current and Future Potential of Metallo Drugs: Revisiting DNA-Binding of Metal Containing Molecules and Their Diverse Mechanism of Action. Inorganica Chim. Acta 2016, 444, 1–22. [Google Scholar] [CrossRef]
- Fanelli, M.; Formica, M.; Fusi, V.; Giorgi, L.; Micheloni, M.; Paoli, P. New Trends in Platinum and Palladium Complexes as Antineoplastic Agents. Coord. Chem. Rev. 2016, 310, 41–79. [Google Scholar] [CrossRef]
- Czarnomysy, R.; Radomska, D.; Szewczyk, O.K.; Roszczenko, P.; Bielawski, K. Platinum and Palladium Complexes as Promising Sources for Antitumor Treatments. Int. J. Mol. Sci. 2021, 22, 8271. [Google Scholar] [CrossRef]
- Azzouzi, A.R.; Vincendeau, S.; Barret, E.; Cicco, A.; Kleinclauss, F.; van Der Poel, H.G.; Stief, C.G.; Rassweiler, J.; Salomon, G.; Solsona, E. Padeliporfin Vascular-Targeted Photodynamic Therapy versus Active Surveillance in Men with Low-Risk Prostate Cancer (CLIN1001 PCM301): An Open-Label, Phase 3, Randomised Controlled Trial. Lancet Oncol. 2017, 18, 181–191. [Google Scholar] [CrossRef]
- Azzouzi, A.; Barret, E.; Moore, C.M.; Villers, A.; Allen, C.; Scherz, A.; Muir, G.; De Wildt, M.; Barber, N.J.; Lebdai, S. TOOKAD® S Oluble Vascular-targeted Photodynamic (VTP) Therapy: Determination of Optimal Treatment Conditions and Assessment of Effects in Patients with Localised Prostate Cancer. BJU Int. 2013, 112, 766–774. [Google Scholar] [CrossRef]
- Gupta, S.; Singh, N.; Khan, T.; Joshi, S. Thiosemicarbazone Derivatives of Transition Metals as Multi-Target Drugs: A Review. Results Chem. 2022, 4, 100459. [Google Scholar] [CrossRef]
- de Siqueira, L.R.P.; de Moraes Gomes, P.A.T.; de Lima Ferreira, L.P.; de Melo Rêgo, M.J.B.; Leite, A.C.L. Multi-Target Compounds Acting in Cancer Progression: Focus on Thiosemicarbazone, Thiazole and Thiazolidinone Analogues. Eur. J. Med. Chem. 2019, 170, 237–260. [Google Scholar] [CrossRef]
- Taylor, S.E.; Behr, S.; Cooper, K.L.; Mahdi, H.; Fabian, D.; Gallion, H.; Ueland, F.; Vargo, J.; Orr, B.; Girda, E. Dose Finding, Bioavailability, and PK-PD of Oral Triapine with Concurrent Chemoradiation for Locally Advanced Cervical Cancer and Vaginal Cancer (ETCTN 9892). Cancer Chemother. Pharmacol. 2025, 95, 4. [Google Scholar] [CrossRef]
- Lima, M.A.; Costa, V.A.; Franco, M.A.; de Oliveira, G.P.; Deflon, V.M.; Rocha, F.V. Palladium (II) Complexes Bearing Thiosemicarbazone and Phosphines as Inhibitors of DNA-Topoisomerase II Enzyme: Synthesis, Characterizations and Biological Studies. Inorg. Chem. Commun. 2020, 112, 107708. [Google Scholar] [CrossRef]
- Lima, M.A. Estudo da Atividade Anticâncer In Vitro para Complexos de Pd(II): Avaliação dos Substituintes dos Ligantes Tiossemicarbazonas. Ph.D. Thesis, Universidade Federal de São Carlos, São Carlos, Brazil, 2023. Available online: https://repositorio.ufscar.br/handle/20.500.14289/18674 (accessed on 15 January 2025).
- Silva, L.T.P.; Pereira, G.B.S.; de Oliveira, G.P.; Lima, M.A.; de Araujo-Neto, J.H.; Akinyemi, A.O.; Vieira, M.A.; Nascimento-Júnior, N.M.; de Farias, R.L.; Ellena, J.A. Synthesis, Characterization, Cytotoxicity Study, Interaction with DNA and Topoisomerase IIα of Square-Planar Complexes with Thiosemicarbazones. Polyhedron 2024, 257, 117021. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT–Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Quiroga, A.G.; Ranninger, C.N. Contribution to the SAR field of metallated and coordination complexes: Studies of the palladium and platinum derivatives with selected thiosemicarbazones as antitumoral drugs. Coord. Chem. Rev. 2004, 248, 119–133. [Google Scholar] [CrossRef]
- Farias, R.L.; Polez, A.M.R.; Silva, D.E.S.; Zanetti, R.D.; Moreira, M.B.; Batista, V.S.; Reis, B.L.; Nascimento-Júnior, N.M.; Rocha, F.V.; Lima, M.A.; et al. In Vitro and in Silico Assessment of Antitumor Properties and Biomolecular Binding Studies for Two New Complexes Based on NiII Bearing K2N,S-Donor Ligands. Mater. Sci. Eng. C 2021, 121, 111815. [Google Scholar] [CrossRef]
- Bakker, J.M.; Besson, T.; Lemaire, J.; Scuderi, D.; Maître, P. Gas-Phase Structure of a π-Allyl-Palladium Complex: Efficient Infrared Spectroscopy in a 7 T Fourier Transform Mass Spectrometer. J. Phys. Chem. A 2007, 111, 13415–13424. [Google Scholar] [CrossRef]
- Kostyukovich, A.Y.; Burykina, J.V.; Eremin, D.B.; Ananikov, V.P. Detection and Structural Investigation of Elusive Palladium Hydride Intermediates Formed from Simple Metal Salts. Inorg. Chem. 2021, 60, 7128–7142. [Google Scholar] [CrossRef]
- Oliveira, G.P.; Lima, M.A.; Pereira, G.B.S.; Costa, A.R.; Batista, A.A.; Forim, M.R.; Cominetti, M.R.; Zanetti, R.D.; Farias, R.L.; Netto, A.V.G.; et al. Thiophene-Based Thiosemicarbazones: Synthesis, Properties, and Anticancer Studies. J. Mol. Struct. 2025, 1322, 140306. [Google Scholar] [CrossRef]
- Dutra, J.L.; Honorato, J.; Graminha, A.; Moraes, C.A.F.; de Oliveira, K.T.; Cominetti, M.R.; Castellano, E.E.; Batista, A.A. Pd (II)/Diphosphine/Curcumin Complexes as Potential Anticancer Agents. Dalt. Trans. 2024, 53, 18902–18916. [Google Scholar] [CrossRef] [PubMed]
- Tonon, G.; Mauceri, M.; Cavarzerani, E.; Piccolo, R.; Santo, C.; Demitri, N.; Orian, L.; Nogara, P.A.; Rocha, J.B.T.; Canzonieri, V. Unveiling the Promising Anticancer Activity of Palladium(II)–Aryl Complexes Bearing Diphosphine Ligands: A Structure–Activity Relationship Analysis. Dalton Trans. 2024, 53, 8463–8477. [Google Scholar] [CrossRef] [PubMed]
- Miyanaga, S.; Sakurai, H.; Saiki, I.; Onaka, H.; Igarashi, Y. Anti-Invasive and Anti-Angiogenic Activities of Naturally Occurring Dibenzodiazepine BU-4664L and Its Derivatives. Bioorg. Med. Chem. Lett. 2010, 20, 963–965. [Google Scholar] [CrossRef] [PubMed]
- Dao, P.; Jarray, R.; Le Coq, J.; Lietha, D.; Loukaci, A.; Lepelletier, Y.; Hadj-Slimane, R.; Garbay, C.; Raynaud, F.; Chen, H. Synthesis of Novel Diarylamino-1, 3, 5-Triazine Derivatives as FAK Inhibitors with Anti-Angiogenic Activity. Bioorg. Med. Chem. Lett. 2013, 23, 4552–4556. [Google Scholar] [CrossRef]
- Chang, T.T.; More, S.V.; Lu, I.H.; Hsu, J.C.; Chen, T.J.; Jen, Y.C.; Lu, C.K.; Li, W.S. Isomalyngamide A, A-1 and Their Analogs Suppress Cancer Cell Migration In Vitro. Eur. J. Med. Chem. 2011, 46, 3810–3819. [Google Scholar] [CrossRef]
- de Oliveira, P.F.; Alves, J.M.; Damasceno, J.L.; Oliveira, R.A.M.; Dias, H.J.; Crotti, A.E.M.; Tavares, D.C. Cytotoxicity Screening of Essential Oils in Cancer Cell Lines. Rev. Bras. Farmacogn. 2015, 25, 183–188. [Google Scholar] [CrossRef]
- Kim, K.; Yoo, H.J.; Jung, J.H.; Lee, R.; Hyun, J.K.; Park, J.H.; Na, D.; Yeon, J.H. Cytotoxic Effects of Plant Sap-Derived Extracellular Vesicles on Various Tumor Cell Types. J. Funct. Biomater. 2020, 11, 22. [Google Scholar] [CrossRef]
- Segun, P.A.; Ogbole, O.O.; Ismail, F.M.D.; Nahar, L.; Evans, A.R.; Ajaiyeoba, E.O.; Sarker, S.D. Resveratrol derivatives from Commiphora africana (A. Rich.) Endl. display cytotoxicity and selectivity against several human cancer cell lines. Phyther Res. 2019, 33, 159–166. [Google Scholar] [CrossRef]
- Sena-Lopes, Â.; Remião, M.H.; Alves, M.S.D.; da Rocha Fonseca, B.; Seixas, F.K.; Collares, T.; Borsuk, S. Cell Viability Analysis of Toxocara Cati Larvae with LIVE/DEAD® Viability/Cytotoxicity Kit. Exp. Parasitol. 2020, 212, 107871. [Google Scholar] [CrossRef]
- Demirbuga, M.; Wink, D.J.; Milligan, R.D. Introduction to Fluorescence in General Chemistry Using the Intercalation of Propidium Iodide with DNA. J. Chem. Educ. 2024, 101, 4327–4332. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.; Zhao, H.; Halicka, H.D.; Rybak, P.; Dobrucki, J.; Wlodkowic, D. DNA Damage Signaling Assessed in Individual Cells in Relation to the Cell Cycle Phase and Induction of Apoptosis. Crit. Rev. Clin. Lab. Sci. 2012, 49, 199–217. [Google Scholar] [CrossRef] [PubMed]
- Brajša, K.; Trzun, M.; Zlatar, I.; Jelić, D. Three-Dimensional Cell Cultures as a New Tool in Drug Discovery. Period. Biol. 2016, 118, 59–65. [Google Scholar] [CrossRef]
- Souza, A.G.; Silva, I.B.B.; Campos-Fernandez, E.; Barcelos, L.S.; Souza, J.B.; Marangoni, K.; Goulart, L.R.; Alonso-Goulart, V. Comparative Assay of 2D and 3D Cell Culture Models: Proliferation, Gene Expression and Anticancer Drug Response. Curr. Pharm. Des. 2018, 24, 1689–1694. [Google Scholar] [CrossRef] [PubMed]
- Foglietta, F.; Serpe, L.; Canaparo, R. The Effective Combination between 3D Cancer Models and Stimuli-Responsive Nanoscale Drug Delivery Systems. Cells 2021, 10, 3295. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R1 | R2 | Complexes | MDA-MB-231 | MCF-7 | SK-BR-3 | MFC-10A |
|---|---|---|---|---|---|---|
| H | PdB1 | 1.45 ± 0.1 | 3.79 ± 0.4 | 1.42 ± 0.07 | 1.58 ± 0.07 | |
| Me | Me | PdB2 | 0.92 ± 0.08 | 1.47 ± 0.2 | 1.48 ± 0.06 | ― |
| Et | PdB3 | 1.04 ± 0.06 | 0.91 ± 0.008 | <2 | ― | |
| H | PdC1 | 5.97 ± 0.1 | 17.68 ± 1 | 2.25 ± 0.3 | 2.64 ± 0.06 | |
| Ph | Me | PdC2 | 9.66 ± 0.4 | >50 | >50 | ― |
| Et | PdC3 | >50 | >50 | >50 | ― | |
| Cisplatin | 33.14 ± 0.3 | 8.64 ± 0.8 | 5.86 ± 0.2 | 23.90 ± 0.7 |
| R1 | R2 | Complexes | A2780 | A2780cis | RF |
|---|---|---|---|---|---|
| H | PdB1 | 0.12 ± 0.01 | 0.20 ± 0.01 | 1.66 | |
| Me | Me | PdB2 | 0.13 ± 0.01 | 0.21 ± 0.01 | 1.62 |
| Et | PdB3 | 0.23 ± 0.01 | 0.26 ± 0.01 | 1.13 | |
| H | PdC1 | 0.40 ± 0.01 | 0.62 ± 0.01 | 1.55 | |
| Ph | Me | PdC2 | 3.93 ± 0.2 | 1.66 ± 0.01 | 0.42 |
| Et | PdC3 | 22.78 ± 1 | 11.1 ± 0.6 | 0.48 | |
| Cisplatin | 14.24 ± 0.05 | 25.09 ± 0.9 | 1.76 |
| R1 | R2 | Complexes | A549 | MRC5 | A375 |
|---|---|---|---|---|---|
| H | PdB1 | 44.8 ± 2 | 1.70 ± 0.2 | 1.34 ± 0.08 | |
| Me | Me | PdB2 | 30.4 ± 0.4 | 6.85 ± 0.3 | ― |
| Et | PdB3 | 12.99 ± 1 | 9.28 ± 0.3 | ― | |
| H | PdC1 | >50 | 46.77 ± 2 | >50 | |
| Ph | Me | PdC2 | >50 | >50 | ― |
| Et | PdC3 | >50 | >50 | ― | |
| Cisplatin | 13.01 ± 0.4 | 21.61 ± 0.8 | 12.21 ± 0.62 |
| Complexes | MDA-MB-231 | MCF-7 | SK-BR-3 | A2780 | A2780cis | A549 |
|---|---|---|---|---|---|---|
| PdB1 | 1.2 | 0.4 | 1.2 | 14.2 | 8.5 | 0.0 |
| PdB2 | 7.4 | 4.7 | 4.6 | 52.7 | 32.6 | 0.2 |
| PdB3 | 8.9 | 10.2 | ― | 40.3 | 35.7 | 0.7 |
| PdC1 | 7.8 | 2.6 | 20.8 | 116.9 | 75.4 | ― |
| Cisplatin | 0.7 | 2.5 | 3.7 | 1.5 | 0.9 | 1.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lima, M.A.; Teixeira, T.; Fortaleza, D.B.; Pereira, G.B.S.; Akinyemi, A.O.; Moraes, C.A.F.; Forim, M.R.; Batista, A.A.; Dutra, J.L.; Araujo-Neto, J.H.; et al. Enhanced Anticancer Potential of Pd(II)-Thiosemicarbazone Complexes: Selectivity, Mechanisms, and 3D Models. Pharmaceutics 2025, 17, 829. https://doi.org/10.3390/pharmaceutics17070829
Lima MA, Teixeira T, Fortaleza DB, Pereira GBS, Akinyemi AO, Moraes CAF, Forim MR, Batista AA, Dutra JL, Araujo-Neto JH, et al. Enhanced Anticancer Potential of Pd(II)-Thiosemicarbazone Complexes: Selectivity, Mechanisms, and 3D Models. Pharmaceutics. 2025; 17(7):829. https://doi.org/10.3390/pharmaceutics17070829
Chicago/Turabian StyleLima, Mauro A., Tamara Teixeira, Dario B. Fortaleza, George B. S. Pereira, Amos O. Akinyemi, Carlos André Ferreira Moraes, Moacir R. Forim, Alzir A. Batista, Jocely L. Dutra, João H. Araujo-Neto, and et al. 2025. "Enhanced Anticancer Potential of Pd(II)-Thiosemicarbazone Complexes: Selectivity, Mechanisms, and 3D Models" Pharmaceutics 17, no. 7: 829. https://doi.org/10.3390/pharmaceutics17070829
APA StyleLima, M. A., Teixeira, T., Fortaleza, D. B., Pereira, G. B. S., Akinyemi, A. O., Moraes, C. A. F., Forim, M. R., Batista, A. A., Dutra, J. L., Araujo-Neto, J. H., Ellena, J. A., & Rocha, F. V. (2025). Enhanced Anticancer Potential of Pd(II)-Thiosemicarbazone Complexes: Selectivity, Mechanisms, and 3D Models. Pharmaceutics, 17(7), 829. https://doi.org/10.3390/pharmaceutics17070829