


A Review on New Frontiers in Drug-Drug Interaction Predictions and Safety Evaluations with In Vitro Cellular Models



Abstract
1. Introduction
2. Reaction Phenotyping Study
2.1. Human Hepatocytes: The Gold Standard Model
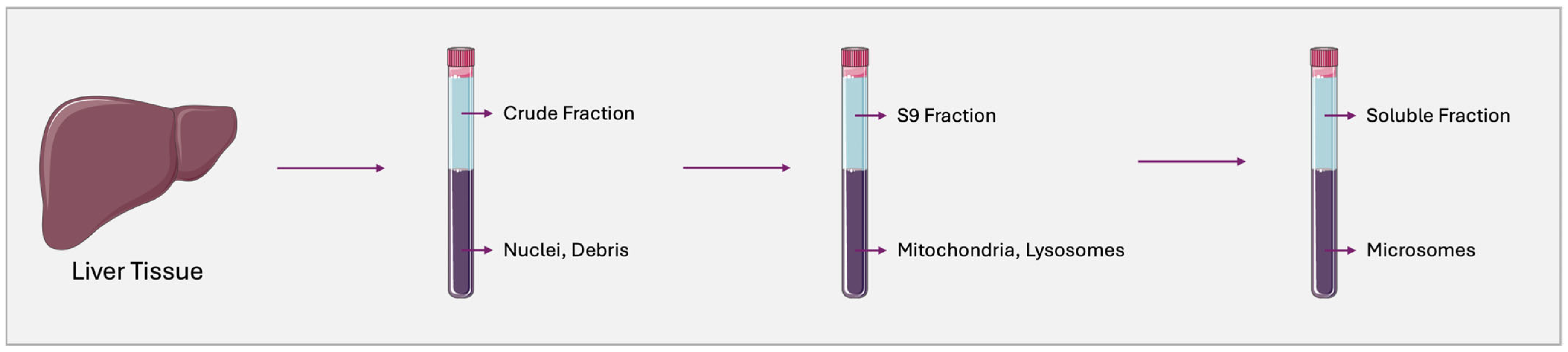
2.2. Human Liver Microsomes
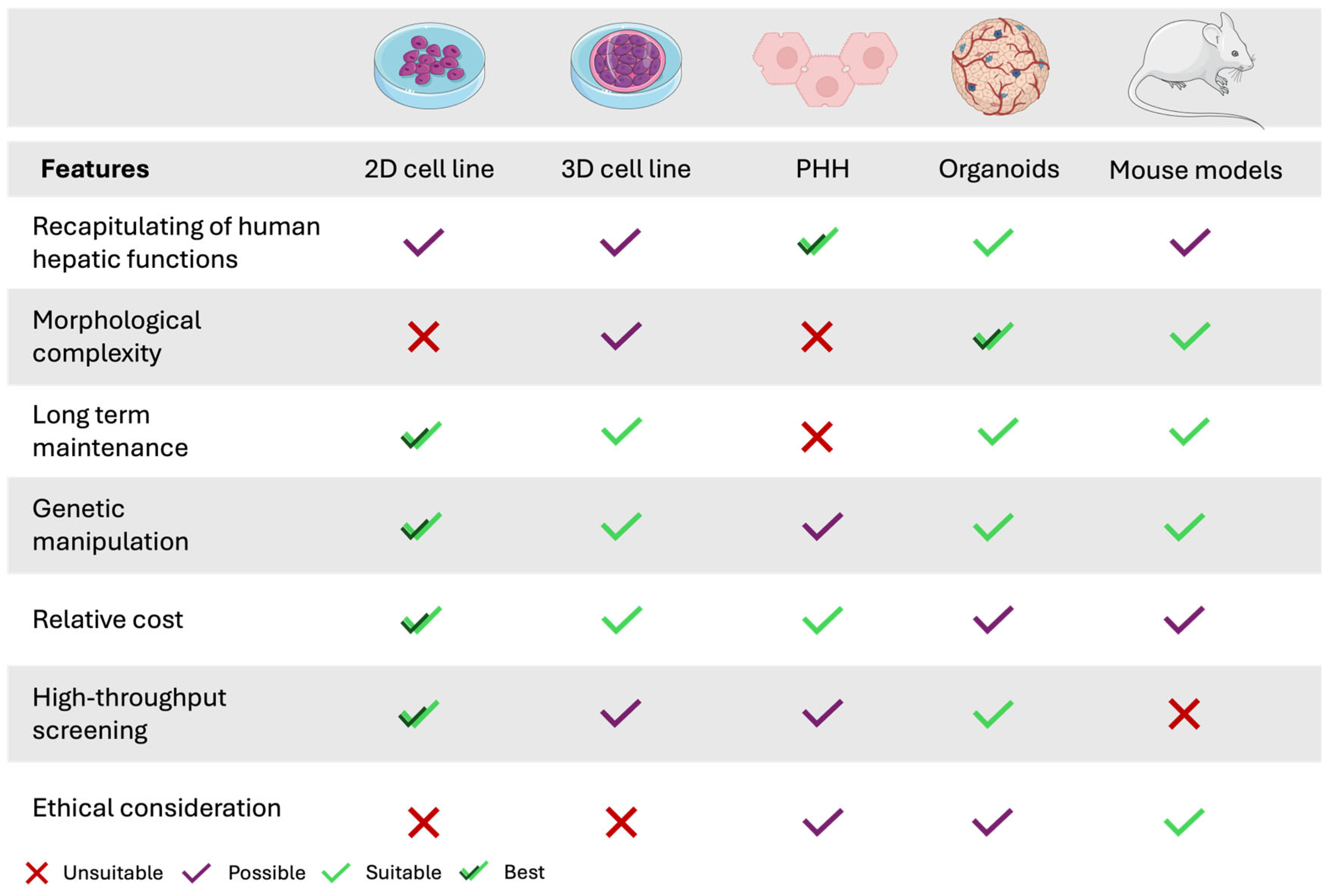
2.3. Human Liver-Derived Cell Lines: Alternatives
2.3.1. HepG2 Cell Line
2.3.2. HepaRG Cell Line
2.3.3. BC2 Cell Line
3. Inhibition Study
3.1. Reversible Inhibition Study
Mechanisms of CYP Reversible Inhibition
3.2. Conventional Models
3.3. Emergent Models
4. Induction Study
5. Future Perspectives: Hybrid In Vitro–In Silico Approaches
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Food and Drug Administration. Guideline for the Format and Content of the Human Pharmacokinetics and Bioavailability Section of an Application; Food and Drug Administration: Silver Spring, MD, USA, 1987.
- European Medicines Agency. Guideline on the Clinical Investigation of the Pharmacokinetics of Therapeutic Proteins; European Medicines Agency: London, UK, 2007.
- Donato, M.T.; Lahoz, A.; Castell, J.V.; Gómez-Lechón, M.J. Cell Lines: A Tool for In Vitro Drug Metabolism Studies. Curr. Drug Metab. 2008, 9, 1–11. [Google Scholar] [PubMed]
- Lin, J.; Sahakian, D.C.; F de Morais, S.M.; Xu, J.J.; Polzer, R.J.; Winter, S.M. The Role of Absorption, Distribution, Metabolism, Excretion and Toxicity in Drug Discovery. Curr. Top. Med. Chem. 2003, 3, 1125–1154. [Google Scholar] [CrossRef] [PubMed]
- Castell, J.V.; Jover, R.; Martínez-Jiménez, C.P.; Gómez-Lechón, M.J. Hepatocyte Cell Lines: Their Use, Scope and Limitations in Drug Metabolism Studies. Expert. Opin. Drug Metab. Toxicol. 2006, 2, 183–212. [Google Scholar] [CrossRef]
- Rendic, S.; Guengerich, F.P. Survey of Human Oxidoreductases and Cytochrome P450 Enzymes Involved in the Metabolism of Xenobiotic and Natural Chemicals. Chem. Res. Toxicol. 2015, 28, 38–42. [Google Scholar] [CrossRef]
- Patel, R.; Barker, J.; Elshaer, A. Pharmaceutical Excipients and Drug Metabolism: A Mini-Review. Int. J. Mol. Sci. 2020, 21, 8224. [Google Scholar] [CrossRef]
- Zhao, M.; Ma, J.; Li, M.; Zhang, Y.; Jiang, B.; Zhao, X.; Huai, C.; Shen, L.; Zhang, N.; He, L.; et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. Int. J. Mol. Sci. 2021, 22, 12808. [Google Scholar] [CrossRef]
- Rendic, S.P. Metabolism and Interactions of Ivermectin with Human Cytochrome P450 Enzymes and Drug Transporters, Possible Adverse and Toxic Effects. Arch. Toxicol. 2021, 95, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.M.; Fernandes, C.; Remião, F.; Tiritan, M.E. Enantioselectivity in Drug Pharmacokinetics and Toxicity: Pharmacological Relevance and Analytical Methods. Molecules 2021, 26, 3113. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome P450s and Other Enzymes in Drug Metabolism and Toxicity. AAPS J. 2006, 8, E101–E111. [Google Scholar] [CrossRef]
- Zahoor, I.; Rui, B.; Khan, J.; Datta, I.; Giri, S. An Emerging Potential of Metabolomics in Multiple Sclerosis: A Comprehensive Overview. Cell. Mol. Life Sci. 2021, 78, 3181–3203. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome P-450 3A4: Regulation and Role in Drug Metabolism. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.J. Molecular Genetics of the P-450 Superfamily. Pharmac. Ther. 1990, 45, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Eichelbaum, M.; Ingelman-Sundberg, M.; Evans, W.E. Pharmacogenomics and Individualized Drug Therapy. Annu. Rev. Med. 2006, 57, 119–137. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Lu, A.Y. Interindividual Variability in Inhibition and Induction of Cytochrome P450 Enzymes. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 535–567. [Google Scholar] [CrossRef]
- Johnell, K.; Klarin, I. The Relationship between Number of Drugs and Potential Drug-Drug Interactions in the Elderly A Study of Over 600 000 Elderly Patients from the Swedish Prescribed Drug Register. Drug Saf. 2007, 30, 911–918. [Google Scholar] [CrossRef]
- Payne, R.A.; Avery, A.J. Polypharmacy: One of the Greatest Prescribing Challenges in General Practice. Br. J. General. Pract. 2011, 61, 83–84. [Google Scholar] [CrossRef]
- Davies, L.E.; Spiers, G.; Kingston, A.; Todd, A.; Adamson, J.; Hanratty, B. Adverse Outcomes of Polypharmacy in Older People: Systematic Review of Reviews. J. Am. Med. Dir. Assoc. 2020, 21, 181–187. [Google Scholar] [CrossRef]
- Onder, G.; Marengoni, A. Polypharmacy. Clin. Pharm. Pharmacol. 2017, 318, 1728. [Google Scholar] [CrossRef]
- Wang, X.; Liu, K.; Shirai, K.; Tang, C.; Hu, Y.; Wang, Y.; Hao, Y.; Dong, J.Y. Prevalence and Trends of Polypharmacy in U.S. Adults, 1999–2018. Glob. Health Res. Policy 2023, 8, 25. [Google Scholar] [CrossRef]
- Delara, M.; Murray, L.; Jafari, B.; Bahji, A.; Goodarzi, Z.; Kirkham, J.; Chowdhury, Z.; Seitz, D.P. Prevalence and Factors Associated with Polypharmacy: A Systematic Review and Meta-Analysis. BMC Geriatr. 2022, 22, 601. [Google Scholar] [CrossRef]
- Charlesworth, C.J.; Smit, E.; Lee, D.S.H.; Alramadhan, F.; Odden, M.C. Polypharmacy among Adults Aged 65 Years and Older in the United States: 1988–2010. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Kantor, E.D.; Rehm, C.D.; Haas, J.S.; Chan, A.T.; Giovannucci, E.L. Trends in Prescription Drug Use among Adults in the United States from 1999-2012. JAMA J. Am. Med. Assoc. 2015, 314, 1818–1831. [Google Scholar] [CrossRef] [PubMed]
- Alhumaidi, R.M.; Bamagous, G.A.; Alsanosi, S.M.; Alqashqari, H.S.; Qadhi, R.S.; Alhindi, Y.Z.; Ayoub, N.; Falemban, A.H. Risk of Polypharmacy and Its Outcome in Terms of Drug Interaction in an Elderly Population: A Retrospective Cross-Sectional Study. J. Clin. Med. 2023, 12, 3960. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Beers, J.L.; Geffert, R.M.; Jackson, K.D. A Review of CYP-Mediated Drug Interactions: Mechanisms and In Vitro Drug-Drug Interaction Assessment. Biomolecules 2024, 14, 99. [Google Scholar] [CrossRef]
- Peng, Y.; Cheng, Z.; Xie, F. Evaluation of Pharmacokinetic Drug-Drug Interactions: A Review of the Mechanisms, in Vitro and in Silico Approaches. Metabolites 2021, 11, 75. [Google Scholar] [CrossRef]
- Tornio, A.; Filppula, A.M.; Niemi, M.; Backman, J.T. Clinical Studies on Drug-Drug Interactions Involving Metabolism and Transport: Methodology, Pitfalls, and Interpretation. Clin. Pharmacol. Ther. 2019, 105, 1345–1361. [Google Scholar] [CrossRef]
- Lu, C.; Di, L. In Vitro and in Vivo Methods to Assess Pharmacokinetic Drug—Drug Interactions in Drug Discovery and Development. Biopharm. Drug Dispos. 2020, 41, 3–31. [Google Scholar] [CrossRef]
- Hughes, J.E.; Moriarty, F.; Bennett, K.E.; Cahir, C. Drug-Drug Interactions and the Risk of Adverse Drug Reaction-Related Hospital Admissions in the Older Population. Br. J. Clin. Pharmacol. 2024, 90, 959–975. [Google Scholar] [CrossRef]
- Aksoy, N.; Ozturk, N. A Meta-Analysis Assessing the Prevalence of Drug-Drug Interactions among Hospitalized Patients. Pharmacoepidemiol. Drug Saf. 2023, 32, 1319–1330. [Google Scholar] [CrossRef]
- Létinier, L.; Bezin, J.; Jarne, A.; Pariente, A. Drug-Drug Interactions and the Risk of Emergency Hospitalizations: A Nationwide Population-Based Study. Drug Saf. 2023, 46, 449–456. [Google Scholar] [CrossRef]
- Wienkers, L.C.; Heath, T.G. Predicting in Vivo Drug Interactions from in Vitro Drug Discovery Data. Nat. Rev. Drug Discov. 2005, 4, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Singer, T. The Application of 3D Cell Models to Support Drug Safety Assessment: Opportunities & Challenges. Adv. Drug Deliv. Rev. 2014, 69–70, 179–189. [Google Scholar]
- Food and Drug Administration (FDA). In Vitro Interaction Drug Interaction Studies-Cytochrome P450 Enzyme and Transporter Mediated Drug Interactions Final-Center for Evaluation and Research (CDER); Food and Drug Administration (FDA): Rockville, MD, USA, 2020.
- Gómez-Lechón, M.J.; Donato, M.T.; Castell, J.V.; Jover, R. Human Hepatocytes as a Tool for Studying Toxicity and Drug Metabolism. Curr. Drug Metab. 2003, 4, 292–312. [Google Scholar] [CrossRef] [PubMed]
- Jaroch, K.; Jaroch, A.; Bojko, B. Cell Cultures in Drug Discovery and Development: The Need of Reliable in Vitro-in Vivo Extrapolation for Pharmacodynamics and Pharmacokinetics Assessment. J. Pharm. Biomed. Anal. 2018, 147, 297–312. [Google Scholar] [CrossRef]
- Zhang, D.; Luo, G.; Ding, X.; Lu, C. Preclinical Experimental Models of Drug Metabolism and Disposition in Drug Discovery and Development. Acta Pharm. Sin. B 2012, 2, 549–561. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H. Application of Pharmaceutical Profiling Assays for Optimization of Drug-like Properties. Curr. Opin. Drug Discov. Devel 2005, 8, 495–504. [Google Scholar] [PubMed]
- European Medicines Agency. Guideline on the Investigation of Drug Interactions; European Medicines Agency: London, UK, 2012.
- Chen, Y.; Mao, J.; Fretland, A.J. Reaction Phenotyping. In Encyclopedia of Drug Metabolism and Interactions; John Wiley and Sons: Hoboken, NJ, USA, 2015; pp. 1–26. [Google Scholar]
- Zientek, M.A.; Youdim, K. Reaction Phenotyping: Advances in the Experimental Strategies Used to Characterize the Contribution of Drug-Metabolizing Enzymes. Drug Metab. Dispos. 2015, 43, 163–181. [Google Scholar] [CrossRef]
- Zhang, H.; Davis, C.D.; Sinz, M.W.; Rodrigues, A.D. Cytochrome P450 Reaction-Phenotyping: An Industrial Perspective. Expert. Opin. Drug Metab. Toxicol. 2007, 3, 667–687. [Google Scholar] [CrossRef]
- Lu, A.Y.H.; Wang, R.W.; Lin, J.H. Cytochrome P450 In Vitro Reaction Phenotyping: A Re-Evaluation of Approaches Used for P450 Isoform Identification. Drug Metab. Dispos. 2003, 31, 345–350. [Google Scholar] [CrossRef]
- Fu, S.; Yu, F.; Hu, Z.; Sun, T. Metabolism-Mediated Drug-Drug Interactions—Study Design, Data Analysis, and Implications for in Vitro Evaluations. Med. Drug Discov. 2022, 14, 100121. [Google Scholar] [CrossRef]
- Li, A.P. Human Hepatocytes: Isolation, Cryopreservation and Applications in Drug Development. Chem. Biol. Interact. 2007, 168, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Hakkola, J.; Hukkanen, J.; Turpeinen, M.; Pelkonen, O. Inhibition and Induction of CYP Enzymes in Humans: An Update. Arch. Toxicol. 2020, 94, 3671–3722. [Google Scholar] [CrossRef] [PubMed]
- Bojar, H.; Basler, M.; Fuchs, F.; Dreyfürst, R.; Staib, W.; Broelsch, C. Preparation of Parenchymal and Non-Parenchymal Cells from Adult Human Liver—Morphological and Biochemical Characteristics. cclm 1976, 14, 527–532. [Google Scholar] [CrossRef]
- Godoy, P.; Hewitt, N.J.; Albrecht, U.; Andersen, M.E.; Ansari, N.; Bhattacharya, S.; Bode, J.G.; Bolleyn, J.; Borner, C.; Böttger, J.; et al. Recent Advances in 2D and 3D in Vitro Systems Using Primary Hepatocytes, Alternative Hepatocyte Sources and Non-Parenchymal Liver Cells and Their Use in Investigating Mechanisms of Hepatotoxicity, Cell Signaling and ADME. Arch. Toxicol. 2013, 87, 1315–1530. [Google Scholar] [PubMed]
- Donato, M.T.; Castell, J.V.; Jose Gomez-Lechon, M. Characterization of Drug Metabolizing Activities in Pig Hepatocytes for Use in Bioartifxial Liver Devices: Comparison with Other Hepatic Cellular Models. J. Hepatol. 1999, 31, 542–549. [Google Scholar] [CrossRef]
- Jigorel, E.; Le Vee, M.; Boursier-Neyret, C.; Bertrand, M.; Fardel, O. Functional Expression of Sinusoidal Drug Transporters in Primary Human and Rat Hepatocytes. Drug Metab. Dispos. 2005, 33, 1418–1422. [Google Scholar] [CrossRef]
- Dvorak, Z. Opportunities and Challenges in Using Human Hepatocytes in Cytochromes P450 Induction Assays. Expert. Opin. Drug Metab. Toxicol. 2016, 12, 169–174. [Google Scholar] [CrossRef]
- Li, A.P.; Maurel, P.; Jose Gomez-Lechon, M.; Cheng, L.C.; Jurima-Romet, M. Preclinical Evaluation of Drug-Drug Interaction Potential: Present Status of the Application of Primary Human Hepatocytes in the Evaluation of Cytochrome P450 Induction. Chem. Biol. Interact. 1997, 107, 5–16. [Google Scholar] [CrossRef]
- Rodríguez-Antona, C.; Donato, M.T.; Boobis, A.; Edwards, R.J.; Watts, P.S.; Castell, J.V.; Gómez-Lechón, M.-J. Cytochrome P450 Expression in Human Hepatocytes and Hepatoma Cell Lines: Molecular Mechanisms That Determine Lower Expression in Cultured Cells. Xenobiotica 2002, 32, 505–520. [Google Scholar] [CrossRef]
- Padgham, C.R.W.; Boyle, C.C.; Wang, X.J.; Raleigh, S.M.; Wright, M.C.; Paine, A.J. Alteration of Transcription Factor MRNAs during the Isolation and Culture of Rat Hepatocytes Suggests the Activation of a Proliferative Mode Underlies Their Dedifferentiation. Biochem. Biophys. Res. Commun. 1993, 197, 599–605. [Google Scholar] [CrossRef]
- Lecluyse, E.; Madan, A.; Hamilton, G.; Carroll, K.; Dehaan, R.; Parkinson, A. Expression and Regulation of Cytochrome P450 Enzymes in Primary Cultures of Human Hepatocytes Magnitude of Induction; John Wiley and Sons: Hoboken, NJ, USA, 2000; Volume 14. [Google Scholar]
- George, J.; Goodwin, B.; Liddle, C.; Tapner, M.; Farrell Westmead, G.C. Time-Dependent Expression of Cytochrome P450 Genes in Primary Cultures of Well-Differentiated Human Hepatocytes. J. Lab. Clin. Med. 1997, 129, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Padgham, C.R.W.; Paine, A.J. Altered Expression of Cytochrome P-450 MRNAs, and Potentially of Other Transcripts Encoding Key Hepatic Functions, Are Triggered during the Isolation of Rat Hepatocytes. Biochem. J. 1993, 289, 621–624. [Google Scholar] [CrossRef]
- Gómez-Lechón, M.J.; Donato, M.T.; Castell, J.V.; Jover, R. Human Hepatocytes in Primary Culture: The Choice to Investigate Drug Metabolism in Man. Curr. Drug Metab. 2004, 5, 443–462. [Google Scholar] [CrossRef]
- Chandra, P.; Lecluyse, E.L.; Brouwer, K.L.R. Optimization of culture conditions for determining hepatobiliary disposition of taurocholate in sandwich-cultured rat hepatocytes. Vitr. Cell. Dev. Biol. Anim. 2001, 37, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Kaur, I.; Vasudevan, A.; Rawal, P.; Tripathi, D.M.; Ramakrishna, S.; Kaur, S.; Sarin, S.K. Primary Hepatocyte Isolation and Cultures: Technical Aspects, Challenges and Advancements. Bioengineering 2023, 10, 131. [Google Scholar] [CrossRef]
- Nelson, L.J.; Treskes, P.; Howie, A.F.; Walker, S.W.; Hayes, P.C.; Plevris, J.N. Profiling the Impact of Medium Formulation on Morphology and Functionality of Primary Hepatocytes in Vitro. Sci. Rep. 2013, 3, 2735. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Liu, H.-L.; Guo, H.-T.; Wen, H.-W.; Liu, J. Primary Hepatocyte Culture in Collagen Gel Mixture and Collagen Sandwich. World J. Gastroenterol. 2004, 10, 699–702. [Google Scholar] [CrossRef]
- Dunn, J.C.Y.; Yarmush, M.L.; Koebe, H.G.; Tompkins, R.G. Hepatocyte Function and Extracellular Matrix Geometry: Long-Term Culture in a Sandwich Configuration. FASEB J. 1989, 3, 174–177. [Google Scholar] [CrossRef]
- Tracy, T.S.; Chaudhry, A.S.; Prasad, B.; Thummel, K.E.; Schuetz, E.G.; Zhong, X.B.; Tien, Y.C.; Jeong, H.; Pan, X.; Shireman, L.M.; et al. Interindividual Variability in Cytochrome P450-Mediated Drug Metabolism. Drug Metab Dispos. 2016, 44, 343–351. [Google Scholar] [CrossRef]
- Lin, J.; Schyschka, L.; Mühl-Benninghaus, R.; Neumann, J.; Hao, L.; Nussler, N.; Dooley, S.; Liu, L.; Stöckle, U.; Nussler, A.K.; et al. Comparative Analysis of Phase i and II Enzyme Activities in 5 Hepatic Cell Lines Identifies Huh-7 and HCC-T Cells with the Highest Potential to Study Drug Metabolism. Arch. Toxicol. 2012, 86, 87–95. [Google Scholar] [CrossRef]
- Sanchez-Quant, E.; Richter, M.L.; Colomé-Tatché, M.; Martinez-Jimenez, C.P. Single-Cell Metabolic Profiling Reveals Subgroups of Primary Human Hepatocytes with Heterogeneous Responses to Drug Challenge. Genome Biol. 2023, 24, 234. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, N.J.; Bühring, K.U.; Dasenbrock, J.; Haunschild, J.; Ladstetter, B.; Utesch, D. Studies Comparing in Vivo:In Vitro Metabolism of Three Pharmaceutical Compounds in Rat, Dog, Monkey, and Human Using Cryopreserved Hepatocytes, Microsomes, and Collagen Gel Immobilized Hepatocyte Cultures. Drug Metab. Dispos. 2001, 29, 1042–1050. [Google Scholar] [PubMed]
- Reinach, B.; de Sousa, G.; Dostert, P.; Ings, R.; Gugenheim, J.; Rahmani, R. Comparative Effects of Rifabutin and Rifampicin on Cytochromes P450 and UDP-Glucuronosyl-Transferases Expression in Fresh and Cryopreserved Human Hepatocytes. Chem. Biol. Interact. 1999, 121, 37–48. [Google Scholar] [CrossRef]
- Samanez, C.H.; Caron, S.; Briand, O.; Dehondt, H.; Duplan, I.; Kuipers, F.; Hennuyer, N.; Clavey, V.; Staels, B. The Human Hepatocyte Cell Lines IHH and HepaRG: Models to Study Glucose, Lipid and Lipoprotein Metabolism. Arch. Physiol. Biochem. 2012, 118, 102–111. [Google Scholar] [CrossRef]
- Rodrigues, A.D. Integrated Cytochrome P450 Reaction Phenotyping attempting to bridge the gap between cdna-expressed cytochromes p450 and native human liver microsomes. Biochem. Pharmacol. 1999, 57, 465–480. [Google Scholar]
- Bjornsson, T.D.; Callaghan, J.T.; Einolf, H.J.; Fischer, V.; Gan, L.; Grimm, S.; Kao, J.; King, S.P.; Miwa, G.; Ni, L.; et al. The Conduct of in Vitro and in Vivo Drug-Drug Interaction Studies: A Pharmaceutical Research and Manufacturers of America (PhRMA) Perspective. Drug Metab. Dispos. 2003, 31, 815–832. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Liu, X. The Conduct of Drug Metabolism Studies Considered Good Practice (II): In Vitro Experiments. Curr. Drug Metab. 2007, 8, 822–829. [Google Scholar] [CrossRef]
- Asha, S.; Vidyavathi, M. Role of Human Liver Microsomes in in Vitro Metabolism of Drugs-A Review. Appl. Biochem. Biotechnol. 2010, 160, 1699–1722. [Google Scholar] [CrossRef]
- Pelkonen, O.; Kaltiala, E.H.; Larmi, T.K.I.; Kärki, N.T. Cytochrome P-450-Linked Monooxygenase System and Drug-Induced Spectral Interactions in Human Liver Microsomes. Chem. Biol. Interact. 1974, 9, 205–216. [Google Scholar] [CrossRef]
- Li, A.P. Preclinical in Vitro Screening Assays for Drug-like Properties. Drug Discov. Today Technol. 2005, 2, 179–185. [Google Scholar] [CrossRef]
- Araya, Z.; Wikvall, K. 6α-Hydroxylation of Taurochenodeoxycholic Acid and Lithocholic Acid by CYP3A4 in Human Liver Microsomes. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 1999, 1438, 47–54. [Google Scholar] [CrossRef]
- Parkinson, A.; Mudra, D.R.; Johnson, C.; Dwyer, A.; Carroll, K.M. The Effects of Gender, Age, Ethnicity, and Liver Cirrhosis on Cytochrome P450 Enzyme Activity in Human Liver Microsomes and Inducibility in Cultured Human Hepatocytes. Toxicol. Appl. Pharmacol. 2004, 199, 193–209. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Hong, Y.; Kleintop, T.A.; Mc Connell, O.J.; Huryn, D.M. Optimization of a Higher Throughput Microsomal Stability Screening Assay for Profiling Drug Discovery Candidates. SLAS Discov. 2003, 8, 453–462. [Google Scholar] [CrossRef]
- Pearce, R.E.; McIntyre, C.J.; Madan, A.; Sanzgiri, U.; Draper, A.J.; Bullock, P.L.; Cook, D.C.; Burton, L.A.; Latham, J.; Nevins, C.; et al. Effects of Freezing, Thawing, and Storing Human Liver Microsomes on Cytochrome P450 Activity. Arch. Biochem. Biophys. 1996, 331, 145–169. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Ryu, J.H.; Chi, Y.H.; Paik, S.H.; Kim, S.K. Cytochrome P450-Mediated Metabolic Interactions between Donepezil and Tadalafil in Human Liver Microsomes. Toxicol. Vitr. 2024, 100, 105922. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, Z.; Chan, E.; Zhao, Y.; Kang, J.; Zhang, X.; Tian, X. Inhibition of Cytochrome P450 Enzymes and Uridine 5′-Diphospho-Glucuronosyltransferases by Vicagrel in Human Liver Microsomes: A Prediction of Potential Drug-Drug Interactions. Chem. Biol. Interact. 2022, 352, 109775. [Google Scholar] [CrossRef]
- Yang, S.; Hu, J.; Li, Y.; Zhao, Z. Evaluation of Pharmacokinetic Interactions between Bicyclol and Co-Administered Drugs in Rat and Human Liver Microsomes in Vitro and in Rats in Vivo. Xenobiotica 2019, 49, 987–994. [Google Scholar] [CrossRef]
- Li, Q.; Li, W.; Chen, J.; Lin, H.; Zhou, C. Inhibitory Effect of Carvedilol on Bedaquiline Metabolism in Vitro and in Vivo. PeerJ 2025, 13, e19313. [Google Scholar] [CrossRef]
- Faison, S.L.; Batonga, J.; Arumugham, T.; Bartkus, A.; Morrison, M.E.; Mullin, M.J.; Tippin, T.; Naderer, O. A Phase 1, Randomized, Crossover Trial to Assess the Effect of Itraconazole on the Pharmacokinetics of Dordaviprone in Healthy Adults. Br. J. Clin. Pharmacol. 2025. [Google Scholar] [CrossRef]
- Jaisupa, N.; Ashton, M.; Birgersson, S. Cannabidiol Metabolism in Vitro: The Role of Antiseizure Medications and CYP2C19 Genotypes. Xenobiotica 2025, 10, 1–10. [Google Scholar] [CrossRef]
- Zhao, C. Cell Culture: In Vitro Model System and a Promising Path to in Vivo Applications. J. Histotechnol. 2023, 46, 1–4. [Google Scholar] [CrossRef]
- Zeilinger, K.; Freyer, N.; Damm, G.; Seehofer, D.; Knöspel, F. Cell Sources for in Vitro Human Liver Cell Culture Models. Exp. Biol. Med. 2016, 241, 1684–1698. [Google Scholar] [CrossRef]
- Steinbrecht, S.; Kammerer, S.; Küpper, J.-H. HepG2 Cells with Recombinant Cytochrome P450 Enzyme Overexpression: Their Use and Limitation as in Vitro Liver Model. J. Cell Biotechnol. 2019, 5, 55–64. [Google Scholar] [CrossRef]
- Olsavsky, K.M.; Page, J.L.; Johnson, M.C.; Zarbl, H.; Strom, S.C.; Omiecinski, C.J. Gene Expression Profiling and Differentiation Assessment in Primary Human Hepatocyte Cultures, Established Hepatoma Cell Lines, and Human Liver Tissues. Toxicol. Appl. Pharmacol. 2007, 222, 42–56. [Google Scholar] [CrossRef]
- Guo, L.; Dial, S.; Shi, L.; Branham, W.; Liu, J.; Fang, J.-L.; Green, B.; Deng, H.; Kaput, J.; Ning, B. Similarities and Differences in the Expression of Drug-Metabolizing Enzymes between Human Hepatic Cell Lines and Primary Human Hepatocytes. Drug Metab. Dispos. 2011, 39, 528–538. [Google Scholar] [CrossRef]
- Dannenberg, L.O.; Edenberg, H.J. Epigenetics of Gene Expression in Human Hepatoma Cells: Expression Profiling the Response to Inhibition of DNA Methylation and Histone Deacetylation. BMC Genom. 2006, 7, 181. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in Human Disease and Prospects for Epigenetic Therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef]
- López-Terrada, D.; Cheung, S.W.; Finegold, M.J.; Knowles, B.B. Hep G2 Is a Hepatoblastoma-Derived Cell Line. Hum. Pathol. 2009, 40, 1512–1515. [Google Scholar] [CrossRef]
- Arzumanian, V.A.; Kiseleva, O.I.; Poverennaya, E.V. The Curious Case of the HepG2 Cell Line: 40 Years of Expertise. Int. J. Mol. Sci. 2021, 22, 13135. [Google Scholar] [CrossRef] [PubMed]
- Donato, M.T.; Tolosa, L.; Gómez-Lechón, M.J. Culture and Functional Characterization of Human Hepatoma HepG2 Cells. In Protocols in In Vitro Hepatocyte Research; Springer: New York, NY, USA, 2015; pp. 77–93. ISBN 9781493920747. [Google Scholar]
- Aden, D.P.; Fogel, A.; Plotkin, S.; Damjanov, I.; Knowles, B.B. Controlled Synthesis of HBsAg in a Differentiated Human Liver Carcinoma-Derived Cell Line. Nature 1979, 282, 615–616. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Hui, L. Progress in Human Liver Organoids. J. Mol. Cell Biol. 2020, 12, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Weir, E.G.; Ali, S.Z. Hepatoblastoma: Cytomorphologic Characteristics in Serious Cavity Fluids. Cancer 2002, 96, 267–274. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Vildhede, A.; Norén, A.; Artursson, P. In-Depth Quantitative Analysis and Comparison of the Human Hepatocyte and Hepatoma Cell Line HepG2 Proteomes. J. Proteom. 2016, 136, 234–247. [Google Scholar] [CrossRef]
- Vildhede, A.; Wiśniewski, J.R.; Norén, A.; Karlgren, M.; Artursson, P. Comparative Proteomic Analysis of Human Liver Tissue and Isolated Hepatocytes with a Focus on Proteins Determining Drug Exposure. J. Proteome Res. 2015, 14, 3305–3314. [Google Scholar] [CrossRef]
- Westerink, W.M.A.; Schoonen, W.G.E.J. Cytochrome P450 Enzyme Levels in HepG2 Cells and Cryopreserved Primary Human Hepatocytes and Their Induction in HepG2 Cells. Toxicol. Vitr. 2007, 21, 1581–1591. [Google Scholar] [CrossRef]
- Westerink, W.M.A.; Schoonen, W.G.E.J. Phase II Enzyme Levels in HepG2 Cells and Cryopreserved Primary Human Hepatocytes and Their Induction in HepG2 Cells. Toxicol. Vitr. 2007, 21, 1592–1602. [Google Scholar] [CrossRef]
- Doostdar, H.; Duthie, S.J.; Burke, M.D.; Melvin, W.T.; Grant, M.H. The Influence of Culture Medium Composition on Drug Metabolising Enzyme Activities of the Human Liver Derived Hep G2 Cell Line. FEBS Lett. 1988, 241, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Wilkening, S.; Bader, A. Influence of Culture Time on the Expression of Drug-metabolizing Enzymes in Primary Human Hepatocytes and Hepatoma Cell Line HepG2. J. Biochem. Mol. Toxicol. 2003, 17, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, N.J.; Hewitt, P. Phase I and II Enzyme Characterization of Two Sources of HepG2 Cell Lines. Xenobiotica 2004, 34, 243–256. [Google Scholar] [CrossRef]
- Aoyama, K.; Yoshinari, K.; Kim, H.-J.; Nagata, K.; Yamazoe, Y. Simultaneous Expression of Plural Forms of Human Cytochrome P450 at Desired Ratios in HepG2 Cells: Adenovirus-Mediated Tool for Cytochrome P450 Reconstitution. Drug Metab. Pharmacokinet. 2009, 24, 209–217. [Google Scholar] [CrossRef]
- Vignati, L.; Turlizzi, E.; Monaci, S.; Grossi, P.; de Kanter, R.; Monshouwer, M. An in Vitro Approach to Detect Metabolite Toxicity Due to CYP3A4-Dependent Bioactivation of Xenobiotics. Toxicology 2005, 216, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Hosomi, H.; Fukami, T.; Iwamura, A.; Nakajima, M.; Yokoi, T. Development of a Highly Sensitive Cytotoxicity Assay System for CYP3A4-Mediated Metabolic Activation. Drug Metab. Dispos. 2011, 39, 1388–1395. [Google Scholar] [CrossRef]
- Iwamura, A.; Fukami, T.; Hosomi, H.; Nakajima, M.; Yokoi, T. CYP2C9-Mediated Metabolic Activation of Losartan Detected by a Highly Sensitive Cell-Based Screening Assay. Drug Metab. Dispos. 2011, 39, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Donato, M.T.; Hallifax, D.; Picazo, L.; Castell, J.V.; Houston, J.B.; Gomez-Lechón, M.J.; Lahoz, A. Metabolite Formation Kinetics and Intrinsic Clearance of Phenacetin, Tolbutamide, Alprazolam, and Midazolam in Adenoviral Cytochrome P450-Transfected HepG2 Cells and Comparison with Hepatocytes and In Vivo. Drug Metab. Dispos. 2010, 38, 1449–1455. [Google Scholar] [CrossRef]
- Bai, J.; Cederbaum, A.I. Adenovirus Mediated Overexpression of CYP2E1 Increases Sensitivity of HepG2 Cells to Acetaminophen Induced Cytotoxicity. Mol. Cell Biochem. 2004, 262, 165–176. [Google Scholar] [CrossRef]
- Tolosa, L.; Gómez-Lechón, M.J.; Pérez-Cataldo, G.; Castell, J.V.; Donato, M.T. HepG2 Cells Simultaneously Expressing Five P450 Enzymes for the Screening of Hepatotoxicity: Identification of Bioactivable Drugs and the Potential Mechanism of Toxicity Involved. Arch. Toxicol. 2013, 87, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.-G.; Shi, J.-G.; Zhang, F.; Zhao, Q.-T.; Pang, X.-W.; Chen, R.; Hu, P.-Z.; Li, Q.-L.; Wang, Z.; Huang, G.-S. Overexpression of CYP2E1 Enhances Sensitivity of HepG2 Cells to Fas-Mediated Cytotoxicity. Cancer Biol. Ther. 2008, 7, 1280–1287. [Google Scholar] [CrossRef]
- Donato, M.; Jover, R.; Gómez-Lechón, M. Hepatic Cell Lines for Drug Hepatotoxicity Testing: Limitations and Strategies to Upgrade Their Metabolic Competence by Gene Engineering. Curr. Drug Metab. 2013, 14, 946–968. [Google Scholar] [CrossRef]
- Tolosa, L.; Donato, M.T.; Pérez-Cataldo, G.; Castell, J.V.; Gómez-Lechón, M.J. Upgrading Cytochrome P450 Activity in HepG2 Cells Co-Transfected with Adenoviral Vectors for Drug Hepatotoxicity Assessment. Toxicol. Vitr. 2012, 26, 1272–1277. [Google Scholar] [CrossRef]
- Chen, S.; Wu, Q.; Li, X.; Li, D.; Mei, N.; Ning, B.; Puig, M.; Ren, Z.; Tolleson, W.H.; Guo, L. Characterization of Cytochrome P450s (CYP)-Overexpressing HepG2 Cells for Assessing Drug and Chemical-Induced Liver Toxicity. J. Environ. Sci. Health Part C 2021, 39, 68–86. [Google Scholar] [CrossRef]
- Bort, R.; Macé, K.; Boobis, A.; Gómez-Lechón, M.-J.; Pfeifer, A.; Castell, J. Hepatic Metabolism of Diclofenac: Role of Human CYP in the Minor Oxidative Pathways. Biochem. Pharmacol. 1999, 58, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Herzog, N.; Katzenberger, N.; Martin, F.; Schmidtke, K.-U.; K, J.-H. Generation of Cytochrome P450 3A4-Overexpressing HepG2 Cell Clones for Standardization of Hepatocellular Testosterone 6β-Hydroxylation Activity. J. Cell Biotechnol. 2015, 1, 15–26. [Google Scholar] [CrossRef]
- Xuan, J.; Chen, S.; Ning, B.; Tolleson, W.H.; Guo, L. Development of HepG2-Derived Cells Expressing Cytochrome P450s for Assessing Metabolism-Associated Drug-Induced Liver Toxicity. Chem. Biol. Interact. 2016, 255, 63–73. [Google Scholar] [CrossRef]
- Jiang, J.; Yang, B.; Ji, L.; Yang, L.; Mao, Y.-C.; Hu, Z.; Wang, Z.; Wang, C. Metabolic-Induced Cytotoxicity of Diosbulbin B in CYP3A4-Expressing Cells. Toxicol. Vitr. 2017, 38, 59–66. [Google Scholar] [CrossRef]
- Chiang, T.-S.; Yang, K.-C.; Chiou, L.-L.; Huang, G.-T.; Lee, H.-S. Enhancement of CYP3A4 Activity in Hep G2 Cells by Lentiviral Transfection of Hepatocyte Nuclear Factor-1 Alpha. PLoS ONE 2014, 9, e94885. [Google Scholar] [CrossRef]
- Nakamura, K.; Aizawa, K.; Aung, K.H.; Yamauchi, J.; Tanoue, A. Zebularine Upregulates Expression of CYP Genes through Inhibition of DNMT1 and PKR in HepG2 Cells. Sci. Rep. 2017, 7, 41093. [Google Scholar] [CrossRef] [PubMed]
- Ruoß, M.; Damm, G.; Vosough, M.; Ehret, L.; Grom-Baumgarten, C.; Petkov, M.; Naddalin, S.; Ladurner, R.; Seehofer, D.; Nussler, A.; et al. Epigenetic Modifications of the Liver Tumor Cell Line HepG2 Increase Their Drug Metabolic Capacity. Int. J. Mol. Sci. 2019, 20, 347. [Google Scholar] [CrossRef]
- Nagai, K.; Fukuno, S.; Miura, T.; Yasuda-Imanishi, E.; Konishi, H. Altered Gene Expression of Cytochrome P450 and ABC Transporter in Human Hepatocellular Carcinoma HepG2 Cells Exposed to Bardoxolone Methyl. Drug Res. 2023, 73, 473–475. [Google Scholar] [CrossRef]
- Prelich, G. Gene Overexpression: Uses, Mechanisms, and Interpretation. Genetics 2012, 190, 841–854. [Google Scholar] [CrossRef]
- Li, X. Notable Drug-Drug Interaction between Omeprazole and Voriconazole in CYP2C19 *1 and *2 (Rs4244285, 681G>A) Alleles in Vitro. Xenobiotica 2024, 54, 847–854. [Google Scholar] [CrossRef]
- Xun, T.; Rong, Y.; Lv, B.; Tian, J.; Zhang, Q.; Yang, X. Interaction and Potential Mechanisms between Atorvastatin and Voriconazole, Agents Used to Treat Dyslipidemia and Fungal Infections. Front. Pharmacol. 2023, 14, 1165950. [Google Scholar] [CrossRef] [PubMed]
- Sager, J.E.; Tripathy, S.; Price, L.S.L.; Nath, A.; Chang, J.; Stephenson-Famy, A.; Isoherranen, N. In Vitro to in Vivo Extrapolation of the Complex Drug-Drug Interaction of Bupropion and Its Metabolites with CYP2D6; Simultaneous Reversible Inhibition and CYP2D6 Downregulation. Biochem. Pharmacol. 2017, 123, 85–96. [Google Scholar] [CrossRef]
- Cui, H.; Wang, J.; Zhang, Q.; Dang, M.; Liu, H.; Dong, Y.; Zhang, L.; Yang, F.; Wu, J.; Tong, X. In Vivo and in Vitro Study on Drug-Drug Interaction of Lovastatin and Berberine from Pharmacokinetic and HepG2 Cell Metabolism Studies. Molecules 2016, 21, 464. [Google Scholar] [CrossRef]
- Andersson, T.B.; Kanebratt, K.P.; Kenna, J.G. The HepaRG Cell Line: A Unique in Vitro Tool for Understanding Drug Metabolism and Toxicology in Human. Expert. Opin. Drug Metab. Toxicol. 2012, 8, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Turpeinen, M.; Tolonen, A.; Chesne, C.; Guillouzo, A.; Uusitalo, J.; Pelkonen, O. Functional Expression, Inhibition and Induction of CYP Enzymes in HepaRG Cells. Toxicol. Vitr. 2009, 23, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a Human Hepatoma Cell Line by Hepatitis B Virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef]
- Aninat, C.; Piton, A.; Glaise, D.; Le Charpentier, T.; Langouët, S.; Morel, F.; Guguen-Guillouzo, C.; Guillouzo, A. Expression of Cytochromes P450, Conjugating Enzymes and Nuclear Receptors in Human Hepatoma HepaRG Cells. Drug Metab. Dispos. 2006, 34, 75–83. [Google Scholar] [CrossRef]
- Cerec, V.; Glaise, D.; Garnier, D.; Morosan, S.; Turlin, B.; Drenou, B.; Gripon, P.; Kremsdorf, D.; Guguen-Guillouzo, C.; Corlu, A. Transdifferentiation of Hepatocyte-like Cells from the Human Hepatoma HepaRG Cell Line through Bipotent Progenitor†. Hepatology 2007, 45, 957–967. [Google Scholar] [CrossRef]
- Anthérieu, S.; Chesné, C.; Li, R.; Guguen-Guillouzo, C.; Guillouzo, A. Optimization of the HepaRG Cell Model for Drug Metabolism and Toxicity Studies. Toxicol. Vitr. 2012, 26, 1278–1285. [Google Scholar] [CrossRef]
- Guillouzo, A.; Corlu, A.; Aninat, C.; Glaise, D.; Morel, F.; Guguen-Guillouzo, C. The Human Hepatoma HepaRG Cells: A Highly Differentiated Model for Studies of Liver Metabolism and Toxicity of Xenobiotics. Chem. Biol. Interact. 2007, 168, 66–73. [Google Scholar] [CrossRef]
- Sison-Young, R.L.C.; Mitsa, D.; Jenkins, R.E.; Mottram, D.; Alexandre, E.; Richert, L.; Aerts, H.; Weaver, R.J.; Jones, R.P.; Johann, E.; et al. Comparative Proteomic Characterization of 4 Human Liver-Derived Single Cell Culture Models Reveals Significant Variation in the Capacity for Drug Disposition, Bioactivation, and Detoxication. Toxicol. Sci. 2015, 147, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Parent, R.; Marion, M.-J.; Furio, L.; Trépo, C.; Petit, M.-A. Origin and Characterization of a Human Bipotent Liver Progenitor Cell Line. Gastroenterology 2004, 126, 1147–1156. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Sasaki, Y.; Terasaki, N.; Kawataki, T.; Takekawa, K.; Iwase, Y.; Shimizu, T.; Sanoh, S.; Ohta, S. Comparison of Drug Metabolism and Its Related Hepatotoxic Effects in HepaRG, Cryopreserved Human Hepatocytes, and HepG2 Cell Cultures. Biol. Pharm. Bull. 2018, 41, 722–732. [Google Scholar] [CrossRef]
- Hart, S.N.; Li, Y.; Nakamoto, K.; Subileau, E.; Steen, D.; Zhong, X. A Comparison of Whole Genome Gene Expression Profiles of HepaRG Cells and HepG2 Cells to Primary Human Hepatocytes and Human Liver Tissues. Drug Metab. Dispos. 2010, 38, 988–994. [Google Scholar] [CrossRef]
- Gerbal-Chaloin, S.; Daujat, M.; Pascussi, J.-M.; Pichard-Garcia, L.; Vilarem, M.-J.; Maurel, P. Transcriptional Regulation of CYP2C9 Gene. J. Biol. Chem. 2002, 277, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Lübberstedt, M.; Müller-Vieira, U.; Mayer, M.; Biemel, K.M.; Knöspel, F.; Knobeloch, D.; Nüssler, A.K.; Gerlach, J.C.; Zeilinger, K. HepaRG Human Hepatic Cell Line Utility as a Surrogate for Primary Human Hepatocytes in Drug Metabolism Assessment in Vitro. J. Pharmacol. Toxicol. Methods 2011, 63, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Kanebratt, K.P.; Andersson, T.B. HepaRG Cells as an in Vitro Model for Evaluation of Cytochrome P450 Induction in Humans. Drug Metab. Dispos. 2008, 36, 137–145. [Google Scholar] [CrossRef]
- Jossé, R.; Aninat, C.; Glaise, D.; Dumont, J.; Fessard, V.; Morel, F.; Poul, J.-M.; Guguen-Guillouzo, C.; Guillouzo, A. Long-Term Functional Stability of Human HepaRG Hepatocytes and Use for Chronic Toxicity and Genotoxicity Studies. Drug Metab. Dispos. 2008, 36, 1111–1118. [Google Scholar] [CrossRef]
- Anthérieu, S.; Chesné, C.; Li, R.; Camus, S.; Lahoz, A.; Picazo, L.; Turpeinen, M.; Tolonen, A.; Uusitalo, J.; Guguen-Guillouzo, C.; et al. Stable Expression, Activity, and Inducibility of Cytochromes P450 in Differentiated HepaRG Cells. Drug Metab. Dispos. 2010, 38, 516–525. [Google Scholar] [CrossRef]
- Dubois-Pot-Schneider, H.; Aninat, C.; Kattler, K.; Fekir, K.; Jarnouen, K.; Cerec, V.; Glaise, D.; Salhab, A.; Gasparoni, G.; Takashi, K.; et al. Transcriptional and Epigenetic Consequences of DMSO Treatment on HepaRG Cells. Cells 2022, 11, 2298. [Google Scholar] [CrossRef]
- Aleksandrova, A.V.; Burmistrova, O.A.; Fomicheva, K.A.; Sakharov, D.A. Maintenance of High Cytochrome P450 Expression in HepaRG Cell Spheroids in DMSO-Free Medium. Bull. Exp. Biol. Med. 2016, 161, 120–124. [Google Scholar] [CrossRef]
- Rebelo, S.P.; Costa, R.; Estrada, M.; Shevchenko, V.; Brito, C.; Alves, P.M. HepaRG Microencapsulated Spheroids in DMSO-Free Culture: Novel Culturing Approaches for Enhanced Xenobiotic and Biosynthetic Metabolism. Arch. Toxicol. 2015, 89, 1347–1358. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-Y.; Li, W.-J.; Li, Q.-G.; Jing, H.-S.; Yuan, T.-J.; Fu, G.-B.; Tang, D.; Zhang, H.-D.; Yan, H.-X.; Zhai, B. A DMSO-Free Hepatocyte Maturation Medium Accelerates Hepatic Differentiation of HepaRG Cells in Vitro. Biomed. Pharmacother. 2019, 116, 109010. [Google Scholar] [CrossRef] [PubMed]
- Sison-Young, R.L.; Lauschke, V.M.; Johann, E.; Alexandre, E.; Antherieu, S.; Aerts, H.; Gerets, H.H.J.; Labbe, G.; Hoët, D.; Dorau, M.; et al. A Multicenter Assessment of Single-Cell Models Aligned to Standard Measures of Cell Health for Prediction of Acute Hepatotoxicity. Arch. Toxicol. 2017, 91, 1385–1400. [Google Scholar] [CrossRef] [PubMed]
- Hammour, M.M.; Othman, A.; Aspera-Werz, R.; Braun, B.; Weis-Klemm, M.; Wagner, S.; Nadalin, S.; Histing, T.; Ruoß, M.; Nüssler, A.K. Optimisation of the HepaRG Cell Line Model for Drug Toxicity Studies Using Two Different Cultivation Conditions: Advantages and Limitations. Arch. Toxicol. 2022, 96, 2511–2521. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Z.; Dong, Z.; Zhu, L.; Xu, J.; Wu, D.; Liang, W. Differentiation of HepaRG Cells into Hepatocytes Based on Substrate Elasticity. Int. J. Clin. Exp. Med. 2018, 11, 10183–10190. [Google Scholar]
- Gomes, J.V.D.; Herz, C.; Helmig, S.; Förster, N.; Mewis, I.; Lamy, E. Drug-Drug Interaction Potential, Cytotoxicity, and Reactive Oxygen Species Production of Salix Cortex Extracts Using Human Hepatocyte-Like HepaRG Cells. Front. Pharmacol. 2021, 12, 779801. [Google Scholar] [CrossRef]
- Meirinho, S.; Rodrigues, M.; Fortuna, A.; Falcão, A.; Alves, G. Study of the Metabolic Stability Profiles of Perampanel, Rufinamide and Stiripentol and Prediction of Drug Interactions Using HepaRG Cells as an in Vitro Human Model. Toxicol. Vitr. 2022, 82, 105389. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Lechón, M.J.; Donato, T.; Jover, R.; Rodriguez, C.; Ponsoda, X.; Glaise, D.; Castell, J.V.; Guguen-Guillouzo, C. Expression and Induction of a Large Set of Drug-metabolizing Enzymes by the Highly Differentiated Human Hepatoma Cell Line BC2. Eur. J. Biochem. 2001, 268, 1448–1459. [Google Scholar] [CrossRef]
- Glaise, D.; Ilyin, G.P.; Loyer, P.; Cariou, S.; Bilodeau, M.; Lucas, J.; Puisieux, A.; Ozturk, M.; Guguen-Guillouzo, C. Cell Cycle Gene Regulation in Reversibly Differentiated New Human Hepatoma Cell Lines. Cell Growth Differ. 1998, 9, 165–176. [Google Scholar]
- Fabre, N.; Arrivet, E.; Trancard, J.; Bichet, N.; Roome, N.O.; Prenez, A.; Vericat, J.-A. A New Hepatoma Cell Line for Toxicity Testing at Repeated Doses. Int. J. Clin. Exp. Med. 2003, 19, 10183–10190. [Google Scholar]
- Fabre, N.; Arrivet, E.; Paillard, F.; Wibaut-Berlaimont, V.; Bichet, N.; Roome, N.O.; Prenez, A.; Vericat, J.-A. A New Human Hepatoma Cell Line to Study Repeated Cell Toxicity. Altern. Lab. Anim. 2004, 32 (Suppl. 1A), 113–116. [Google Scholar] [PubMed]
- Knake, C.; Stamp, L.; Bahn, A. Molecular Mechanism of an Adverse Drug-Drug Interaction of Allopurinol and Furosemide in Gout Treatment. Biochem. Biophys. Res. Commun. 2014, 452, 157–162. [Google Scholar] [CrossRef]
- Ferreira, A.; Rodrigues, M.; Silvestre, S.; Falcão, A.; Alves, G. HepaRG Cell Line as an in Vitro Model for Screening Drug-Drug Interactions Mediated by Metabolic Induction: Amiodarone Used as a Model Substance. Toxicol. Vitr. 2014, 28, 1531–1535. [Google Scholar] [CrossRef]
- Ramachandran, S.D.; Vivarès, A.; Klieber, S.; Hewitt, N.J.; Muenst, B.; Heinz, S.; Walles, H.; Braspenning, J. Applicability of Second-generation Upcyte ® Human Hepatocytes for Use in CYP Inhibition and Induction Studies. Pharmacol. Res. Perspect. 2015, 3, e00161. [Google Scholar] [CrossRef] [PubMed]
- Harati, R.; Vandamme, M.; Blanchet, B.; Bardin, C.; Praz, F.; Hamoudi, R.A.; Desbois-Mouthon, C. Drug-Drug Interaction between Metformin and Sorafenib Alters Antitumor Effect in Hepatocellular Carcinoma Cells. Mol. Pharmacol. 2021, 100, 32–45. [Google Scholar] [CrossRef]
- Tian, S.; Su, R.; Wu, K.; Zhou, X.; Vadgama, J.V.; Wu, Y. Diaporine Potentiates the Anticancer Effects of Oxaliplatin and Doxorubicin on Liver Cancer Cells. J. Pers. Med. 2022, 12, 1318. [Google Scholar] [CrossRef]
- Kwon, S.J.; Lee, D.W.; Shah, D.A.; Ku, B.; Jeon, S.Y.; Solanki, K.; Ryan, J.D.; Clark, D.S.; Dordick, J.S.; Lee, M.-Y. High-Throughput and Combinatorial Gene Expression on a Chip for Metabolism-Induced Toxicology Screening. Nat. Commun. 2014, 5, 3739. [Google Scholar] [CrossRef]
- Tassaneeyakul, W.; Birkett, D.J.; Veronese, M.E.; McManus, M.E.; Tukey, R.H.; Quattrochi, L.C.; Gelboin, H.V.; Miners, J.O. Specificity of Substrate and Inhibitor Probes for Human Cytochromes P450 1A1 and 1A2. J. Pharmacol. Exp. Ther. 1993, 265, 401–407. [Google Scholar] [CrossRef]
- Tsyrlov, I.B.; Goldfarb, I.S.; Gelboin, H.V. Enzyme-Kinetic and Immunochemical Characteristics of Mouse CDNA-Expressed, Microsomal, and Purified CYP1A1 and CYP1A2. Arch. Biochem. Biophys. 1993, 307, 259–266. [Google Scholar] [CrossRef]
- Sesardic, D.; Pasanen, M.; Pelkonen, O.; Boobis, A.R. Differential Expression and Regulation of Members of the Cytochrome P450IA Gene Subfamily in Human Tissues. Carcinogenesis 1990, 11, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- Kunze, K.L.; Trager, W.F. Isoform-Selective Mechanism-Based Inhibition of Human Cytochrome P450 1A2 by Furafylline. Chem. Res. Toxicol. 1993, 6, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.E.; Ayrton, A.D.; Chenery, R.J. Characterization of the Inhibition of P4501A2 by Furafylline. Xenobiotica 1994, 24, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.E.; Baldwin, S.J.; Bloomer, J.C.; Ayrton, A.D.; Sozio, R.S.; Chenery, R.J. Lauric Acid as a Model Substrate for the Simultaneous Determination of Cytochrome P450 2E1 and 4A in Hepatic Microsomes. Chem. Res. Toxicol. 1994, 7, 836–842. [Google Scholar] [CrossRef]
- Richter, T.; Mürdter, T.E.; Heinkele, G.; Pleiss, J.; Tatzel, S.; Schwab, M.; Eichelbaum, M.; Zanger, U.M. Potent Mechanism-Based Inhibition of Human CYP2B6 by Clopidogrel and Ticlopidine. J. Pharmacol. Exp. Ther. 2004, 308, 189–197. [Google Scholar] [CrossRef]
- Turpeinen, M.; Tolonen, A.; Uusitalo, J.; Jalonen, J.; Pelkonen, O.; Laine, K. Effect of Clopidogrel and Ticlopidine on Cytochrome P450 2B6 Activity as Measured by Bupropion Hydroxylation. Clin. Pharmacol. Ther. 2005, 77, 553–559. [Google Scholar] [CrossRef]
- Backman, J.T.; Filppula, A.M.; Niemi, M.; Neuvonen, P.J. Role of Cytochrome P450 2C8 in Drug Metabolism and Interactions. Pharmacol. Rev. 2016, 68, 168–241. [Google Scholar] [CrossRef]
- Zhou, Q.; Chen, M.; Zhu, L.; Yu, L.; Zeng, S.; Xiang, M.; Wang, Z.-Y. Pharmacokinetic Drug Interactions with Clopidogrel: Updated Review and Risk Management in Combination Therapy. Ther. Clin. Risk Manag. 2015, 11, 449–467. [Google Scholar] [CrossRef]
- Hesse, L.M.; Venkatakrishnan, K.; Court, M.H.; von Moltke, L.L.; Duan, S.X.; Shader, R.I.; Greenblatt, D.J. CYP2B6 Mediates the in Vitro Hydroxylation of Bupropion: Potential Drug Interactions with Other Antidepressants. Drug Metab. Dispos. 2000, 28, 1176–1183. [Google Scholar] [CrossRef]
- Palacharla, R.C.; Nirogi, R.; Uthukam, V.; Manoharan, A.; Ponnamaneni, R.K.; Kalaikadhiban, I. Quantitative in Vitro Phenotyping and Prediction of Drug Interaction Potential of CYP2B6 Substrates as Victims. Xenobiotica 2018, 48, 663–675. [Google Scholar] [CrossRef]
- Rae, J.M.; Soukhova, N.V.; Flockhart, D.A.; Desta, Z. Triethylenethiophosphoramide Is a Specific Inhibitor of Cytochrome P450 2B6: Implications for Cyclophosphamide Metabolism. Drug Metab. Dispos. 2002, 30, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Harleton, E.; Webster, M.; Bumpus, N.N.; Kent, U.M.; Rae, J.M.; Hollenberg, P.F. Metabolism of N,N′,N″-Triethylenethiophosphoramide by CYP2B1 and CYP2B6 Results in the Inactivation of Both Isoforms by Two Distinct Mechanisms. J. Pharmacol. Exp. Ther. 2004, 310, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Turpeinen, M.; Nieminen, R.; Juntunen, T.; Taavitsainen, P.; Raunio, H.; Pelkonen, O. Selective inhibition of cyp2b6-catalyzed bupropion hydroxylation in human liver microsomes in vitro. Drug Metab. Dispos. 2004, 32, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Richter, T.; Schwab, M.; Eichelbaum, M.; Zanger, U.M. Inhibition of Human CYP2B6 by N,N′,N″-Triethylenethiophosphoramide Is Irreversible and Mechanism-Based. Biochem. Pharmacol. 2005, 69, 517–524. [Google Scholar] [CrossRef]
- Bae, S.H.; Kwon, M.J.; Choi, E.J.; Zheng, Y.F.; Yoon, K.D.; Liu, K.-H.; Bae, S.K. Potent Inhibition of Cytochrome P450 2B6 by Sibutramine in Human Liver Microsomes. Chem. Biol. Interact. 2013, 205, 11–19. [Google Scholar] [CrossRef]
- Turpeinen, M.; Jouko, U.; Jorma, J.; Olavi, P. Multiple P450 Substrates in a Single Run: Rapid and Comprehensive in Vitro Interaction Assay. Eur. J. Pharm. Sci. 2005, 24, 123–132. [Google Scholar] [CrossRef]
- Wang, J.-S.; Neuvonen, M.; Wen, X.; Backman, J.T.; Neuvonen, P.J. Gemfibrozil Inhibits CYP2C8-Mediated Cerivastatin Metabolism in Human Liver Microsomes. Drug Metab. Dispos. 2002, 30, 1352–1356. [Google Scholar] [CrossRef]
- Kajosaari, L.I.; Laitila, J.; Neuvonen, P.J.; Backman, J.T. Metabolism of Repaglinide by CYP2C8 and CYP3A4 In Vitro: Effect of Fibrates and Rifampicin. Basic. Clin. Pharmacol. Toxicol. 2005, 97, 249–256. [Google Scholar] [CrossRef]
- Ogilvie, B.W.; Zhang, D.; Li, W.; Rodrigues, A.D.; Gipson, A.E.; Holsapple, J.; Toren, P.; Parkinson, A. Glucuronidation converts gemfibrozil to a potent, metabolism-dependent inhibitor of cyp2c8: Implications for drug-drug interactions. Drug Metab. Dispos. 2006, 34, 191–197. [Google Scholar] [CrossRef]
- Kahma, H.; Filppula, A.M.; Launiainen, T.; Viinamäki, J.; Neuvonen, M.; Evangelista, E.A.; Totah, R.A.; Backman, J.T. Critical Differences between Enzyme Sources in Sensitivity to Detect Time-Dependent Inactivation of CYP2C8. Drug Metab. Dispos. 2019, 47, 436–443. [Google Scholar] [CrossRef]
- Walsky, R.L.; Gaman, E.A.; Obach, R.S. Examination of 209 Drugs for Inhibition of Cytochrome P450 2C8. J. Clin. Pharmacol. 2005, 45, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Walsky, R.L.; Obach, R.S.; Gaman, E.A.; Gleeson, J.-P.R.; Proctor, W.R. Selective inhibition of human cytochrome p4502c8 by montelukast. Drug Metab. Dispos. 2005, 33, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, T.; Patel, A.; Templeton, I.; Chen, Y.; Lu, C.; Lai, G.; Leung, L.; Tse, S.; Einolf, H.J.; Wang, Y.-H.; et al. Evaluation of a New Molecular Entity as a Victim of Metabolic Drug-Drug Interactions—An Industry Perspective. Drug Metab. Dispos. 2016, 44, 1399–1423. [Google Scholar] [CrossRef]
- Back, D.; Tjia, J.; Karbwang, J.; Colbert, J. In Vitro Inhibition Studies of Tolbutamide Hydroxylase Activity of Human Liver Microsomes by Azoles, Sulphonamides and Quinolines. Br. J. Clin. Pharmacol. 1988, 26, 23–29. [Google Scholar] [CrossRef]
- Bourrié, M.; Meunier, V.; Berger, Y.; Fabre, G. Cytochrome P450 Isoform Inhibitors as a Tool for the Investigation of Metabolic Reactions Catalyzed by Human Liver Microsomes. J. Pharmacol. Exp. Ther. 1996, 277, 321–332. [Google Scholar] [CrossRef]
- Baldwin, S.J.; Bloomer, J.C.; Smith, G.J.; Ayrton, A.D.; Clarke, S.E.; Chenery, R.J. Ketoconazole and Sulphaphenazole as the Respective Selective Inhibitors of P4503A and 2C9. Xenobiotica 1995, 25, 261–270. [Google Scholar] [CrossRef]
- Hutzler, J.M.; Balogh, L.M.; Zientek, M.; Kumar, V.; Tracy, T.S. Mechanism-Based Inactivation of Cytochrome P450 2C9 by Tienilic Acid and (±)-Suprofen: A Comparison of Kinetics and Probe Substrate Selection. Drug Metab. Dispos. 2009, 37, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kneller, M.B.; Haining, R.L.; Trager, W.F.; Rettie, A.E. (+)-N-3-Benzyl-Nirvanol and (−)-N-3-Benzyl-Phenobarbital: New Potent and Selective in Vitro Inhibitors of CYP2C19. Drug Metab. Dispos. 2002, 30, 235–239. [Google Scholar] [CrossRef]
- Barecki, M.E.; Casciano, C.N.; Johnson, W.W.; Clement, R.P. In Vitro Characterization of the Inhibition Profile of Loratadine, Desloratadine, and 3-OH-Desloratadine for Five Human Cytochrome P-450 Enzymes. Drug Metab. Dispos. 2001, 29, 1173–1175. [Google Scholar]
- Tassaneeyakul, W.; Guo, L.-Q.; Fukuda, K.; Ohta, T.; Yamazoe, Y. Inhibition Selectivity of Grapefruit Juice Components on Human Cytochromes P450. Arch. Biochem. Biophys. 2000, 378, 356–363. [Google Scholar] [CrossRef]
- Ha-Duong, N.-T.; Dijols, S.; Macherey, A.-C.; Goldstein, J.A.; Dansette, P.M.; Mansuy, D. Ticlopidine as a Selective Mechanism-Based Inhibitor of Human Cytochrome P450 2C19. Biochemistry 2001, 40, 12112–12122. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.W.; Desta, Z.; Soukhova, N.V.; Tracy, T.; Flockhart, D.A. In Vitro Inhibition of the Cytochrome P450 (CYP450) System by the Antiplatelet Drug Ticlopidine: Potent Effect on CYP2C19 and CYP2D6. Br. J. Clin. Pharmacol. 2000, 49, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Turpeinen, M.; Raunio, H.; Pelkonen, O. The Functional Role of CYP2B6 in Human Drug Metabolism: Substrates and Inhibitors In Vitro, In Vivo and In Silico. Curr. Drug Metab. 2006, 7, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Crewe, H.; Lennard, M.; Tucker, G.; Woods, F.; Haddock, R. The Effect of Selective Serotonin Re-uptake Inhibitors on Cytochrome P4502D6 (CYP2D6) Activity in Human Liver Microsomes. Br. J. Clin. Pharmacol. 1992, 34, 262–265. [Google Scholar] [CrossRef]
- Kobayashi, K.; Yamamoto, T.; Chiba, K.; Tani, M.; Ishizaki, T.; Kuroiwa, Y. The Effects of Selective Serotonin Reuptake Inhibitors and Their Metabolites on S-mephenytoin 4′-hydroxylase Activity in Human Liver Microsomes. Br. J. Clin. Pharmacol. 1995, 40, 481–485. [Google Scholar] [CrossRef]
- Schmider, J.; Greenblatt, D.J.; Fogelman, S.M.; von Moltke, L.L.; Shader, R.I. Metabolism of Dextromethorphanin Vitro: Involvement of Cytochromes P450 2D6 AND 3A3/4, with a Possible Role of 2E1. Biopharm. Drug Dispos. 1997, 18, 227–240. [Google Scholar] [CrossRef]
- Broly, F.; Libersa, C.; Lhermitte, M.; Bechtel, P.; Dupuis, B. Effect of Quinidine on the Dextromethorphan O-demethylase Activity of Microsomal Fractions from Human Liver. Br. J. Clin. Pharmacol. 1989, 28, 29–36. [Google Scholar] [CrossRef]
- Otton, S.V.; Inaba, T.; Kalow, W. Competitive Inhibition of Sparteine Oxidation in Human Liver by β-Adrenoceptor Antagonists and Other Cardiovascular Drugs. Life Sci. 1984, 34, 73–80. [Google Scholar] [CrossRef]
- Otton, S.V.; Brinn, R.U.; Gram, L.F. In Vitro Evidence against the Oxidation of Quinidine by the Sparteine/Debrisoquine Monooxygenase of Human Liver. Drug Metab. Dispos. 1988, 16, 15–17. [Google Scholar] [CrossRef]
- Parmentier, Y.; Pothier, C.; Delmas, A.; Caradec, F.; Trancart, M.-M.; Guillet, F.; Bouaita, B.; Chesne, C.; Brian Houston, J.; Walther, B. Direct and Quantitative Evaluation of the Human CYP3A4 Contribution (f m) to Drug Clearance Using the In Vitro SILENSOMES Model. Xenobiotica 2017, 47, 562–575. [Google Scholar] [CrossRef]
- Stresser, D.M.; Broudy, M.I.; Ho, T.; Cargill, C.E.; Blanchard, A.P.; Sharma, R.; Dandeneau, A.A.; Goodwin, J.J.; Turner, S.D.; Erve, J.C.L.; et al. Highly selective inhibition of human cyp3a in vitro by azamulin and evidence that inhibition is irreversible. Drug Metab. Dispos. 2004, 32, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Isoherranen, N.; Kunze, K.L.; Allen, K.E.; Nelson, W.L.; Thummel, K.E. Role of itraconazole metabolites in cyp3a4 inhibition. Drug Metab. Dispos. 2004, 32, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- von Moltke, L.L.; Greenblatt, D.J.; Harmatz, J.S.; Duan, S.X.; Harrel, L.M.; Cotreau-Bibbo, M.M.; Pritchard, G.A.; Wright, C.E.; Shader, R.I. Triazolam Biotransformation by Human Liver Microsomes in Vitro: Effects of Metabolic Inhibitors and Clinical Confirmation of a Predicted Interaction with Ketoconazole. J. Pharmacol. Exp. Ther. 1996, 276, 370–379. [Google Scholar] [CrossRef]
- Niwa, T.; Shiraga, T.; Takagi, A. Effect of Antifungal Drugs on Cytochrome P450 (CYP) 2C9, CYP2C19, and CYP3A4 Activities in Human Liver Microsomes. Biol. Pharm. Bull. 2005, 28, 1805–1808. [Google Scholar] [CrossRef]
- Yoshida, K.; Maeda, K.; Konagaya, A.; Kusuhara, H. Accurate Estimation of In Vivo Inhibition Constants of Inhibitors and Fraction Metabolized of Substrates with Physiologically Based Pharmacokinetic Drug-Drug Interaction Models Incorporating Parent Drugs and Metabolites of Substrates with Cluster Newton Method. Drug Metab. Dispos. 2018, 46, 1805–1816. [Google Scholar] [CrossRef] [PubMed]
- Newton, D.J.; Wang, R.W.; Lu, A.Y. Cytochrome P450 Inhibitors. Evaluation of Specificities in the in Vitrometabolism of Therapeutic Agents by Human Liver Microsomes. Drug Metab. Dispos. 1995, 23, 154–158. [Google Scholar] [CrossRef]
- Yamazaki, H.; Inoue, K.; Chiba, K.; Ozawa, N.; Kawai, T.; Suzuki, Y.; Goldstein, J.A.; Guengerich, F.P.; Shimada, T. Comparative Studies on the Catalytic Roles of Cytochrome P450 2C9 and Its Cys- and Leu-Variants in the Oxidation of Warfarin, Flurbiprofen, and Diclofenac by Human Liver Microsomes. Biochem. Pharmacol. 1998, 56, 243–251. [Google Scholar] [CrossRef]
- Soars, M.G.; Grime, K.; Riley, R.J. Comparative Analysis of Substrate and Inhibitor Interactions with CYP3A4 and CYP3A5. Xenobiotica 2006, 36, 287–299. [Google Scholar] [CrossRef]
- Yeo, K.R.; Yeo, W.W. Inhibitory Effects of Verapamil and Diltiazem on Simvastatin Metabolism in Human Liver Microsomes. Br. J. Clin. Pharmacol. 2001, 51, 461–470. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Jones, D.R.; Hall, S.D. DIFFERENTIAL MECHANISM-BASED INHIBITION OF CYP3A4 AND CYP3A5 BY VERAPAMIL. Drug Metab. Dispos. 2005, 33, 664–671. [Google Scholar] [CrossRef]
- Palleria, C.; Di Paolo, A.; Giofrè, C.; Caglioti, C.; Leuzzi, G.; Siniscalchi, A.; De Sarro, G.; Gallelli, L. Pharmacokinetic Drug-Drug Interaction and Their Implication in Clinical Management. J. Res. Med. Sci. 2013, 18, 601–610. [Google Scholar] [PubMed]
- Døssing, M.; Pilsgaard, H.; Rasmussen, B.; Poulsen, H.E. Time Course of Phenobarbital and Cimetidine Mediated Changes in Hepatic Drug Metabolism. Eur. J. Clin. Pharmacol. 1983, 25, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Murray, M. Drug-Mediated Inactivation of Cytochrome P450. Clin. Exp. Pharmacol. Physiol. 1997, 24, 465–470. [Google Scholar] [CrossRef]
- European Medicines Agency. ICH M12 Guideline on Drug Interaction Studies; European Medicines Agency: London, UK, 2024.
- Deodhar, M.; Al Rihani, S.B.; Arwood, M.J.; Darakjian, L.; Dow, P.; Turgeon, J.; Michaud, V. Mechanisms of CYP450 Inhibition: Understanding Drug-Drug Interactions Due to Mechanism-Based Inhibition in Clinical Practice. Pharmaceutics 2020, 12, 846. [Google Scholar] [CrossRef]
- Ring, B.; Wrighton, S.A.; Mohutsky, M. Reversible Mechanisms of Enzyme Inhibition and Resulting Clinical Significance. Methods Mol. Biol. 2014, 1113, 37–56. [Google Scholar]
- Lin, J.H.; Lu, A.Y.H. Inhibition and Induction of Cytochrome P450 and the Clinical Implications. Clin. Pharmacokinet. 1998, 35, 361–390. [Google Scholar] [CrossRef] [PubMed]
- Seibert, E.; Tracy, T.S. Fundamentals of Enzyme Kinetics. Methods Mol. Biol. 2014, 1113, 9–22. [Google Scholar]
- Doehmer, J.; Tewes, B.; Klein, K.-U.; Gritzko, K.; Muschick, H.; Mengs, U. Assessment of Drug-Drug Interaction for Silymarin. Toxicol. Vitr. 2008, 22, 610–617. [Google Scholar] [CrossRef]
- Rioux, N.; Batonga, J.; Colombo, F.; Massé, J.; Zouki, C.; Ribadeneira, M.D.; Duan, J.; Bethell, R.C. A Simplified Approach to Predict CYP3A-Mediated Drug-Drug Interactions at Early Drug Discovery: Validation with Clinical Data. Xenobiotica 2013, 43, 592–597. [Google Scholar] [CrossRef]
- Ishigam, M.; Uchiyama, M.; Kondo, T.; Iwabuchi, H.; Inoue, S.; Takasaki, W.; Ikeda, T.; Komai, T.; Ito, K.; Sugiyama, Y. Inhibition of in Vitro Metabolism of Simvastatin by Itraconazole in Humans and Prediction of in Vivo Drug-Drug Interactions. Pharm. Res. 2001, 18, 622–631. [Google Scholar] [CrossRef]
- Chen, N.; Cui, D.; Wang, Q.; Wen, Z.; Finkelman, R.D.; Welty, D. In Vitro Drug-Drug Interactions of Budesonide: Inhibition and Induction of Transporters and Cytochrome P450 Enzymes. Xenobiotica 2018, 48, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.-L.T.; McAnulty, M.J.; Evers, D.L.; Reinhardt, B.J.; Weina, P.J. Pre-Clinical Drug-Drug Interaction (DDI) of Gefitinib or Erlotinib with Cytochrome P450 (CYP) Inhibiting Drugs, Fluoxetine and/or Losartan. Curr. Res. Toxicol. 2021, 2, 217–224. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, D.; Raghavan, N.; Yao, M.; Ma, L.; Frost, C.A.; Maxwell, B.D.; Chen, S.; He, K.; Goosen, T.C.; et al. In Vitro Assessment of Metabolic Drug-Drug Interaction Potential of Apixaban through Cytochrome P450 Phenotyping, Inhibition, and Induction Studies. Drug Metab. Dispos. 2010, 38, 448–458. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, J.; Kirk, C.; Fang, Y.; Alsina, M.; Badros, A.; Papadopoulos, K.; Wong, A.; Woo, T.; Bomba, D.; et al. Clinical Pharmacokinetics, Metabolism, and Drug-Drug Interaction of Carfilzomib. Drug Metab. Dispos. 2013, 41, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Li, A.P.; Jurima-Romet, M. Applications of Primary Human Hepatocytes in the Evaluation of Pharmacokinetic Drug-Drug Interactions: Evaluation of Model Drugs Terfenadine and Rifampin. Cell Biol. Toxicol. 1997, 13, 365–374. [Google Scholar] [CrossRef]
- Beumer, J.H.; Pillai, V.C.; Parise, R.A.; Christner, S.M.; Kiesel, B.F.; Rudek, M.A.; Venkataramanan, R. Human Hepatocyte Assessment of Imatinib Drug-Drug Interactions—Complexities in Clinical Translation. Br. J. Clin. Pharmacol. 2015, 80, 1097–1108. [Google Scholar] [CrossRef]
- Paine, M.F.; Hart, H.L.; Ludington, S.S.; Haining, R.L.; Rettie, A.E.; Zeldin, D.C. The Human Intestinal Cytochrome P450 “Pie”. Drug Metab. Dispos. 2006, 34, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Keefer, C.; Scott, D.O.; Strelevitz, T.J.; Chang, G.; Bi, Y.-A.; Lai, Y.; Duckworth, J.; Fenner, K.; Troutman, M.D.; et al. Mechanistic Insights from Comparing Intrinsic Clearance Values between Human Liver Microsomes and Hepatocytes to Guide Drug Design. Eur. J. Med. Chem. 2012, 57, 441–448. [Google Scholar] [CrossRef]
- Keefer, C.; Chang, G.; Carlo, A.; Novak, J.J.; Banker, M.; Carey, J.; Cianfrogna, J.; Eng, H.; Jagla, C.; Johnson, N.; et al. Mechanistic Insights on Clearance and Inhibition Discordance between Liver Microsomes and Hepatocytes When Clearance in Liver Microsomes Is Higher than in Hepatocytes. Eur. J. Pharm. Sci. 2020, 155, 105541. [Google Scholar] [CrossRef]
- Stringer, R.A.; Strain-Damerell, C.; Nicklin, P.; Houston, J.B. Evaluation of Recombinant Cytochrome P450 Enzymes as an in Vitro System for Metabolic Clearance Predictions. Drug Metab. Dispos. 2009, 37, 1025–1034. [Google Scholar] [CrossRef]
- Alalyani, M.Q.; Al dosari, Y.M.; Oteif, R.M.; Hilbah, M.A.E.; Hakami, E.A.A.; Zedin, N.A.; Alqahl, S.A.; Alfaifi, Y.M.; Almutairi, A.N.; Alsahali, F.F.; et al. Biochemical Pathways in Drug-Drug Interactions: A Pharmacological Perspective for Enhanced Drug Safety and Efficacy-Cytochrome Inhibition Mechanisms. Egypt. J. Chem. 2024, 67, 1507–1517. [Google Scholar] [CrossRef]
- Parmentier, Y.; Bossant, M.-J.; Bertrand, M.; Walther, B. In Vitro Studies of Drug Metabolism. In Comprehensive Medicinal Chemistry II; Elsevier: Amsterdam, The Netherlands, 2007; pp. 231–257. [Google Scholar]
- José Gómez-Lechón, M.; Castell, J.V.; María, T.D. An Update on Metabolism Studies Using Human Hepatocytes in Primary Culture. Expert. Opin. Drug Metab. Toxicol. 2008, 4, 837–854. [Google Scholar] [CrossRef]
- Bouwmeester, M.C.; Tao, Y.; Proença, S.; van Steenbeek, F.G.; Samsom, R.-A.; Nijmeijer, S.M.; Sinnige, T.; van der Laan, L.J.W.; Legler, J.; Schneeberger, K.; et al. Drug Metabolism of Hepatocyte-like Organoids and Their Applicability in In Vitro Toxicity Testing. Molecules 2023, 28, 621. [Google Scholar] [CrossRef] [PubMed]
- Yadav, J.; El Hassani, M.; Sodhi, J.; Lauschke, V.M.; Hartman, J.H.; Russell, L.E. Recent Developments in in Vitro and in Vivo Models for Improved Translation of Preclinical Pharmacokinetics and Pharmacodynamics Data. Drug Metab. Rev. 2021, 53, 207–233. [Google Scholar] [CrossRef] [PubMed]
- Kostadinova, R.; Boess, F.; Applegate, D.; Suter, L.; Weiser, T.; Singer, T.; Naughton, B.; Roth, A. A Long-Term Three Dimensional Liver Co-Culture System for Improved Prediction of Clinically Relevant Drug-Induced Hepatotoxicity. Toxicol. Appl. Pharmacol. 2013, 268, 1–16. [Google Scholar] [CrossRef]
- Meli, L.; Jordan, E.T.; Clark, D.S.; Linhardt, R.J.; Dordick, J.S. Influence of a Three-Dimensional, Microarray Environment on Human Cell Culture in Drug Screening Systems. Biomaterials 2012, 33, 9087–9096. [Google Scholar] [CrossRef]
- Scalise, M.; Marino, F.; Salerno, L.; Cianflone, E.; Molinaro, C.; Salerno, N.; De Angelis, A.; Viglietto, G.; Urbanek, K.; Torella, D. From Spheroids to Organoids: The Next Generation of Model Systems of Human Cardiac Regeneration in a Dish. Int. J. Mol. Sci. 2021, 22, 13180. [Google Scholar] [CrossRef]
- Sang, C.; Lin, J.; Ji, S.; Gao, Q. Progress, Application and Challenges of Liver Organoids. Clin. Cancer Bull. 2024, 3, 7. [Google Scholar] [CrossRef]
- Prior, N.; Inacio, P.; Huch, M. Liver Organoids: From Basic Research to Therapeutic Applications. Gut 2019, 68, 2228–2237. [Google Scholar] [CrossRef]
- Krewski, D.; Acosta, D.; Andersen, M.; Anderson, H.; Bailar, J.C.; Boekelheide, K.; Brent, R.; Charnley, G.; Cheung, V.G.; Green, S.; et al. Toxicity Testing in the 21st Century: A Vision and a Strategy. J. Toxicol. Environ. Health Part. B 2010, 13, 51–138. [Google Scholar] [CrossRef]
- Zink, D.; Chuah, J.K.C.; Ying, J.Y. Assessing Toxicity with Human Cell-Based In Vitro Methods. Trends Mol. Med. 2020, 26, 570–582. [Google Scholar] [CrossRef]
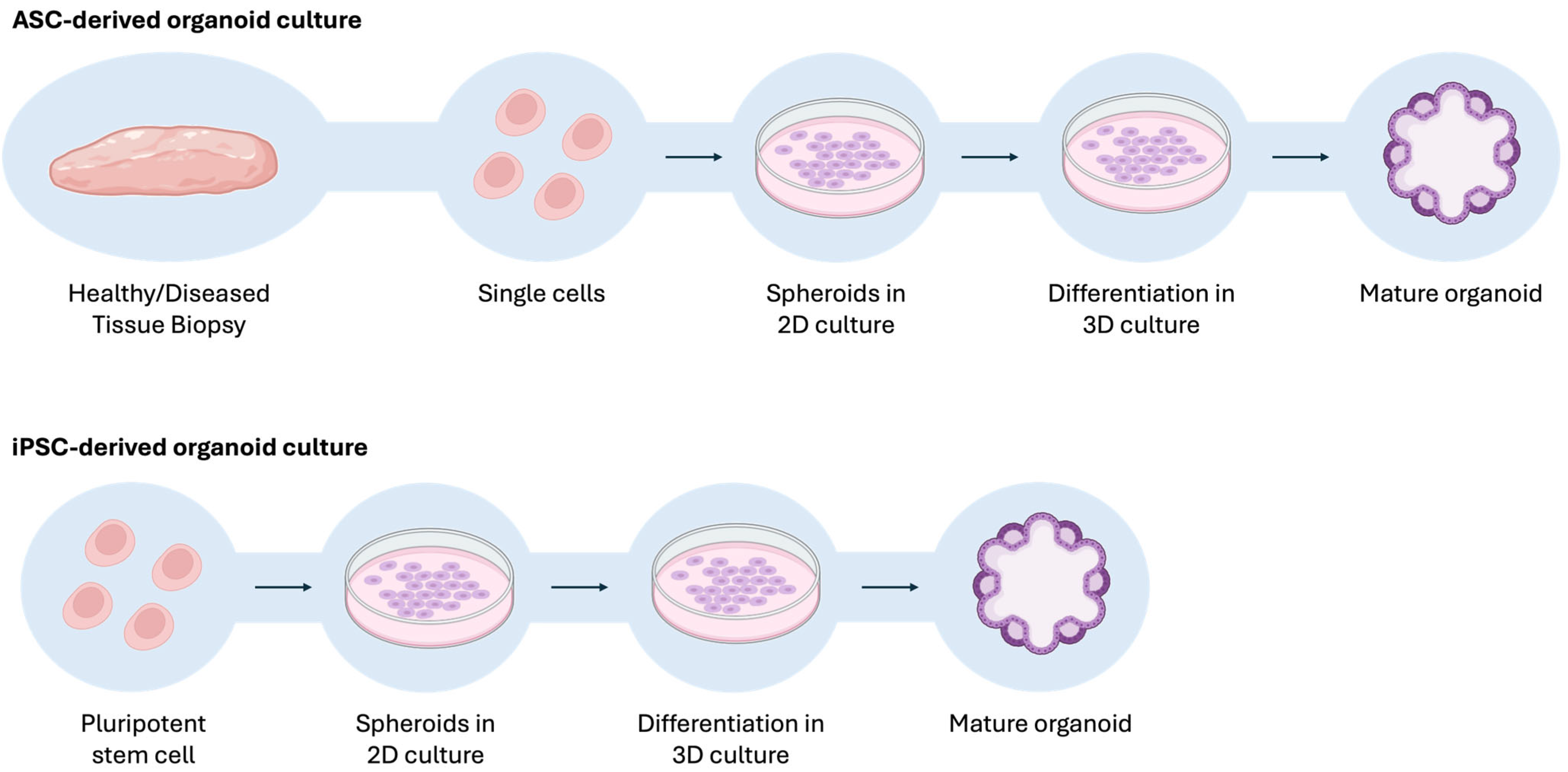
- Afonso, M.B.; Marques, V.; van Mil, S.W.C.; Rodrigues, C.M.P. Human Liver Organoids: From Generation to Applications. Hepatology 2024, 79, 1432–1451. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science (1979) 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Hu, H.; Kung, H.; Zou, R.; Dai, Y.; Hu, Y.; Wang, T.; Lv, T.; Yu, J.; Li, F. Organoids: The Current Status and Biomedical Applications. MedComm 2023, 4, e274. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a Dish: Modeling Development and Disease Using Organoid Technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Koo, B.-K. Modeling Mouse and Human Development Using Organoid Cultures. Development 2015, 142, 3113–3125. [Google Scholar] [CrossRef]
- Huch, M.; Gehart, H.; van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.A.; Ellis, E.; van Wenum, M.; Fuchs, S.A.; de Ligt, J.; et al. Long-Term Culture of Genome-Stable Bipotent Stem Cells from Adult Human Liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef]
- Tong, Y.; Ueyama-Toba, Y.; Yokota, J.; Matsui, H.; Kanai, M.; Mizuguchi, H. Efficient Hepatocyte Differentiation of Primary Human Hepatocyte-Derived Organoids Using Three Dimensional Nanofibers (HYDROX) and Their Possible Application in Hepatotoxicity Research. Sci. Rep. 2024, 14, 10846. [Google Scholar] [CrossRef]
- Brooks, A.; Liang, X.; Zhang, Y.; Zhao, C.-X.; Roberts, M.S.; Wang, H.; Zhang, L.; Crawford, D.H.G. Liver Organoid as a 3D in Vitro Model for Drug Validation and Toxicity Assessment. Pharmacol. Res. 2021, 169, 105608. [Google Scholar] [CrossRef]
- Heinzelmann, E.; Piraino, F.; Costa, M.; Roch, A.; Norkin, M.; Garnier, V.; Homicsko, K.; Brandenberg, N. IPSC-Derived and Patient-Derived Organoids: Applications and Challenges in Scalability and Reproducibility as Pre-Clinical Models. Curr. Res. Toxicol. 2024, 7, 100197. [Google Scholar] [CrossRef]
- Rowe, R.G.; Daley, G.Q. Induced Pluripotent Stem Cells in Disease Modelling and Drug Discovery. Nat. Rev. Genet. 2019, 20, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-Y.; Charton, C.; Shim, J.H.; Lim, S.Y.; Kim, J.; Lee, S.; Ohn, J.H.; Kim, B.K.; Heo, C.Y. Patient-Derived Organoids Recapitulate Pathological Intrinsic and Phenotypic Features of Fibrous Dysplasia. Cells 2024, 13, 729. [Google Scholar] [CrossRef] [PubMed]
- Palakkan, A.A.; Nanda, J.; Ross, J.A. Pluripotent Stem Cells to Hepatocytes, the Journey so Far. Biomed. Rep. 2017, 6, 367–373. [Google Scholar] [CrossRef]
- Baker, B.M.; Chen, C.S. Deconstructing the Third Dimension—How 3D Culture Microenvironments Alter Cellular Cues. J. Cell Sci. 2012, 125, 3015–3024. [Google Scholar] [CrossRef]
- Akbari, S.; Sevinç, G.G.; Ersoy, N.; Basak, O.; Kaplan, K.; Sevinç, K.; Ozel, E.; Sengun, B.; Enustun, E.; Ozcimen, B.; et al. Robust, Long-Term Culture of Endoderm-Derived Hepatic Organoids for Disease Modeling. Stem Cell Rep. 2019, 13, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Kiecker, C.; Bates, T.; Bell, E. Molecular Specification of Germ Layers in Vertebrate Embryos. Cell. Mol. Life Sci. 2016, 73, 923–947. [Google Scholar] [CrossRef]
- Lewandowski, J.; Kolanowski, T.J.; Kurpisz, M. Techniques for the Induction of Human Pluripotent Stem Cell Differentiation towards Cardiomyocytes. J. Tissue Eng. Regen. Med. 2017, 11, 1658–1674. [Google Scholar] [CrossRef]
- Park, E.; Kim, H.K.; Jee, J.; Hahn, S.; Jeong, S.; Yoo, J. Development of Organoid-Based Drug Metabolism Model. Toxicol. Appl. Pharmacol. 2019, 385, 114790. [Google Scholar] [CrossRef]
- Dwyer, B.J.; Tirnitz-Parker, J.E.E. Patient-Derived Organoid Models to Decode Liver Pathophysiology. Trends Endocrinol. Metab. 2025, 36, 235–248. [Google Scholar] [CrossRef]
- Mun, S.J.; Ryu, J.-S.; Lee, M.-O.; Son, Y.S.; Oh, S.J.; Cho, H.-S.; Son, M.-Y.; Kim, D.-S.; Kim, S.J.; Yoo, H.J.; et al. Generation of Expandable Human Pluripotent Stem Cell-Derived Hepatocyte-like Liver Organoids. J. Hepatol. 2019, 71, 970–985. [Google Scholar] [CrossRef]
- Luo, Q.; Wang, N.; Que, H.; Mai, E.; Hu, Y.; Tan, R.; Gu, J.; Gong, P. Pluripotent Stem Cell-Derived Hepatocyte-like Cells: Induction Methods and Applications. Int. J. Mol. Sci. 2023, 24, 11592. [Google Scholar] [CrossRef] [PubMed]
- Shinozawa, T.; Kimura, M.; Cai, Y.; Saiki, N.; Yoneyama, Y.; Ouchi, R.; Koike, H.; Maezawa, M.; Zhang, R.-R.; Dunn, A.; et al. High-Fidelity Drug-Induced Liver Injury Screen Using Human Pluripotent Stem Cell–Derived Organoids. Gastroenterology 2021, 160, 831–846.e10. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, S.K.; Oelgeschläger, M.; Park, H. Prediction of Acute Hepatotoxicity With Human Pluripotent Stem Cell-Derived Hepatic Organoids. Curr. Protoc. 2024, 4, e1015. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Huang, Z.; Tang, Z.; Chen, Y.; Huang, M.; Liu, H.; Huang, W.; Ye, Q.; Jia, B. Research Progress, Challenges, and Breakthroughs of Organoids as Disease Models. Front. Cell Dev. Biol. 2021, 9, 740574. [Google Scholar] [CrossRef]
- Ahammed, B.; Kalangi, S.K. A Decade of Organoid Research: Progress and Challenges in the Field of Organoid Technology. ACS Omega 2024, 9, 30087–30096. [Google Scholar] [CrossRef]
- Malki, M.A.; Pearson, E.R. Drug-Drug-Gene Interactions and Adverse Drug Reactions. Pharmacogenom. J. 2020, 20, 355–366. [Google Scholar] [CrossRef]
- Furuta, T.; Iwaki, T.; Umemura, K. Influences of Different Proton Pump Inhibitors on the Anti-platelet Function of Clopidogrel in Relation to CYP2C19 Genotypes. Br. J. Clin. Pharmacol. 2010, 70, 383–392. [Google Scholar] [CrossRef]
- Stanke-Labesque, F.; Gautier-Veyret, E.; Chhun, S.; Guilhaumou, R. Inflammation Is a Major Regulator of Drug Metabolizing Enzymes and Transporters: Consequences for the Personalization of Drug Treatment. Pharmacol. Ther. 2020, 215, 107627. [Google Scholar] [CrossRef]
- Safdari, R.; Ferdousi, R.; Aziziheris, K.; Niakan-Kalhori, S.R.; Omidi, Y. Computerized Techniques Pave the Way for Drug-Drug Interaction Prediction and Interpretation. BioImpacts 2016, 6, 71–78. [Google Scholar] [CrossRef]
- Qiu, Y.; Zhang, Y.; Deng, Y.; Liu, S.; Zhang, W. A Comprehensive Review of Computational Methods For Drug-Drug Interaction Detection. IEEE ACM Trans. Comput. Biol. Bioinform. 2022, 19, 1968–1985. [Google Scholar] [CrossRef]
- Percha, B.; Altman, R.B. Informatics Confronts Drug-Drug Interactions. Trends Pharmacol. Sci. 2013, 34, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Fowler, S.; Zhang, H. In Vitro Evaluation of Reversible and Irreversible Cytochrome P450 Inhibition: Current Status on Methodologies and Their Utility for Predicting Drug-Drug Interactions. AAPS J. 2008, 10, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yao, X.; Zhang, M.; Liu, D.; Gao, Y.; Sahasranaman, S.; Ou, Y.C. Comprehensive PBPK Model to Predict Drug Interaction Potential of Zanubrutinib as a Victim or Perpetrator. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-M.; Yoon, J.-H.; Maeng, H.-J.; Kim, Y.C. Physiologically Based Pharmacokinetic (PBPK) Modeling to Predict CYP3A-Mediated Drug Interaction between Saxagliptin and Nicardipine: Bridging Rat-to-Human Extrapolation. Pharmaceutics 2024, 16, 280. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Che, C.; Jiang, H.; Xu, J.; Yin, J.; Zhong, Z. SSF-DDI: A Deep Learning Method Utilizing Drug Sequence and Substructure Features for Drug-Drug Interaction Prediction. BMC Bioinform. 2024, 25, 39. [Google Scholar] [CrossRef]
- Lewis, D.F.V.; Modi, S.; Dickins, M. Structure–Activity Relationship for Human Cytochrome P450 Substrates and Inhibitors. Drug Metab. Rev. 2002, 34, 69–82. [Google Scholar] [CrossRef]
- Mei, S.; Zhang, K. A Machine Learning Framework for Predicting Drug-Drug Interactions. Sci. Rep. 2021, 11, 17619. [Google Scholar] [CrossRef]
- Generaux, G.T. In Vitro Modeling of Drug-Drug Interactions. In Drug Interactions in Infectious Diseases: Mechanisms and Models of Drug Interactions; Springer International Publishing: Cham, Switzerland, 2018; pp. 243–257. [Google Scholar]
- Tiryannik, I.; Heikkinen, A.T.; Gardner, I.; Onasanwo, A.; Jamei, M.; Polasek, T.M.; Rostami-Hodjegan, A. Static Versus Dynamic Model Predictions of Competitive Inhibitory Metabolic Drug-Drug Interactions via Cytochromes P450: One Step Forward and Two Steps Backwards. Clin. Pharmacokinet. 2025, 64, 155–170. [Google Scholar] [CrossRef]
- Rostami-Hodjegan, A.; Tucker, G. ‘In Silico’ Simulations to Assess the ‘in Vivo’ Consequences of ‘in Vitro’ Metabolic Drug-Drug Interactions. Drug Discov. Today Technol. 2004, 1, 441–448. [Google Scholar] [CrossRef]
- Jiang, P.; Chen, T.; Chu, L.-F.; Xu, R.; Gao, J.-T.; Wang, L.; Liu, Q.; Tang, L.; Wan, H.; Li, M.; et al. Enhancing Drug-Drug Interaction Prediction by Integrating Physiologically-Based Pharmacokinetic Model with Fraction Metabolized by CYP3A4. Expert Opin. Drug Metab. Toxicol. 2023, 19, 721–731. [Google Scholar] [CrossRef]
- Lin, W.; Chen, Y.; Unadkat, J.D.; Zhang, X.; Wu, D.; Heimbach, T. Applications, Challenges, and Outlook for PBPK Modeling and Simulation: A Regulatory, Industrial and Academic Perspective. Pharm. Res. 2022, 39, 1701–1731. [Google Scholar] [CrossRef] [PubMed]
- Spanakis, M.; Tzamali, E.; Tzedakis, G.; Koumpouzi, C.; Pediaditis, M.; Tsatsakis, A.; Sakkalis, V. Artificial Intelligence Models and Tools for the Assessment of Drug–Herb Interactions. Pharmaceuticals 2025, 18, 282. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Study Example | Description |
|---|---|
| Yu et al. [81] | A study between donepezil and tadalafil, both primarily metabolized by CYP3A, using pooled HLMs. Tadalafil was found to concentration-dependently inhibit donepezil metabolism. |
| Liu et al. [82] | The DDI potential of vicagrel was investigated using pooled HLMs and PBPK modeling. Vicagrel potently inhibited CYP2B6 and CYP2C19 and showed mixed-type and noncompetitive inhibition for bupropion and S-mephenytoin metabolism, respectively. PBPK simulations suggest vicagrel poses low DDI risk with these substrates. |
| Yang et al. [83] | Potential PK interactions between bicyclol and commonly co-administered agents were evaluated using rat liver microsomes (RLMs) and HLMs. Bicyclol was notably inhibited by pioglitazone, fenofibrate, tacrolimus, and cyclosporin A. However, as the selected inhibitory drug concentrations in vitro exceeded clinically relevant levels and maximum inhibition remained below 50% the risk of clinically meaningful DDIs involving bicyclol in humans appears low. |
| Li et al. [84] | The study assessed the impact of carvedilol on the metabolism of bedaquiline using in vitro systems, including RLMs and HLMs, a recombinant CYP3A4 system, and in vivo rat models. Their findings suggest that carvedilol can inhibit bedaquiline metabolism. |
| Faison et al. [85] | Evaluation of the PKs and safety of dordaviprone (ONC201), a novel antitumor agent, when administered alone and with itraconazole. It represents an integrated approach combining in vitro experiments with clinical investigation. In vitro assays using HLMs and rCYP enzymes demonstrated that CYP3A4 is the primary enzyme involved in dordaviprone metabolism. In healthy participants, co-administration with itraconazole significantly increased dordaviprone maximum plasma concentration and area under the curve, confirming a CYP3A4-mediated drug interaction. |
| Jaisupa et al. [86] | The study investigated the metabolic interaction between cannabidiol (CBD) and commonly co-administered antiseizure medications, as well as the influence of intermediate-activity CYP2C19 genotypes. Using pooled HLMs, the intrinsic clearance of CBD was reduced when combined with antiseizure medications. No significant difference in CBD metabolism was observed between HLMs from CYP2C19*1/*2 and *1/*4 donors. |
| Study Example | Description |
|---|---|
| Xue Li [127] | The influence of CYP2C19 genetic polymorphism on the DDI between voriconazole and omeprazole was investigated using lentivirus-engineered HepG2 cell lines expressing either CYP2C19*1 or CYP2C19*2. Although omeprazole inhibited voriconazole in both genotypes, the IC50 for CYP2C19*1 was slightly lower, suggesting a marginally stronger inhibitory effect. |
| Xun et al. [128] | This study examined the PK interaction between voriconazole and atorvastatin using a comprehensive approach that included clinical data, in vivo experiments in rat models, and in vitro models. Among the in vitro systems, HepG2 cells were employed to assess the metabolic profile of atorvastatin in the presence of voriconazole. |
| Sager et al. [129] | Using HepG2 cells and plated PHHs it was demonstrated that bupropion and its metabolites significantly downregulate CYP2D6 mRNA expression in a concentration-dependent manner. |
| Cui et al. [130] | The effects of berberine on lovastatin PKs were analyzed using both in vivo (rats) and in vitro (HepG2 cells) models. Berberine pretreatment significantly decreased lovastatin plasma exposure in rats, indicating enhanced metabolism. Correspondingly, berberine induced increased metabolic activity and altered kinetic parameters of lovastatin in HepG2 cells. |
| Cell Line | Origin | Advantages | Disadvantages | Applications |
|---|---|---|---|---|
| HepG2 | Human hepatoblastoma |
|
| [127,130,160] |
| HepaRG | Human hepatocellular carcinoma |
|
| [155,161] |
| BC2 | Human hepatoblastoma |
|
| [156] |
| Huh-7 | Human hepatocellular carcinoma |
|
| - |
| PH5CH | Immortalized human fetal hepatocytes |
|
| - |
| Upcyte | Genetically modified human hepatocytes |
|
| [162] |
| PLC | Human hepatocellular carcinoma |
|
| [163] |
| SNU-182 | Human hepatocellular carcinoma |
|
| - |
| SNU-449 | Human hepatocellular carcinoma |
|
| - |
| Hep3B | Human hepatocellular carcinoma |
|
| [164] |
| THLE-2 | Immortalized human normal liver epithelial cells |
|
| [165] |
| THLE-3 | Immortalized normal liver cells |
|
| - |
| CYP Enzyme | Inhibitor | Ki/IC50 (Μm) in In Vitro | References |
|---|---|---|---|
| CYP1A2 | α-naphthoflavone | 0.01 | [166,167] |
| Furafylline (1) | 0.6–0.7 | [168,169,170,171] | |
| CYP2B6 | Clopidogrel (1) | 1.1 | [172,173,174,175] |
| Sertraline | 3.2 | [176,177] | |
| Thiotepa (1) | 2.8–3.8 | [178,179,180,181,182] | |
| Ticlopidine (1) | 0.2–0.8 | [172,173,177,180,183] | |
| CYP2C8 | Gemfibrozil glucuronide (1) | 52–75 | [184,185,186,187] |
| Montelukast | 0.009–0.15 | [188,189,190] | |
| Phenelzine (1) | 1.2 | [187] | |
| CYP2C9 | Sulfaphenazole | 0.3 | [191,192,193] |
| Tienilic acid (1) | 5 | [194] | |
| CYP2C19 | N-3-benzyl-nirvanol | 0.079–0.12 | [195] |
| Loratadine | 0.76 | [196] | |
| Nootkatone | 0.5 | [197] | |
| Ticlopidine (1) | 1.1 | [198,199,200] | |
| CYP2D6 | Paroxetine (1) | 0.15 | [201,202,203] |
| Quinidine | 0.018–0.06 | [204,205,206] | |
| CYP3A4 | Azamulin (1) | 0.03–0.24 | [207,208] |
| Itraconazole | 0.013–0.27 | [191,209,210,211,212] | |
| Ketoconazole | 0.0037–0.028 | [173,191,193,210] | |
| Troleandomycin (1) | 0.26 | [213,214,215] | |
| Verapamil (1) | 2.3–2.9 | [216,217] |
| CYP Enzyme | Inducer | Class of Drugs | Mechanism (Receptor) |
|---|---|---|---|
| CYP1A2 | Omeprazole | Proton pump inhibitors | AHR |
| CYP2B6 | Phenobarbital | Barbiturates | CAR and PXR |
| CYP2C8 | Rifampicin | Antibiotics | PXR |
| CYP2C9 | |||
| CYP2C19 | |||
| CYP2D6 | |||
| CYP3A4 |
| Type of Study | Major Findings of the Study | Reference |
|---|---|---|
| PBPK model | A PBPK model was developed using in vitro, physicochemical, and clinical data to predict DDIs involving zanubrutinib. The model evaluated the effects of CYP3A inhibitors and inducers on zanubrutinib exposure, its impact on CYP3A4, CYP2C8, and CYP2B6 substrates, and the influence of gastric pH changes. This model was validated using clinical DDI data. It accurately predicted plasma concentrations and DDI outcomes. | [281] |
| PBPK model | This study aimed to predict the CYP3A-mediated DDI between saxagliptin and nicardipine using a PBPK model, incorporating in silico and in vitro data. PBPK models for both drugs were constructed using parameters derived from in vitro experiments and literature, and validated in rats, where co-administration resulted in 2.6-fold increase in saxagliptin exposure. The model was then extrapolated to humans, with simulations predicting only a minimal AUC increase (1.05-fold) indicating no clinically significant interaction. This study demonstrates the value of in vitro-informed PBPK modeling in assessing DDIs. | [282] |
| Deep learning method | The study presents a DDI prediction based on sequence and substructure features (SSF-DDI). By integrating these complementary data types, the model offers a more comprehensive molecular representation. Experimental results and case studies show that SSF-DDI significantly outperforms existing models, particularly in predicting DDIs involving previously unseen drugs, with a 5.67% improvement in accuracy over state-of-the-art approaches. | [283] |
| Quantitative structure-activity relationship (QSAR) model | QSAR models were developed using 11, 6, 10, 8, 8, 10, 10, and 10 substrates of CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4, respectively. | [284] |
| ML method | The study introduces an ML framework for predicting DDI using simple rug target profile representations and an L2-regularized logistic regression model. This approach emphasizes biological interpretability by examining the gene-level relationships between drug targets. New statistical metrics are proposed to quantify DDI intensity, efficacy, and action range within protein–protein interaction (PPI) networks and signaling pathways. Empirical validation demonstrates the model outperforms existing models. Results reveal that DDIs are more likely when drugs share targets, their targets are closely connected in PPI networks, or they are involved in interacting pathways—offering mechanistic insights into potential adverse reactions. | [285] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marques, L.; Vale, N. A Review on New Frontiers in Drug-Drug Interaction Predictions and Safety Evaluations with In Vitro Cellular Models. Pharmaceutics 2025, 17, 747. https://doi.org/10.3390/pharmaceutics17060747
Marques L, Vale N. A Review on New Frontiers in Drug-Drug Interaction Predictions and Safety Evaluations with In Vitro Cellular Models. Pharmaceutics. 2025; 17(6):747. https://doi.org/10.3390/pharmaceutics17060747
Chicago/Turabian StyleMarques, Lara, and Nuno Vale. 2025. "A Review on New Frontiers in Drug-Drug Interaction Predictions and Safety Evaluations with In Vitro Cellular Models" Pharmaceutics 17, no. 6: 747. https://doi.org/10.3390/pharmaceutics17060747
APA StyleMarques, L., & Vale, N. (2025). A Review on New Frontiers in Drug-Drug Interaction Predictions and Safety Evaluations with In Vitro Cellular Models. Pharmaceutics, 17(6), 747. https://doi.org/10.3390/pharmaceutics17060747