
Application of Theoretical Solubility Calculations and Thermal and Spectroscopic Measurements to Guide the Processing of Triamcinolone Acetonide by Hot-Melt Extrusion

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Material
2.2. Theoretical Screening of Polymers and Plasticizers
2.3. Sample Preparation
2.4. Differential Scanning Calorimetry Analyses
2.5. Thermogravimetric Analyses
2.6. Fourier Transform Infrared Spectroscopy
3. Results and Discussion
3.1. Theoretical Screening for Polymer and Plasticizer Selection
3.2. Differential Scanning Calorimetry Analyses
3.3. Thermogravimetric Analyses
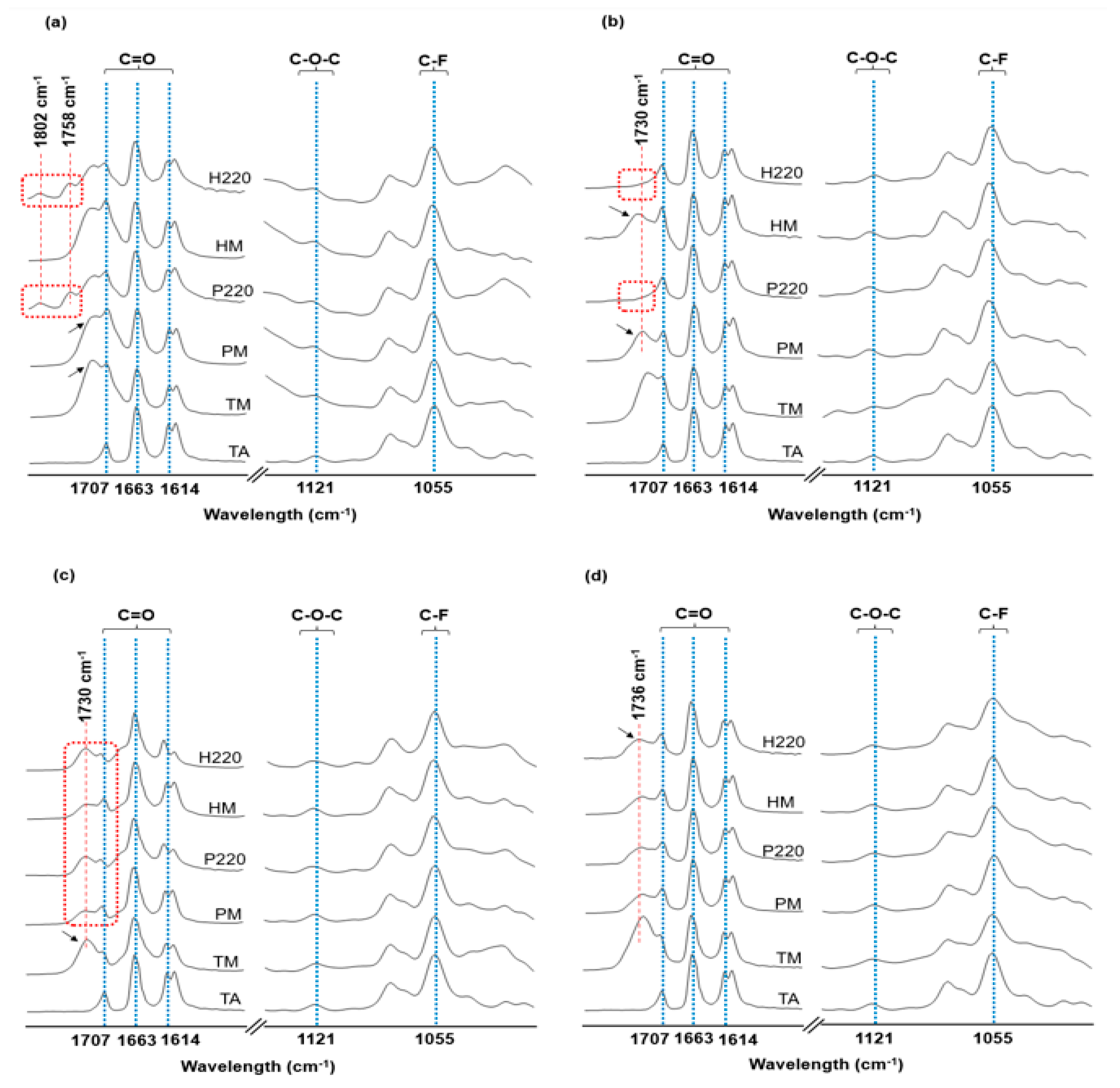
3.4. Fourier Transform Infrared Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TA | Triamcinolone acetonide |
| HME | Hot-melt extrusion |
| HSP | Hansen solubility parameters |
| EUD | Eudragit® L100 |
| PVA | Parteck® MXP |
| PVPVA | Plasdone® S-630 |
| HPMCAS | Aquasolve™ AS-MG |
| TEC | Triethyl citrate |
| DSC | Differential scanning calorimetry |
| TGA | Thermogravimetry |
| FTIR | Fourier transform infrared spectroscopy |
| BM | Binary physical mixtures |
| PM | Ternary physical mixtures |
| HM | Heat-stressed mixtures |
| Tg | Glass transition temperature |
| TM | Theoretical profile |
| P220 | PM with additional heating cycle |
| H220 | HM with additional heating cycle |
References
- Abou-ElNour, M.; Ishak, R.A.; Tiboni, M.; Bonacucina, G.; Cespi, M.; Casettari, L.; Soliman, M.E.; Geneidi, A.S. Triamcinolone acetonide-loaded PLA/PEG-PDL microparticles for effective intra-articular delivery: Synthesis, optimization, in vitro and in vivo evaluation. JCR 2019, 309, 125–144. [Google Scholar] [CrossRef] [PubMed]
- Fredman, R.; Tenenhaus, M. Cushing’s syndrome after intralesional triamcinolone acetonide: A systematic review of the literature and multinational survey. Burns 2013, 39, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Obata, R.; Iriyama, A.; Inoue, Y.; Takahashi, H.; Tamaki, Y.; Yanagi, Y. Triamcinolone acetonide suppresses early proangiogenic response in retinal pigment epithelial cells after photodynamic therapy in vitro. Br. J. Opfthalmol. 2007, 91, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Formica, M.L.; Ullio Gamboa, G.V.; Tártara, L.I.; Luna, J.D.; Benoit, J.P.; Palma, S.D. Triamcinolone acetonide-loaded lipid nanocapsules for ophthalmic applications. Int. J. Pharm. 2020, 573, 118795. [Google Scholar] [CrossRef]
- Granados, P.A.; Pinho, L.A.G.; Sa-Barreto, L.L.; Gratieri, T.; Gelfuso, G.M.; Cunha-Filho, M. Application of hot-melt extrusion in the complexation of naringenin with cyclodextrin using hydrophilic polymers. Adv. Powder Technol. 2022, 33, 103380. [Google Scholar] [CrossRef]
- Patil, H.; Vemula, S.K.; Narala, S.; Lakkala, P.; Munnangi, S.R.; Narala, N.; Jara, M.O.; Williams, R.O.; Terefe, H.; Repka, M.A. Hot-melt extrusion: From theory to application in pharmaceuticalformulation—Where are we now? AAPS PharmSciTech 2024, 25, 37. [Google Scholar] [CrossRef]
- Forster, S.P.; Dippold, E.; Chiang, T. Twin-screw melt granulation for oral solid pharmaceutical products. Pharmaceutics 2021, 13, 665. [Google Scholar] [CrossRef]
- Thakore, S.D.; Akhtar, J.; Jain, R.; Paudel, A.; Bansal, A.K. Analytical and computational methods for the determination of drug-polymer solubility and miscibility. Mol. Pharm. 2021, 18, 2835–2866. [Google Scholar] [CrossRef]
- Lima, A.L.; Gross, I.P.; Sá-Barreto, L.L.; Gratieri, T.; Gelfuso, G.M.; Cunha-Filho, M. Extrusion-based systems for topical and transdermal drug delivery. Expert. Opin. Drug Deliv. 2023, 20, 979–992. [Google Scholar] [CrossRef]
- Maniruzzaman, M.; Boateng, J.S.; Snowden, M.J.; Douroumis, D. A review of Hot-melt extrusion: Process technology to pharmaceutical products. ISRN Pharm. 2012, 2012, 436763. [Google Scholar] [CrossRef]
- Maximiano, F.P.; Novack, K.M.; Bahia, M.T.; de Sá-Barreto, L.L.; da Cunha-Filho, M.S.S. Polymorphic screen and drug–excipient compatibility studies of the antichagasic benznidazole. J. Therm. Anal. Calorim. 2011, 106, 819–824. [Google Scholar] [CrossRef]
- Ricarte, R.G.; Van Zee, N.J.; Li, Z.; Johnson, L.M.; Lodge, T.P.; Hillmyer, M.A. Recent advances in understanding the micro- and nanoscale phenomena of amorphous solid dispersions. Mol. Pharm. 2019, 16, 4089–4103. [Google Scholar] [CrossRef] [PubMed]
- Jelić, D. Thermal stability of amorphous solid dispersions. Molecules 2021, 26, 238. [Google Scholar] [CrossRef]
- Hansen, C.M. Hansen Solubility Parameters: A User’s Handbook, 2nd ed.; Taylor & Francis Group: Abingdon, UK, 2007. [Google Scholar]
- Van Krevelen, D.W.; Te Nijenhuis, K. Properties of Polymers: Their Correlation with Chemical Structure; Their Numerical Estimation and Prediction from Additive Group Contributions, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Pinho, L.A.G.; Lima, A.L.; Sa-Barreto, L.L.; Gratieri, T.; Gelfuso, G.M.; Marreto, R.N.; Cunha-Filho, M. Preformulation studies to guide the production of medicines by fused deposition modeling 3D printing. AAPS PharmSciTech 2021, 22, 263. [Google Scholar] [CrossRef]
- Malaquias, L.F.; Schulte, H.L.; Chaker, J.A.; Karan, K.; Durig, T.; Marreto, R.N.; Gratieri, T.; Gelfuso, G.M.; Cunha-Filho, M. Hot melt extrudates formulated using design space: One simple process for both palatability and dissolution rate improvement. J. Pharm. Sci. 2018, 107, 286–296. [Google Scholar] [CrossRef]
- Alves-Silva, I.; Sá-Barreto, L.C.L.; Lima, E.M.; Cunha-Filho, M.S.S. Preformulation studies of itraconazole associated with benznidazole and pharmaceutical excipients. Thermochim. Acta 2014, 575, 29–33. [Google Scholar] [CrossRef]
- Pires, F.Q.; Gross, I.P.; Sa-Barreto, L.L.; Gratieri, T.; Gelfuso, G.M.; Bao, S.N.; Cunha-Filho, M. In-situ formation of nanoparticles from drug-loaded 3D polymeric matrices. Eur. J. Pharm. Sci. 2023, 188, 106517. [Google Scholar] [CrossRef]
- Moseson, D.E.; Jordan, M.A.; Shah, D.D.; Corum, I.D.; Alvarenga, B.R.; Taylor, L.S. Application and limitations of thermogravimetric analysis to delineate the hot melt extrusion chemical stability processing window. Int. J. Pharm. 2020, 590, 119916. [Google Scholar] [CrossRef]
- van Heugten, A.J.P.; de Vries, W.S.; Markesteijn, M.M.A.; Pieters, R.J.; Vromans, H. The role of excipients in the stability of triamcinolone acetonide in ointments. AAPS PharmSciTech 2018, 19, 1448–1453. [Google Scholar] [CrossRef]
- van Heugten, A.J.P.; de Boer, W.; de Vries, W.S.; Markesteijn, C.M.A.; Vromans, H. Development and validation of a stability-indicating HPLC-UV method for the determination of triamcinolone acetonide and its degradation products in an ointment formulation. J. Pharm. Biomed. Anal. 2018, 149, 265–270. [Google Scholar] [CrossRef]
- Hilgeroth, P.S.; Thümmler, J.F.; Binder, W.H. 3D printing of triamcinolone acetonide in triblock copolymers of styrene-isobutylene-styrene as a slow-release system. Polymers 2022, 14, 3742. [Google Scholar] [CrossRef] [PubMed]
- Medarevic, D.; Djuriš, J.; Barmpalexis, P.; Kachrimanis, K.; Ibric, S. Analytical and computational methods for the estimation of drug-polymer solubility and miscibility in solid dispersions development. Pharmaceutics 2019, 11, 372. [Google Scholar] [CrossRef] [PubMed]
- Louwerse, M.J.; Maldonado, A.; Rousseau, S.; Moreau-Masselon, C.; Roux, B.; Rothenberg, G. Revisiting Hansen solubility parameters by including thermodynamics. ChemPhysChem 2017, 18, 2999–3006. [Google Scholar] [CrossRef]
- Xu, L.; Chu, Z.; Zhang, J.; Cai, T.; Zhang, X.; Li, Y.; Wang, H.; Shen, X.; Cai, R.; Shi, H.; et al. Steric effects in the deposition mode and drug-delivering efficiency of nanocapsule-based multilayer films. ACS Omega 2022, 7, 30321–30332. [Google Scholar] [CrossRef]
- Zheng, G.-Q.; Wu, J.-N.; Zhang, L.; Zhang, Q.; Chen, L.; Wang, X.-L.; Zhao, H.-B.; Liu, B.-W.; Wang, Y.-Z. Tailoring cross-linking temperature via hydrogen bonding and steric hindrance towards high-performance fire-safe polymer. Polymer 2024, 298, 126908. [Google Scholar] [CrossRef]
- Tran, P.H.L.; Lee, B.J.; Tran, T.T.D. Recent studies on the processes and formulation impacts in the development of solid dispersions by hot-melt extrusion. Eur. J. Pharm. Biopharm. 2021, 164, 13–19. [Google Scholar] [CrossRef]
- Greenhalgh, D.J.; Williams, A.C.; Timmins, P.; York, P. Solubility parameters as predictors of miscibility in solid dispersions. J. Pharm. Sci. 1999, 88, 1182–1190. [Google Scholar] [CrossRef]
- Ponnammal, P.; Kanaujia, P.; Yani, Y.; Ng, W.K.; Tan, R.B.H. Orally disintegrating tablets containing melt extruded amorphous solid dispersion of tacrolimus for dissolution enhancement. Pharmaceutics 2018, 10, 35. [Google Scholar] [CrossRef]
- Jankovic, S.; Tsakiridou, G.; Ditzinger, F.; Koehl, N.J.; Price, D.J.; Ilie, A.-R.; Kalantzi, L.; Kimpe, K.; Holm, R.; Nair, A.; et al. Application of the solubility parameter concept to assist with oral delivery of poorly water-soluble drugs—A PEARRL review. JPP 2019, 71, 441–463. [Google Scholar] [CrossRef]
- Thakkar, R.; Thakkar, R.; Pillai, A.; Ashour, E.A.; Repka, M.A. Systematic screening of pharmaceutical polymers for hot melt extrusion processing: A comprehensive review. Int. J. Pharm. 2020, 576, 118989. [Google Scholar] [CrossRef]
- Klar, F.; Urbanetz, N.A. Solubility parameters of hypromellose acetate succinate and plasticization in dry coating procedures. Drug Dev. Ind. Pharm. 2016, 42, 1621–1635. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, J.; da Silva, G.S.; Velho, M.C.; Beck, R.C.R. Eudragit®: A versatile family of polymers for hot melt extrusion and 3D printing processes in pharmaceutics. Pharmaceutics 2021, 13, 1424. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, R.; Chen, Y.; Xue, K.; Danrong, H.; Miaomiao, H. Application of carrier and plasticizer to improve the dissolution and bioavailability of poorly water-soluble baicalein by hot-melt extrusion. AAPS PharmSciTech 2014, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Maniruzzaman, M.; Bonnefille, M.; Aranyos, A.; Snowden, M.J.; Douroumis, D. An in-vivo and in-vitro taste masking evaluation of bitter melt-extruded drugs. JPP 2014, 66, 323–337. [Google Scholar] [CrossRef]
- Maniruzzaman, M.; Pang, Y.; Morgan, D.; Douroumis, D. Molecular modeling as a predictive tool for the development of solid dispersions. Mol. Pharm. 2015, 12, 1040–1049. [Google Scholar] [CrossRef]
- Alqarni, M.H.; Haq, N.; Alam, P.; Abdel-Kader, M.S.; Foudah, A.I.; Shakeel, F. Solubility data, Hansen solubility parameters and thermodynamic behavior of pterostilbene in some pure solvents and different (PEG-400 + water) cosolvent compositions. J. Mol. Liq. 2021, 331, 115700. [Google Scholar] [CrossRef]
- Ghosh, I.; Snyder, J.; Vippagunta, R.; Alvine, M.; Vakil, R.; Tong, W.Q.; Vippagunta, S. Comparison of HPMC based polymers performance as carriers for manufacture of solid dispersions using the melt extruder. Int. J. Pharm. 2011, 419, 12–19. [Google Scholar] [CrossRef]
- Bharti, K.; Dubey, G.; Kumar, M.; Jha, A.; Manjit; Upadhyay, M.; Mali, P.S.; Kumar, A.; Bharatam, P.V.; Mishra, B. A multifaceted approach for grading of polymers for the development of stable amorphous solid dispersion of Riluzole. J. Drug Deliv. Sci. Technol. 2023, 90, 105158. [Google Scholar] [CrossRef]
- Luo, Y.; Hong, Y.; Shen, L.; Wu, F.; Lin, X. Multifunctional role of polyvinylpyrrolidone in pharmaceutical formulations. AAPS PharmSciTech 2021, 22, 34. [Google Scholar] [CrossRef]
- Cardoso, G.; Gonzalez, C.A.G.; Santos-Rosales, V.; Taveira, S.F.; Cunha-Filho, M.; Concheiro, A.; Alvarez-Lorenzo, C.; Marreto, R.N. Supercritical fluid (SCF)-assisted preparation of cyclodextrin-based poly(pseudo)rotaxanes for transdermal purposes. DDTR 2024, 14, 103–115. [Google Scholar] [CrossRef]
- Araújo, J.; Gonzalez-Mira, E.; Egea, M.A.; Garcia, M.L.; Souto, E.B. Optimization and physicochemical characterization of a triamcinolone acetonide-loaded NLC for ocular antiangiogenic applications. Int. J. Pharm. 2010, 393, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Näther, C.; Jeß, I. New news about an old drug: Investigations on the polymorphism of triamcinolone acetonide. Angew. Chem. Int. Ed. Engl. 2006, 45, 6381–6383. [Google Scholar] [CrossRef] [PubMed]
- Kramarczyk, D.; Knapik-Kowalczuk, J.; Kurek, M.; Jamróz, W.; Jachowicz, R.; Paluch, M. Hot melt extruded posaconazole-based amorphous solid dispersions—The effect of different types of polymers. Pharmaceutics 2023, 15, 799. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.A.D.; Cintra, E.R.; Alonso, E.C.P.; Alves, G.L.; Lima, E.M.; Taveira, S.F.; da Cunha-Filho, M.S.S.; Marreto, R.N. Selection of excipients for the development of carvedilol loaded lipid-based drug delivery systems. J. Therm. Anal. Calorim. 2017, 130, 1593–1604. [Google Scholar] [CrossRef]
- Pires, F.Q.; Pinho, L.A.; Freire, D.O.; Silva, I.C.R.; Sa-Barreto, L.L.; Cardozo-Filho, L.; Gratieri, T.; Gelfuso, G.M.; Cunha-Filho, M. Thermal analysis used to guide the production of thymol and Lippia origanoides essential oil inclusion complexes with cyclodextrin. J. Therm. Anal. Calorim. 2019, 137, 543–553. [Google Scholar] [CrossRef]
- Daniel, J.S.P.; Cruz, J.C.; Catelani, T.A.; Garcia, J.S.; Trevisan, M.G. Erythromycin-excipients compatibility studies using the thermal analysis and dynamic thermal infrared spectroscopy coupled with chemometrics. J. Therm. Anal. Calorim. 2021, 143, 3127–3135. [Google Scholar] [CrossRef]
- Cunha-Filho, M.S.; Martinez-Pacheco, R.; Landin, M. Dissolution rate enhancement of the novel antitumoral β-lapachone by solvent change precipitation of microparticles. Eur. J. Pharm. Biopharm. 2008, 69, 871–877. [Google Scholar] [CrossRef]
- Teixeira, S.C.; Silva, R.R.A.; de Oliveira, T.V.; Stringheta, P.C.; Pinto, M.R.M.R.; Soares Nde, F.F. Glycerol and triethyl citrate plasticizer effects on molecular, thermal, mechanical, and barrier properties of cellulose acetate films. Food Biosci. 2021, 42, 101202. [Google Scholar] [CrossRef]
- Andrews, G.P.; Jones, D.S.; Abu Diak, O.; McCoy, C.P.; Watts, A.B.; McGinity, J.W. The manufacture and characterisation of hot-melt extruded enteric tablets. Eur. J. Pharm. Biopharm. 2008, 69, 264–273. [Google Scholar] [CrossRef]
- Ponsar, H.; Quodbach, J. Customizable 3D printed Implants containing triamcinolone acetonide: Development, analysis, modification, and modeling of drug release. Pharmaceutics 2023, 15, 2097. [Google Scholar] [CrossRef]
- Low, A.Q.J.; Parmentier, J.; Khong, Y.M.; Chai, C.C.E.; Tun, T.Y.; Berania, J.E.; Liu, X.; Gokhale, R.; Chan, S.Y. Effect of type and ratio of solubilising polymer on characteristics of hot-melt extruded orodispersible films. Int. J. Pharm. 2013, 455, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, F.P.; Fortes, A.C.; Da Cruz Fonseca, S.G.; Breitkreutz, J.; Ferraz, H.G. Manufacture and characterization of mucoadhesive buccal films based on pectin and gellan gum containing triamcinolone acetonide. Int. J. Polym. Sci. 2018, 2018, 2403802. [Google Scholar] [CrossRef]
- Cilurzo, F.; Minghetti, P.; Selmin, F.; Casiraghi, A.; Montanari, L. Polymethacrylate salts as new low-swellable mucoadhesive materials. JCR 2003, 88, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Teodorescu, M.; Bercea, M.; Morariu, S. Biomaterials of PVA and PVP in medical and pharmaceutical applications: Perspectives and challenges. Biotechnol. Adv. 2019, 37, 109–131. [Google Scholar] [CrossRef]
- Alvarenga BRde Moseson, D.E.; Carneiro, R.L.; Taylor, L.S. Impact of polymer type on thermal degradation of amorphous solid dispersions containing ritonavir. Mol. Pharm. 2022, 19, 332–344. [Google Scholar] [CrossRef]
- Butreddy, A.; Sarabu, S.; Bandari, S.; Batra, A.; Lawal, K.; Chen, N.N.; Bi, V.; Durig, T.; Repka, M.A. Influence of PlasdoneTM S630 ultra—An improved copovidone on the processability and oxidative degradation of quetiapine fumarate amorphous solid dispersions prepared via hot-melt extrusion technique. AAPS PharmSciTech 2021, 22, 196. [Google Scholar] [CrossRef]
- Payab, S.; Davaran, S.; Tanhaei, A.; Fayyazi, B.; Jahangiri, A.; Farzaneh, A.; Adibkia, K. Triamcinolone acetonide-Eudragit® RS100 nanofibers and nanobeads: Morphological and physicochemical characterization. Artif. Cells Nanomed. Biotechnol. 2016, 44, 362–369. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Liao, C.-M.; Hsiue, G.-H. A reflectance FTIR/DSC microspectroscopic study of the nonisothermal kinetics of anhydride formation in Eudragit L-100 films. Polym. Deqrad. Stab. 1995, 47, 299–303. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Liao, C.-M.; Hsiue, G.-H. Isothermal kinetics of anhydride formation in Eudragit L-100 films determined by reflectance FTIR/DSC microspectroscopy. Polymer 1996, 37, 269–273. [Google Scholar] [CrossRef]
- Hermanson, G.T. Bioconjugate Techniques, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 229–258. [Google Scholar]
- Katopodis, K.; Kapourani, A.; Vardaka, E.; Karagianni, A.; Chorianopoulou, C.; Kontogiannopoulos, K.N.; Bikiaris, D.N.; Kachrimanis, K.; Barmpalexis, P. Partially hydrolyzed polyvinyl alcohol for fusion-based pharmaceutical formulation processes: Evaluation of suitable plasticizers. Int. J. Pharm. 2020, 578, 119121. [Google Scholar] [CrossRef]
- Hartauer, K.J.; Arbuthnot, G.N.; Baertschi, S.W.; Johnson, R.A.; Luke, W.D.; Pearson, N.G.; Rickard, E.C.; Tingle, C.A.; Tsang, P.K.S.; Wiens, R.E. Influence of peroxide impurities in povidone and crospovidone on the stability of raloxifene hydrochloride in tablets: Identification and control of an oxidative degradation product. Pharm. Dev. Technol. 2000, 5, 303–310. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Material | δt (MPa1/2) | δt Components (MPa1/2) | Δδt (MPa1/2) | Tg (°C) | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δd | δp | δh | δv | |TA-P| | |TA-Pl| | |P-TEC| | ||||
| TA | 21.2 | 18.7 | 7.7 | 6.4 | 20.22 | --- | --- | --- | --- | Author |
| HPMC | 29.1 | --- | --- | --- | 7.9 | --- | 8.12 | 117 | [32] | |
| HPMCAS | 25.7 | 20.5 | 5.1 | 14.6 | 21.12 | 4.5 | --- | 4.72 | 135 | [19,33] |
| E-E100 | 18.9 | --- | --- | --- | 2.3 | --- | 2.08 | 48 | [34,35] | |
| E-L100 | 22.75 | 19.31 | 0.41 | 12.03 | 19.31 | 1.55 | --- | 1.77 | 150 | [34,36] |
| E-S100 | 18.38 | --- | --- | --- | 2.82 | --- | 2.6 | 150 | [32] | |
| PVA | 21.17 | 11.2 | 12.40 | 13.0 | 16.44 | 0.03 | --- | 0.19 | 45 | [5,19] |
| PVPVA | 19.60 | 0.64 | 18.0 | 18.01 | 7.73 | 1.60 | --- | 1.38 | 105 | [37] |
| Glycerin | 34.34 | --- | --- | --- | --- | 13.14 | --- | --- | [14] | |
| PEG400 | 18.9 | --- | --- | --- | --- | 2.3 | --- | --- | [38] | |
| PPG | 29.9 | --- | --- | --- | --- | 8.7 | --- | --- | [14] | |
| TEC | 20.98 | 16.5 | 4.90 | 12.0 | 17.21 | --- | 0.22 | --- | --- | [14,19] |
| Sample | PM | HM | ||||
|---|---|---|---|---|---|---|
| Drug Crystallinity (%) | (Jg−1) | TA Melting Peak (°C) | Drug Crystallinity (%) | (Jg−1) | TA Melting Peak (°C) | |
| TA-EUD-TEC | 15.3 | 23.9 | 255 | 18.9 | 29.5 | 255 |
| TA-PVA-TEC | 34.7 | 54.1 | 269 | 32.5 | 50.6 | 263 |
| TA-PVPVA-TEC | 96.9 | 151.1 | 287 | 54.4 | 84.9 | 286 |
| TA-HPMCAS-TEC | 66.1 | 103.1 | 267 | 91.5 | 142.8 | 268 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Granados, P.A.; Gross, I.P.; Medeiros-Souza, P.; Sá-Barreto, L.L.; Gelfuso, G.M.; Gratieri, T.; Cunha-Filho, M. Application of Theoretical Solubility Calculations and Thermal and Spectroscopic Measurements to Guide the Processing of Triamcinolone Acetonide by Hot-Melt Extrusion. Pharmaceutics 2025, 17, 586. https://doi.org/10.3390/pharmaceutics17050586
Granados PA, Gross IP, Medeiros-Souza P, Sá-Barreto LL, Gelfuso GM, Gratieri T, Cunha-Filho M. Application of Theoretical Solubility Calculations and Thermal and Spectroscopic Measurements to Guide the Processing of Triamcinolone Acetonide by Hot-Melt Extrusion. Pharmaceutics. 2025; 17(5):586. https://doi.org/10.3390/pharmaceutics17050586
Chicago/Turabian StyleGranados, Pedro A., Idejan P. Gross, Patrícia Medeiros-Souza, Livia L. Sá-Barreto, Guilherme M. Gelfuso, Tais Gratieri, and Marcilio Cunha-Filho. 2025. "Application of Theoretical Solubility Calculations and Thermal and Spectroscopic Measurements to Guide the Processing of Triamcinolone Acetonide by Hot-Melt Extrusion" Pharmaceutics 17, no. 5: 586. https://doi.org/10.3390/pharmaceutics17050586
APA StyleGranados, P. A., Gross, I. P., Medeiros-Souza, P., Sá-Barreto, L. L., Gelfuso, G. M., Gratieri, T., & Cunha-Filho, M. (2025). Application of Theoretical Solubility Calculations and Thermal and Spectroscopic Measurements to Guide the Processing of Triamcinolone Acetonide by Hot-Melt Extrusion. Pharmaceutics, 17(5), 586. https://doi.org/10.3390/pharmaceutics17050586