1. Introduction
Tay–Sachs disease (TSD) is a monogenic lysosomal storage disease (LSD) caused by mutations in the
HEXA gene, which encodes the α-subunit of the lysosomal enzyme β-hexosaminidase A (HexA). HexA is a heterodimeric enzyme constituted by the α- and β-subunits, the latest encoded by the
HEXB gene [
1,
2]. Mutations in the
HEXA gene result in the loss of function of HexA, preventing the degradation of GM2 gangliosides. Lysosomal GM2 ganglioside accumulation in the central nervous system (CNS) leads to neurodegenerative symptoms in patients [
1,
3]. The excessive accumulation of GM2 gangliosides disrupts calcium homeostasis in the rough endoplasmic reticulum by interfering with the SERCA pump, affecting chaperones like BiP and triggering the unfolded protein response. This leads to the activation of IRE1α and PERK, which phosphorylate eIF2α, inducing ATF4 and CHOP/GADD153 expression [
4], whereas CHOP promotes ERO1 activity, increasing reactive oxygen species (ROS) production. Mitochondrial proteins like BAX facilitate cytochrome C release, activating caspase 9 and the intrinsic apoptosis pathway [
5,
6]. In early apoptosis, phosphatidylserine externalization alters membrane asymmetry and promotes microglial recruitment [
7]. Astrocyte-derived cytokines (CCL2, CXCL10) further enhance microglial activation [
1,
8]. These cellular changes can lead to defects in autophagy, increased lysosomal mass, ROS, synaptic dysfunction, inflammation, and ultimately cell death [
1,
3].
Currently, there is no specific treatment for TSD. Therefore, different therapeutic strategies are being investigated, including enzyme replacement therapy, substrate reduction therapy, and pharmacological chaperones [
1,
9,
10]. One of the main limitations of these therapies is the blood–brain barrier (BBB), which restricts the movement of almost all molecules > 1 kDa, including recombinant proteins and gene-based drugs, preventing therapeutic molecules from crossing and performing their function [
11]. Given these restrictions and the monogenic nature of this disease, different studies have focused on evaluating gene therapy (GT) alternatives for treating TSD [
1]. Among the different evaluated GT strategies, genome editing approaches, such as those based on the CRISPR/Cas9 system, have emerged during the last years for the treatment of TSD [
12,
13].
Although the CRISPR/Cas9 system has demonstrated significant potential for biomedical applications, a critical aspect is the selection of the delivery vehicle, which prevents DNA degradation and ensures targeted delivery [
14]. Various methods exist for GT delivery, yet limitations remain regarding the efficiency and safety of the vectors used. Viral vectors, such as lentivirus, adeno-associated virus, retrovirus, and adenovirus, are the most used, both in vivo and in vitro, due to their high transfection efficiency (TE) and stability. However, these vectors present challenges, including high immunogenicity, safety risks, and complex production processes [
15,
16]. On the other hand, non-viral vectors offer several advantages, including a high gene cargo capacity, low immunogenicity, cost-effectiveness, high safety for large-scale manufacturing, and broad cellular compatibility [
17]. Common non-viral vectors used in GT include liposomes, cationic polymers, micelles, inorganic nanoparticles, and DNA nanoclews [
15,
18,
19]. Non-viral vectors, such as polymeric or lipid nanoparticles, function by condensing and protecting DNA, facilitating its cellular delivery while preventing extracellular degradation. Electrostatic interactions between the cationic vector and DNA phosphate groups enable nanoparticle formation, enhancing cellular uptake. Polymeric nanoparticles provide greater stability and efficiency in vitro models, whereas lipid-based complexes (e.g., liposomes) enable transient transfection with diminishing gene expression over time. Understanding these differences is crucial when designing GT strategies based on the need for sustained or temporary gene expression [
19,
20].
Previously, we described a polymer-based non-viral vector named PP6D5 [
21,
22,
23]. This polymer is composed of a 2 kDa segment of methoxy-poly(ethylene glycol) (mPEG), which improves the biocompatibility and colloidal stability of polymer–DNA complexes. This polymer also includes a hydrophobic backbone of poly(ε-caprolactone-
co-propargyl carbonate) (P(CL-
co-MPC)), which strengthens DNA interactions through hydrophobic forces. This backbone was grafted with five cationic segments of 4.4 kDa poly(2-(dimethylamino) ethyl methacrylate) (PDMAEMA) for DNA condensation [
21]. PP6D5 interacts with DNA through electrostatic forces between amines, forming a complex that facilitates the delivery of GT drugs to the target cell [
15]. PP6D5 formed particles with a positive surface at low N/P ratios (≥3) due to the use of several low-molecular-weight PDMAEMA grafts, which increased the charge density and DNA compaction ability. Specifically, the PP6D5 polymer complexed with DNA, at an N/P ratio of 5, showed a zeta potential of 6.6 ± 0.2 mV. It also showed a hydrodynamic diameter of 291 ± 30 nm [
21,
22,
23]. PDMAEMA further promotes endosomal escape by protonating amines and activating the proton pump, leading to acidification of the medium, activation of enzymes and counterions, increased ionic strength, and osmotic swelling. This process ultimately disrupts the endosomal membrane, releasing DNA into the cytosol [
21,
22,
23]. We demonstrated that the PP6D5 polymer has the capacity to transfect HEK293 and “difficult-to-transfect” Jurkat cells, achieving high expression levels of a reporter gene (i.e., EGFP) [
21]. Despite its promising performance, the capacity of this polymer to mediate gene transfer within a disease context has not been evaluated, reinforcing the relevance of our study in further evaluating its potential applications.
Recently, we reported the use of a CRISPR/Cas9-based gene editing strategy that relies on a Cas9 nickase (nCas9) and the use of non-viral vectors as a potential approach for treating GM2 gangliosidoses (TS and Sandhoff diseases) [
13]. The results showed the potential of CRISPR/nCas9 as a new alternative for treating GM2 gangliosidoses, as well as the capacity of non-viral vectors to mediate gene transfer for this group of disorders and for other LSDs [
22,
23]. In this study, we expanded our GT approach through the evaluation of the potential of the PP6D5 polymer as a non-viral vector for TSD. We evaluated the TE in several cell models, including NIH-3T3 mouse fibroblasts, U87MG astrocytoma cells, SHSY5Y neuroblastoma cells, and TSD fibroblasts, as well as the potential to correct enzyme deficiency and cellular parameters such as lysosomal mass, total reactive oxygen species, polar and neutral/apolar lipid accumulation, and autophagy. While CRISPR/nCas9-based genome editing has been demonstrated in previous studies [
13,
24,
25], our study demonstrates the application of the PP6D5 polymer, a promising non-viral vector, for CRISPR/nCas9-based genome editing in human fibroblasts derived from TSD patients and three other different cellular models, providing a direct comparison with a gold-standard non-viral vector (Lipofectamine 3000). This comparative analysis provides valuable insights into the potential applications of PP6D5 for the development of GT for TSD.
2. Materials and Methods
2.1. PP6D5 Polymer and Plasmid DNA Preparation
The PP6D5 polymer was synthesized and characterized as previously described by Diaz et al. [
21]. The PP6D5 polymer complexed with DNA, at an N/P ratio of 5, showed a zeta potential of 6.6 ± 0.2 mV and a hydrodynamic diameter of 291 ± 30 nm [
21,
22,
23]. Six previously constructed plasmids [
13,
24,
25,
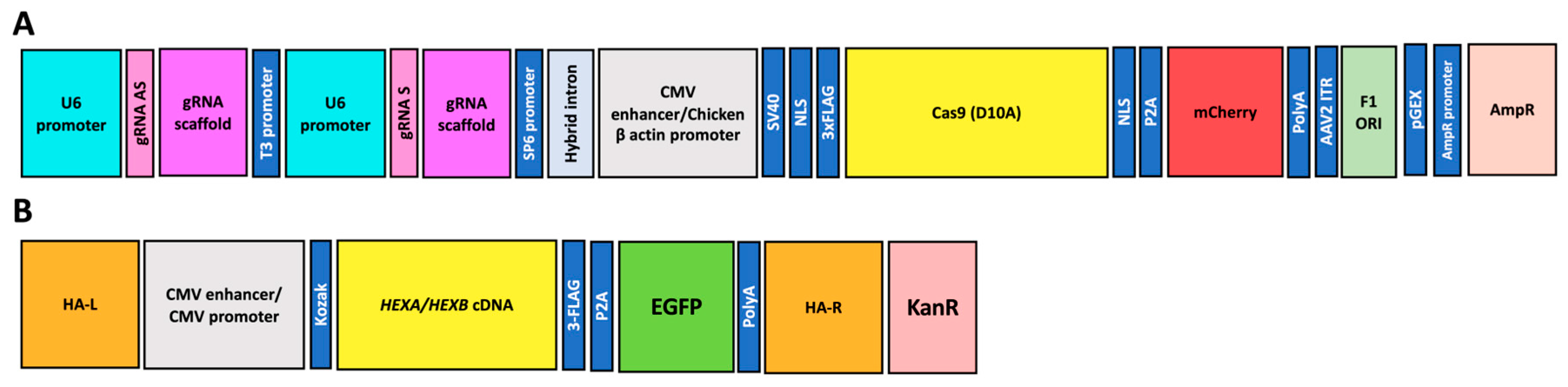
26] were used in this study. Two plasmids were based on the AIO-mCherry plasmid (Addgene: #74120), with one carrying Cas9 nickase (nCas9), Cas9 with a mutation in the RuvC domain (D10A), cloning sites for the sgRNA pair, an mCherry fluorescent marker, and an ampicillin resistance gene (
Figure 1A). For the ROSA26 locus, gRNAs with the Sense TAAGCATGCTCTAACAGGCC and the Antisense CACAAGAGTAGTTACTTGGC were cloned onto the AIO-mCherry plasmid (hereafter, gRNA ROSA26 plasmid) [
22]; meanwhile, for the AAVS1
locus, an sgRNA pair with the Sense ACAGACTAGAGAGGTAAAGG and the Antisense CCTGTCACGGCATCTTCCAG was cloned (hereafter, gRNA AAVS1 plasmid) [
24,
26]. Four plasmids containing
HEXA or
HEXB cDNAs, the homologous recombination arms either for ROSA26 (mouse cells, hereafter HEXA or HEXB ROSA26 plasmids) or AAVS1 (human cells, hereafter HEXA or HEXB AAVS1 plasmid) loci, an EGFP fluorescent marker, and a kanamycin resistance gene were also previously constructed (
Figure 1B) [
13,
24,
25,
26]. Plasmids were prepared using the ZymoPURE II Plasmid Midiprep Kit (Zymo Research, Irvine, CA, USA), following the manufacturer’s recommendations.
2.2. Cell Culture
NIH-3T3 mouse embryonic fibroblasts, U87MG astrocytoma cells, a human glioblastoma cell line derived from a malignant astrocytoma in the brain of a patient, and HEK293FT were cultured in high-glucose DMEM (Biowest, Nuaillé, France) supplemented with 10% FBS and 1% antibiotics. Neuroblastoma SHSY5Y cells, a human cell line derived from a bone marrow biopsy of a neuroblastoma patient, were cultured in DMEM-F12 (Biowest) with 10% FBS and 1% antibiotics. TSD fibroblasts were obtained from the Coriell Institute, Camden, NJ, USA (GM00515); these have a c.1274_1277dupTATC mutation in the HEXA gene, a characteristic pathogenic insertion in exon 11 responsible for the TSD phenotype. TSD and wild-type (WT) fibroblasts, from a healthy donor, were cultured in high-glucose DMEM (Biowest, Nuaillé, France) supplemented with 15% FBS and 1% antibiotics. All cells were cultured at 37 °C with 5% CO2, between passages 4, 26, 30, 9, and 8 for NIH-3T3 fibroblasts, U87MG astrocytoma cells, SHSY5Y neuroblastoma cells, TSD fibroblasts, and WT fibroblasts, respectively, and were periodically tested for Mycoplasma spp. contamination using PCR and staining with Hoechst 33258.
2.3. On-Target and Homologous Recombinantion Assays
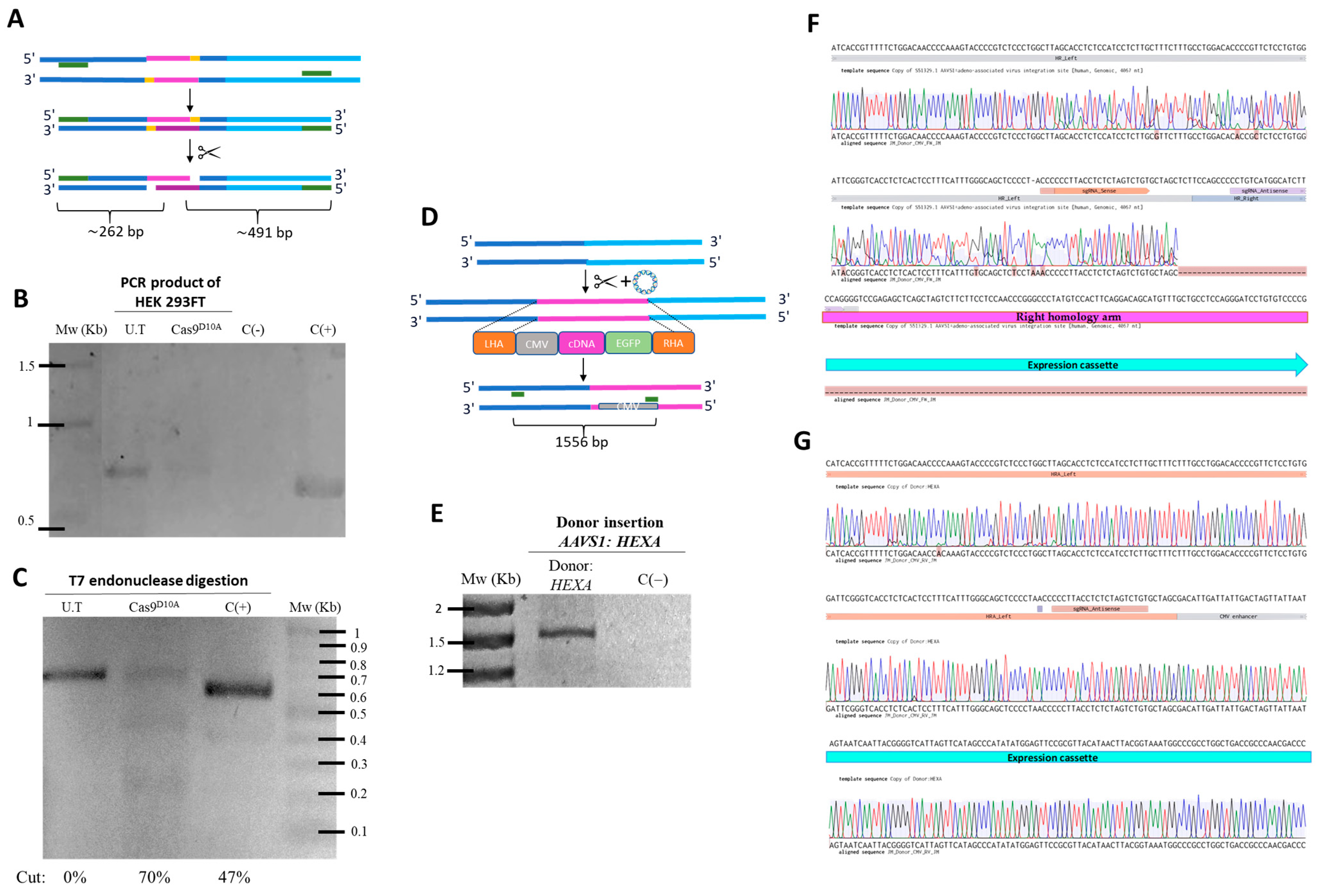
HEK 293FT cells were transfected in 24-well plates with 500 ng of each plasmid using Lipofectamine 3000 (LP; Thermo Fisher Scientific, Waltham, MA, USA), according to manufacturer’s protocol. After medium replacement, successful transfection was confirmed at 48 h via mCherry or EGFP expression using fluorescence microscopy. Genomic DNA was then extracted, with concentration, purity, and integrity assessed by NanoDrop (Thermo Fisher Scientific, Waltham, MA, USA) and 1% agarose gel electrophoresis (80 V, 1 h). The experimental design compared untransfected cells, cells with gRNA AAVS1 plasmids, or cells co-transfected with both sgRNA and HEXA donor plasmids (
Figure 2).
2.3.1. On-Target Assay
Genomic DNA (gDNA) was used for T7 endonuclease assays (EnGen
® Mutation Detection Kit, NEB, Ipswich, MA, USA), following the manufacturer’s protocol (
Figure 2A). The assay was performed with gDNA from cells transfected with the gRNA AAVS1 plasmid and from untransfected cells (negative control) to detect nCas9-mediated cleavage at the AAVS1 locus. PCR amplification used the forward primer 5′-GGCCTTCTCCGACGGATGTCTCCC-3′ and the reverse primer 5′-CCGGGGGCAGGTCACGCATCCC-3′, with the following conditions: 98 °C for 30 s; 35 cycles of 98 °C for 5 s, 72 °C for 10 s, and 72 °C for 20 s/kb; and a final extension at 72 °C for 2 min. PCR products were digested with T7 endonuclease and resolved on a 2% agarose gel (80 V, 1 h) after ethidium bromide staining. Band intensities were analyzed using GelAnalyzer 19.1 to estimate cleavage efficiency by densitometry.
2.3.2. Homologous Recombination Assay
To confirm HEXA cDNA insertion, PCR was performed using gDNA from cells co-transfected with both sgRNA and HEXA donor plasmids. PCR reactions used the Q5
® Hot Start High-Fidelity Master Mix (NEB, Ipswich, MA, USA) and primers flanking the AAVS1 locus: a forward primer targeting a region absent in the left homology arm (5′-TTCGATTGGAGTCGCTTTAACTG-3′), and reverse primer within the CMV promoter (5′-GCGGGGCCGCAGCTCTCTCTGCTTATATATAGACCTCC-3′) (
Figure 2D). The PCR conditions were as follows: 98 °C for 30 s; 30 cycles of 98 °C for 10 s, 66 °C for 30 s, and 72 °C for 30 s/kb; and a final extension at 72 °C for 2 min. Amplification was confirmed on a 1% agarose gel (80 V, 1 h) with ethidium bromide staining. PCR products were Sanger sequenced (Macrogen, Seoul, Republic of Korea) and aligned to the AAVS1 locus (forward) and AAVS1 donor plasmid (reverse) using Benchling and electropherogram analysis.
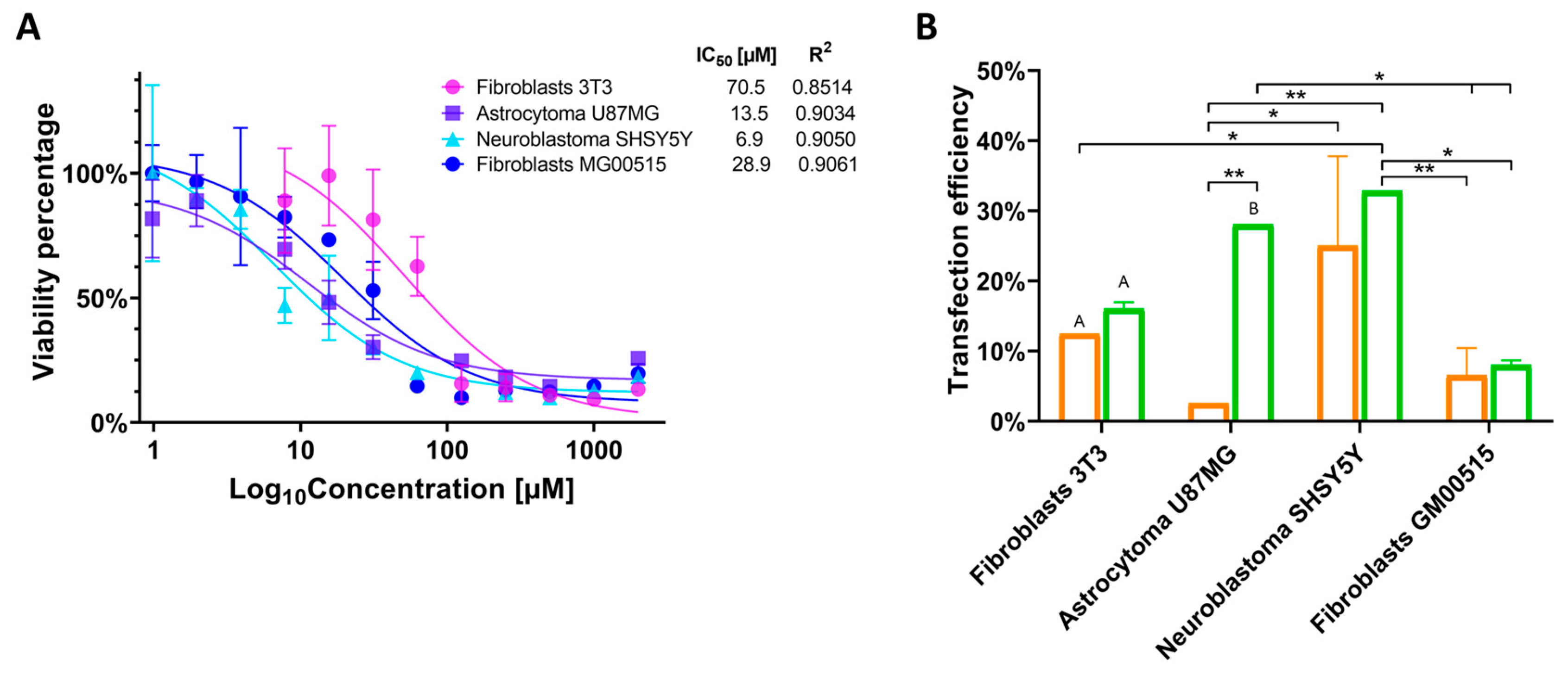
2.4. Cell Viability Measurement with the PP6D5 Polymer
To evaluate the impact of the PP6D5 polymer on cell viability, 1 × 10
4 cells per well were seeded in a 96-well plate. After 24 h, the PP6D5 polymer was added at concentrations ranging from 0.976 to 2000 µM and incubated for another 24 h. Then, 10 µL of MTT reagent was added to each well. After 4 h of incubation, 100 µL of dimethyl sulfoxide was added to resuspend the formazan crystals. The plate was then read in an Anthos 2020 microplate reader at 540/630 nm. IC
50 was estimated using non-linear regression with GraphPad Prism 9.3.1 software and used to estimate IC
80, which represents the concentration at which 80% of the maximum TE is achieved with minimal cytotoxic effects and no significant cell death, as reported previously [
13,
25]. All experiments were conducted in triplicate.
2.5. Transfection Efficiency
To evaluate the PP6D5-mediated CRISPR/nCas9 system’s TE, 5 × 10
4 NIH-3T3 and TSD fibroblasts, and 1 × 10
5 U87MG and SHSY5Y cells, were seeded in 24-well plates and cultured for 24 h before treatment. Untreated cells and cells transfected with empty donors served as controls. The ROSA26 sgRNA:AIO-mCherry plasmid was used for mouse cells, while the AAVS1 sgRNA:AIO-mCherry plasmid was used for human cells. Transfection was performed at a nitrogen-to-phosphate (N/P) ratio of 5:1, with 1 µg of plasmid DNA [
21]. The PP6D5 polymer was prepared at 100 mM (11.7 mg in 500 µL ultrapure water), with the required amount calculated based on 5 nmol of nitrogen per 3 nmol of phosphate. Components were added sequentially (150 mM saline, DNA, polymer), vortexed for 10 s, and incubated for 20 min at room temperature [
21]. The PP6D5/DNA complex was then diluted with OptiMEM™ (Gibco, Waltham, MA, USA) and added to cells after 10 min of incubation. The medium of each well was replaced with the PP6D5/DNA solution. LP transfection served as a control, following the manufacturer’s instructions. TE was assessed after 48 h by mCherry expression using an AxioScope A1 microscope (ZEISS, Goettingen, Germany). Cells were then trypsinized, centrifuged, washed with 1X PBS, and resuspended in HBSS with 1 µM propidium iodide (PI) before analysis on a BD FACSAria II cytometer (BD Biosciences, Auckland, New Zealand) using a PerCP filter (exc/emi: 670/630 nm) for mCherry and a PE filter (exc/emi: 596/578 nm) for PI detection. Data were analyzed using FlowJo vX.0.7 software. All experiments were performed in duplicate.
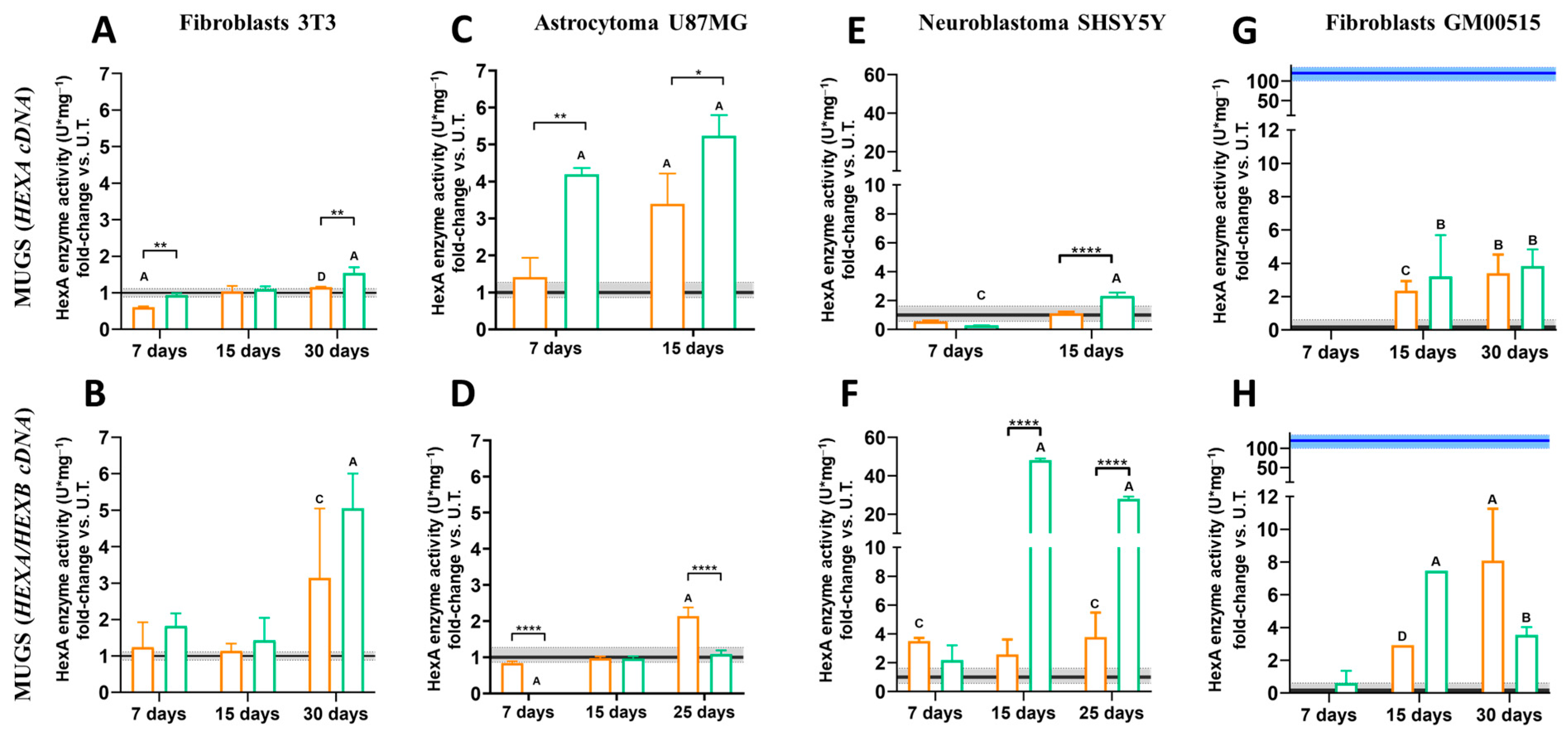
2.6. β-Hexosaminidase A Enzymatic Activity
To evaluate the effect of CRISPR/nCas9 treatment on HexA activity, 5 × 10
4 cells/well (NIH-3T3, U87MG, SHSY5Y, and GM00515) were transfected in 24-well plates using either gRNAs and HEXA or HEXB plasmids (
Figure 1). Cells treated with LP or the PP6D5 polymer without DNA, as well as with the non-viral vectors conjugated with empty donor plasmids, were included as controls. After 7, 15, and 25/30 days post transfection, cells were lysed with 150 μL of lysis buffer (0.01 M citrate-phosphate, pH 4.4, with 0.5% Triton X-100). The supernatant was collected and centrifuged for 10 min at 1600 rpm [
13]. Protein quantification was performed using the BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). To determine the specific HexA activity, 50 µL of cell lysate supernatant was mixed with 20 µL of 3.2 μM MUGS substrate (Merck, Darmstadt, Germany) and incubated for 20 min at 37 °C. The reaction was stopped with 150 µL of stop solution (glycine-carbonate, pH 9.8). A standard curve was prepared with 4-methylumbelliferone from 0.007 to 2 µM. Fluorescence was measured at 360/445 nm (excitation/emission) using the Twinkle LB970 a fluorometer (Interchim Berthold Technologies, Montluçon, France). One unit of enzymatic activity was defined as the amount of enzyme hydrolyzing 1 nmol of substrate per hour. The specific enzymatic activity was reported as U/mg of total protein. All experiments were conducted in triplicate.
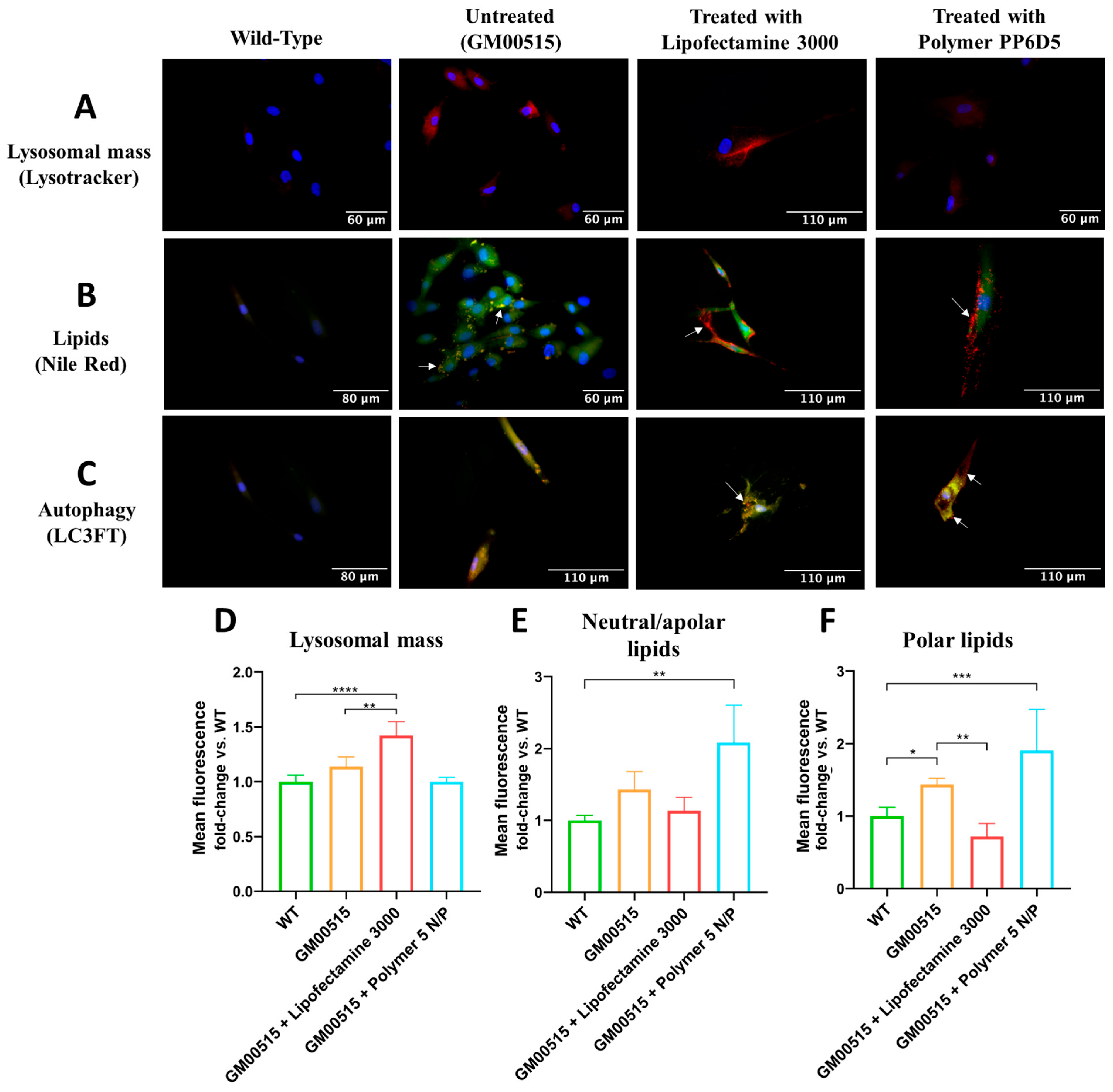
2.7. Measurement of Physiological Parameters by Fluorescence Microscopy
To evaluate the changes in some physiological parameters by fluorescence microscopy, coverslips were sterilized, treated with poly-D-lysine, and washed with 1X PBS. The following day, 3.3 × 10
3 TSD fibroblasts were seeded per coverslip, incubated for 24 h, and transfected using LP or the PP6D5 polymer conjugated with nCas9 and Donor plasmids for the AAVS1 locus. Non-affected human fibroblasts and untreated TSD fibroblasts were used as controls. After 30 days, cells were stained with specific probes or transfected with reporter plasmids. To evaluate the lysosomal mass, cells were incubated with 50 nM LysoTracker™ Deep Red (Thermo Fisher Scientific, Waltham, MA, USA) in complete DMEM at 37 °C for 1 h [
27]. Hoechst 33342 (Biotium, Fremont, CA, USA) (1/2000 dilution) was added and incubated for 10 min at RT. Cells were washed twice with 1X PBS and fixed with 4% paraformaldehyde, incubated at RT for 15 min, and then washed again. Cells were mounted on coverslips with Fluoroshield and observed with DAPI (Hoechst; exc/emi: 358/461 nm) and DsRed (exc/emi: 558/583 nm) filters under an AxioScope A1 microscope (ZEISS, Goettingen, Germany) with a 40X objective. To evaluate lipid accumulation, cells were incubated with 1 µM Nile Red (Thermo Fisher Scientific, Waltham, MA, USA) and Hoechst 33342 (Biotium, Fremont, CA, USA) (1/2000 dilution). Cells were treated as described for LysoTracker, and observed under an AxioScope A1 epifluorescence microscope (ZEISS, Goettingen, Germany) with a 40X objective using DAPI (nucleus), DsRed (polar lipids), and eGFP (neutral and nonpolar lipids; exc/emi: 488/509 nm) filters. Finally, to evaluate the autophagy flux, cells were transfected with the pMXs GFP-LC3-RFP plasmid (Addgene #117413). This plasmid expresses the LC3 protein fused with GFP and RFP proteins. Transfection was performed with LP 3000, and 48 h later, the wells were washed with 1X PBS, stained with Hoechst, and treated as described previously. Observations were conducted using an AxioScope A1 epifluorescence microscope with DAPI, DsRed, and eGFP filters. Autofluorescence was controlled by establishing the exposure time based on untreated cells.
2.8. Measurement of Physiological Parameters by Flow Cytometry
To evaluate changes in physiological parameters using flow cytometry, 5 × 103 TSD fibroblasts per well were seeded in 24-well plates and transfected the following day with CRISPR/nCas9 and donor AAVS1 plasmids. After 30 days post transfection, cells were stained with probes to assess functional parameters. For lysosomal mass measurement, cells were incubated with 50 nM LysoTracker™ Deep Red (Thermo Fisher Scientific, Waltham, MA, USA) in complete DMEM at 37 °C for 1 h. For total ROS measurement, cells were incubated with 1 µM H2DCFDA (Invitrogen, Waltham, MA, USA) in complete DMEM at 37 °C for 45 min. For lipid analysis, cells were treated with 1 µM Nile Red (Invitrogen, Waltham, MA, USA) for 10 min at 37 °C. The cells were washed twice with 1X PBS, trypsinized, centrifuged, and resuspended in HBSS buffer with 1 µM PI (Thermo Fisher Scientific, Waltham, MA, USA). Fluorescence was measured using a BD FACSAria II flow cytometer (BD Biosciences, East Rutherford, NJ, USA) with PerCP (Lysotracker, Exc/Emi: 670/30 nm), FITC (ROS and neutral/apolar lipids, Exc/Em: 494/519 nm), and PE (polar lipids, Exc/Emi: 596/578 nm) filters. WT and untreated TSD fibroblasts were used as controls. Data analysis was performed using FlowJo vX.0.7 software, and all experiments were conducted in triplicate.
2.9. Statistical Analysis
Normality and homogeneity were assessed using the Shapiro–Wilk test. Welch’s t-test was used for comparisons between two treatments. One-way ANOVA with Sidak’s test was used for multiple comparisons when analyzing a single factor. For one-way ANOVA that included a control group compared with the other groups, Dunnett’s test was used instead. Two-way ANOVA with Sidak’s test was applied for comparisons involving two factors. If the data did not meet the assumptions, the Kruskal–Wallis test was used. All data were analyzed using GraphPad Prism 9.3.1 with a significance level of p < 0.05.
4. Discussion
We have previously explored CRISPR/nCas9-based GT approaches for LSD using non-viral vectors [
13,
24,
25,
26]. In the case of TSD, patients’ fibroblasts were transfected with HEXA cDNA by using a magnetoliposomes-based vector, and we observed not only a significant increase in β-hexosaminidase activity, but also changes in glycosaminoglycan levels, lysosome mass, and oxidative stress [
13]. In this study, we expanded our GT approach through the following methods: (1) the use of the PP6D5 polymer as a non-viral vector for gene delivery in the context of TSD; (2) the evaluation of our CRISPR/nCas9-based approach in TSD relevant cells, such as astrocytoma and neuroblastoma cells; and (3) the co-transfection of HEXA and HEXB cDNAs, which was not evaluated in our previous study [
13]. We investigated the use of the PP6D5 polymer, since it may offer several advantages, such as a reduction in the cytotoxicity due to the presence of PEG segments and a grafted structure with lower charge density, and a transfection improvement due to the combination of hydrophobic and cationic segments for DNA interaction [
20,
21]. Diaz et al. [
21] demonstrated that compared to the gold-standard 25 kDa linear poly(ethylenimine), the PP6D5 polymer showed superior TE in HEK293 cells and “difficult-to-transfect” Jurkat cells. While increasing N/P ratios enhanced transgene expression, PP6D5 showed increased cytotoxicity in Jurkat suspension cells at concentrations above 20 μg/mL, with viability ranging from 35% to 80%. In these cell models, the TE never exceeded 40%, and was positively correlated with the N/P ratio and polymer concentration, but negatively correlated with the available plasmid DNA molecules, suggesting different intracellular processing mechanisms compared to adherent cells [
21]. PP6D5’s superior performance over LP is likely related to its electrostatic and hydrophobic interactions, which enhance DNA binding and cellular delivery. The polymer’s flexible structure and ability to form stable DNA complexes appear to be key factors, highlighting the advantages of PP6D5 over other vectors such as LP.
Initially we demonstrated a high on-target efficiency (up to 70%) induced by CRISPR/nCas9 in cells treated with the respective plasmid, exceeding results achieved by Leal et al. [
13] (36.8%) and Chiang et al. [
34] (20.2%). The homologous recombination assay confirmed the successful insertion of the expression cassette into an nCas9-dependent vector, verified by PCR and Sanger sequencing, results that align with those reported previously [
13,
26].
Based on the IC
50 values and TE results, incubation with the DNA–polymer complex affected the IC
50 values differently depending on the cell type. Particularly in the U87MG astrocytoma model, the cytotoxicity increased significantly with the complete complex (polymer-CRISPR/nCas9-donor), resulting in greater cell loss than with the polymer alone. Although enzyme activity was still detected, many cells did not survive transfection, as indicated by a decrease in cell viability, whereas surviving cells retained the capacity to proliferate over time. In contrast, NIH-3T3 fibroblasts showed higher resistance, as the CRISPR/nCas9 system was less cytotoxic and did not significantly impact the initial cell population. These results suggest that both the polymer and CRISPR transfection may influence cell viability, aligning with previous studies in human pluripotent stem cells, where Cas9-induced toxicity, mediated by p53, affects cell viability [
35]. Additionally, it has been reported that the cytotoxicity of cationic polymers correlates with molecular weight and charge [
36]. Additionally, when forming a complex with DNA, the system’s apparent molecular weight increases, potentially affecting cytotoxicity [
15,
37]. Furthermore, EGFP reporter proteins in the CRISPR/nCas9-polymer complex can generate free radicals and oxidative stress, with immature eGFP producing superoxide anions and H
2O
2 in the presence of NADH, leading to alterations in biological pathways, including decreased HIF1α stabilization and activity [
38]. Because of this, it is important to evaluate the combined effects of both the polymer and CRISPR system to optimize transfection protocols while reducing cytotoxicity.
Transfection with LP was lower across all four cellular models, and has been reported to be possibly toxic to specific cell types, leading to increased background cell death [
39]. NIH-3T3 fibroblasts showed lower cytotoxicity to the PP6D5 polymer, possibly due to their stiffer extracellular matrix [
36]. However, their TE was only 16.06%, lower than that of U87MG astrocytoma (28.05%) and SHSY5Y neuroblastoma (32.85%). O’Keefe et al. [
37] consider a TE of ≥11.5% to be adequate, which suggests that in the present study, most cell lines were successfully transfected, except for TSD fibroblasts, which showed the lowest efficiency (8.05%), due to their primary nature limiting exogenous DNA integration and expression, though they better simulate in vivo conditions [
40]. The accumulation of gangliosides in TSD cells may affect cell membrane and endocytosis mechanisms [
41,
42]. In contrast, Diaz et al. [
21] achieved 80% transfection with PP6D5 in HEK 293FT cells, due to their more permeable membrane and faster cell cycle [
43]. Since cellular characteristics influence transfection outcomes, is crucial to standardize conditions according to vector type and concentrations. For TSD, intrathecal or intracerebroventricular administration could be more effective due to the blood–brain barrier [
1,
44]. However, alternative strategies, like molecular Trojan horses (including engineered proteins, cell-penetrating peptides, monoclonal antibodies, and extracellular vesicles), also show potential to improve therapeutic delivery to the central nervous system [
44]. The results from SHSY5Y and U87MG cell lines can help to determine appropriate polymer concentrations to minimize the risk of damage to central nervous system cells.
We evaluated HexA activity using the MUGS substrate in cells either transfected with HEXA or co-transfected with HEXA/HEXB cDNAs. When only HEXA cDNA was transfected, the heterodimer HexA (αβ) was predominantly produced, due to limited β-subunit availability, which prevented excessive formation of the more stable HexB (ββ) isoenzyme [
45]. However, transfection with both HEXA and HEXB cDNAs, driven by the CMV promoter’s constitutive expression, ensured sufficient β-subunit supply, allowing balanced production of both isoenzymes [
13]. At 30 days post transfection, the enzyme activity in NIH-3T3 and SHSY5Y cells was significantly increased by using the polymer and co-transfection of the HEXA and HEXB cDNAs, outperforming the results from the cells edited only with HEXA cDNA. In U87MG cells, transfection with HEXA cDNA led to the highest HexA activity levels when using both the polymer and LP, with PP6D5-transfected cells showing the highest enzyme activity during the first 7–15 days. However, by day 25, the cells transfected with HEXA/HEXB cDNAs using the polymer showed no increase in activity compared to untreated cells, suggesting possible editing inefficiency. In TSD fibroblasts, transfection with HEXA/HEXB cDNAs using LP and the PP6D5 polymer achieved 11.24% and 2.92% of the activity levels of WT fibroblasts, respectively, while the activity after transfection with HEXA cDNA alone reached 2.80% and 3.16%. In this sense, Picache et al. [
46] indicate that 10–15% WT enzyme activity can reduce GM2 gangliosidosis complications. Leal et al. [
13] reported an improvement in lysosomal function upon partial restoration of β-hexosaminidase A activity, reaching 10% of WT levels. This suggest therapeutic benefits from modest enzyme activity increases [
47]. In SHSY5Y cells and TSD fibroblasts, transfection with HEXA/HEXB cDNAs using the PP6D5 polymer resulted in sustained enzymatic activity over time, in contrast to the previously observed decline from 15 to 30 days post transfection [
13]. This persistent enzyme activity could be attributed to either stabilization of gene expression or potential genomic integration through homologous recombination. Regardless of the non-viral vector employed, the CRISPR/nCas9 system with donor DNA functioned effectively across all studied models, achieving significant increases in enzymatic activity compared to untreated cells, and demonstrating successful genomic editing.
To evaluate the impact of genomic editing on cellular alterations associated with TSD, some physiological parameters were analyzed in patient fibroblasts. Proper lysosomal function is essential for maintaining cellular homeostasis, particularly for the degradation and recycling of macromolecules. In TSD, dysfunction of the β-hexosaminidase A enzyme leads to accumulation of GM2 gangliosides, which disrupts various lysosome-related processes, including autophagy, and causes lysosomal stress [
48].
The increase in lysosomal mass fluorescence in TSD fibroblasts transfected with LP may have resulted from sustained lysosomal biogenesis. This could be explained by persistent activation of TFEB, a key regulator that translocates to the nucleus under stress and upregulates lysosomal genes [
47]. Wang et al. [
6] observed increased nuclear TFEB in TSD models, correlating with dysfunctional lysosome accumulation, though this hypothesis requires further investigation. Previous research has demonstrated recovery of lysosomal mass in TSD fibroblasts after CRISPR/nCas9 with HEXA cDNA using LPs, but not when using MLPs, indicating that restoring lysosomal mass requires higher intracellular HexA activity production [
13].
In untreated TSD fibroblasts, GM2 gangliosides and other lipids, like phospholipids and cholesterol, accumulate [
1]. Nile Red (NR) staining was used to assess whether the CRISPR/nCas9 system could correct this lipid dysregulation, revealing, through fluorescence microscopy, that both polar and apolar lipids accumulate in the same cellular compartments. Previous research showed that enzyme replacement therapy with rhHex-A produced in
Komagataella phaffii reduced lipid accumulation [
28]. In the current study, lipid accumulation was evaluated after treatment with CRISPR/nCas9 using LP and the PP6D5 polymer as non-viral vectors. Cells transfected with LP showed decreased neutral/apolar lipids, likely due to partial restoration of lysosomal membrane dynamics to a level more similar to that of unaffected cells, and improved fusion with autophagosomes [
48], as indicated by the lower RFP signal in
Figure 5C. Meanwhile, cells transfected with the polymer showed significantly increased neutral/apolar lipids compared to WT cells. This could be related to a continuous accumulation of lipids during the period evaluated. An alternative explanation involves NR affinity for various hydrophobic structures, potentially interacting with transfection polymer residues or aggregates and overestimating lipid accumulation. This raises the possibility of interference, where NR fluorescence could be enhanced not only by lipid deposits, but also by residual polymer structures with hydrophobic characteristics [
49]. These results indicate that while the polymer promotes lipid accumulation, LP facilitates the restoration of lipid balance to a level closer to that of WT cells, highlighting the importance of understanding how gene delivery strategies affect not only TE, but also broader metabolic processes, including lipid homeostasis and lysosomal dynamics.
Autophagy, which is essential for differentiation, development, and cellular homeostasis, can be compromised in neurodegenerative diseases like TSD, resulting in intracellular molecule accumulation and disrupted cellular homeostasis [
50]. This study observed greater autophagosome accumulation in TSD fibroblasts, indicating disrupted lysosomal-autophagosome fusion. Settembre et al. [
48] corroborate that this may result from altered lysosomal membrane lipid composition, with cholesterol and ganglioside accumulation affecting membrane dynamics and fusion capability. Similarly, increased cholesterol in late endosomes, as seen in Niemann–Pick disease, disrupts intraendosomal trafficking and membrane properties [
51]. At 30 days post transfection, partial autophagy restoration was observed. Cells transfected with the PP6D5 polymer displayed higher red fluorescence signals, suggesting increased autolysosome formation and decreased lysosomal mass. This effect may be linked to the high TE achieved with the polymer, leading to enhanced therapeutic construct expression and subsequent restoration of autophagic regulation [
51].



 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}