
Biosimilars Targeting Pathogens: A Comprehensive Review of Their Role in Bacterial, Fungal, Parasitic, and Viral Infections

 ,
, 
Abstract

1. Introduction
1.1. The Emergence of Biosimilars
1.2. Economic Viability of Biosimilars and Impact on Healthcare Systems
2. Types of Therapeutic Proteins Against Pathogens
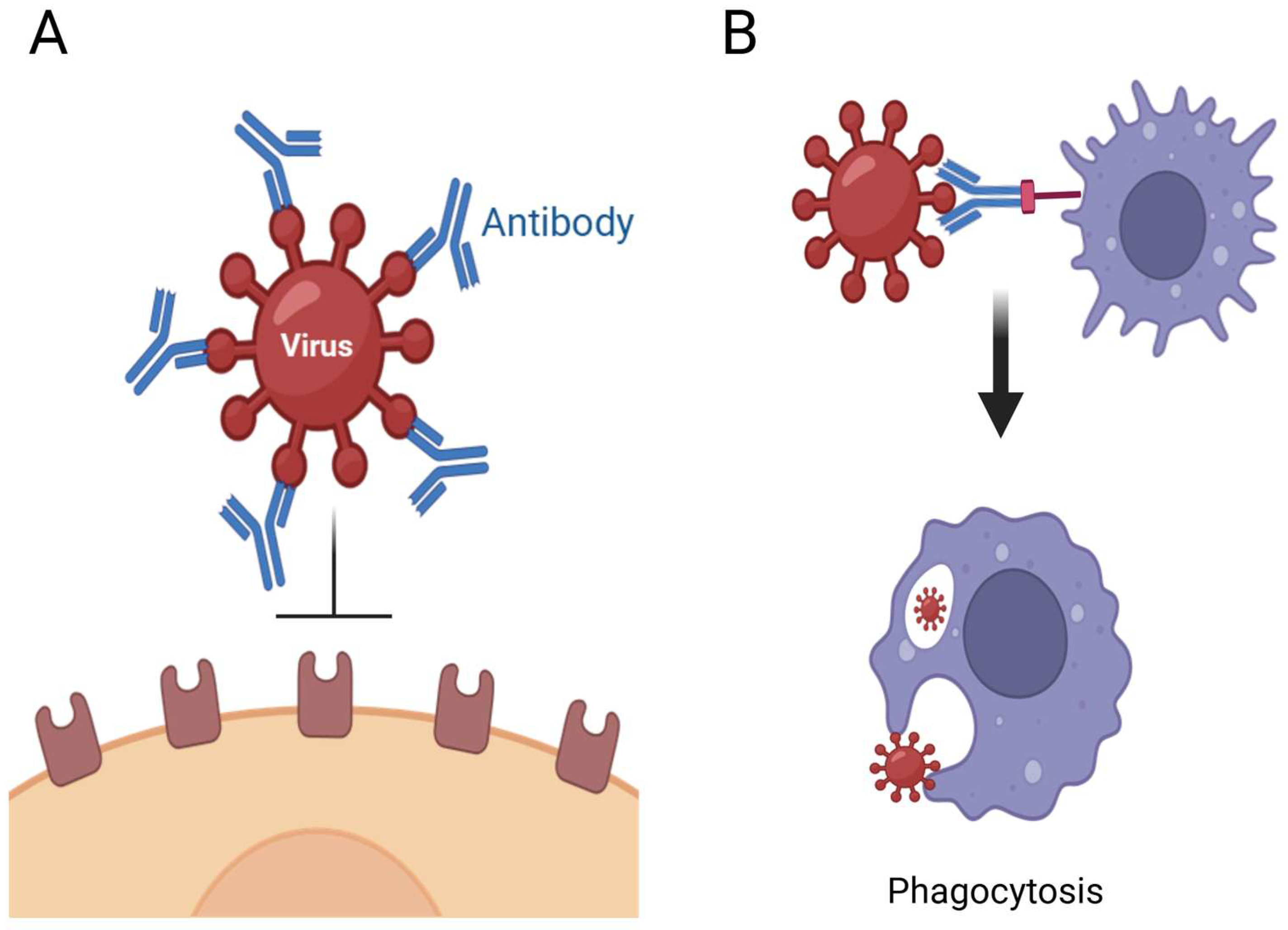
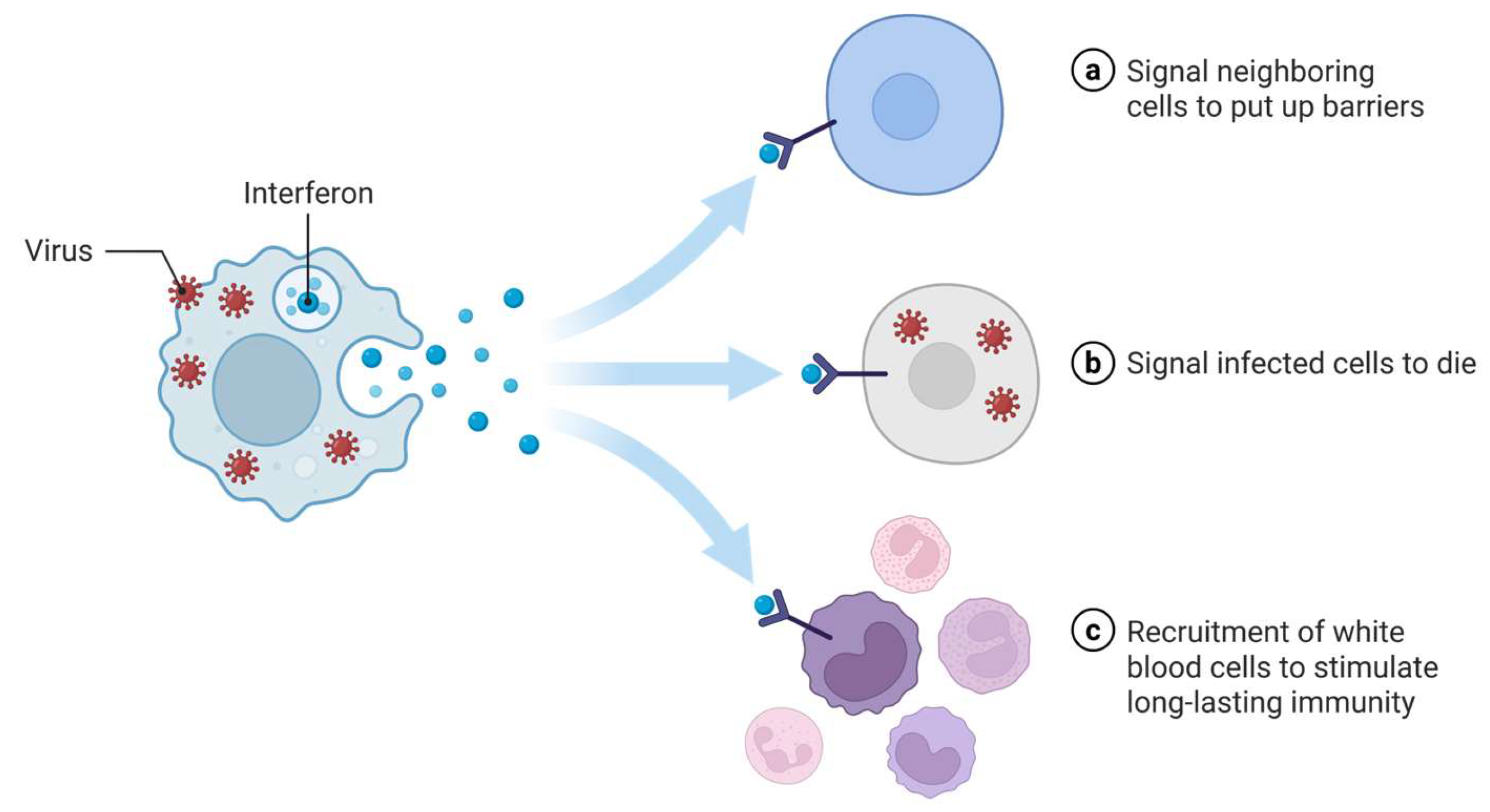
2.1. Antiviral Therapeutic Proteins
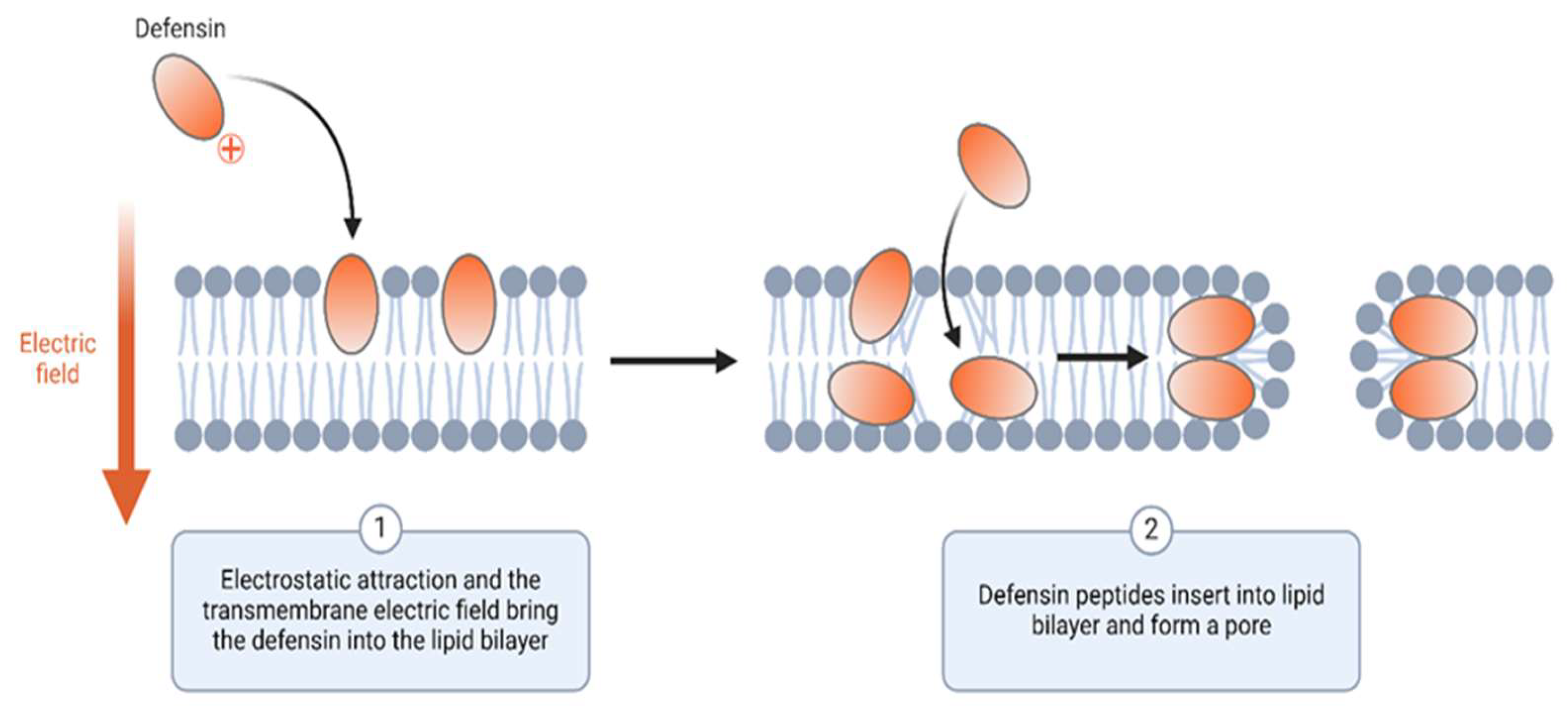
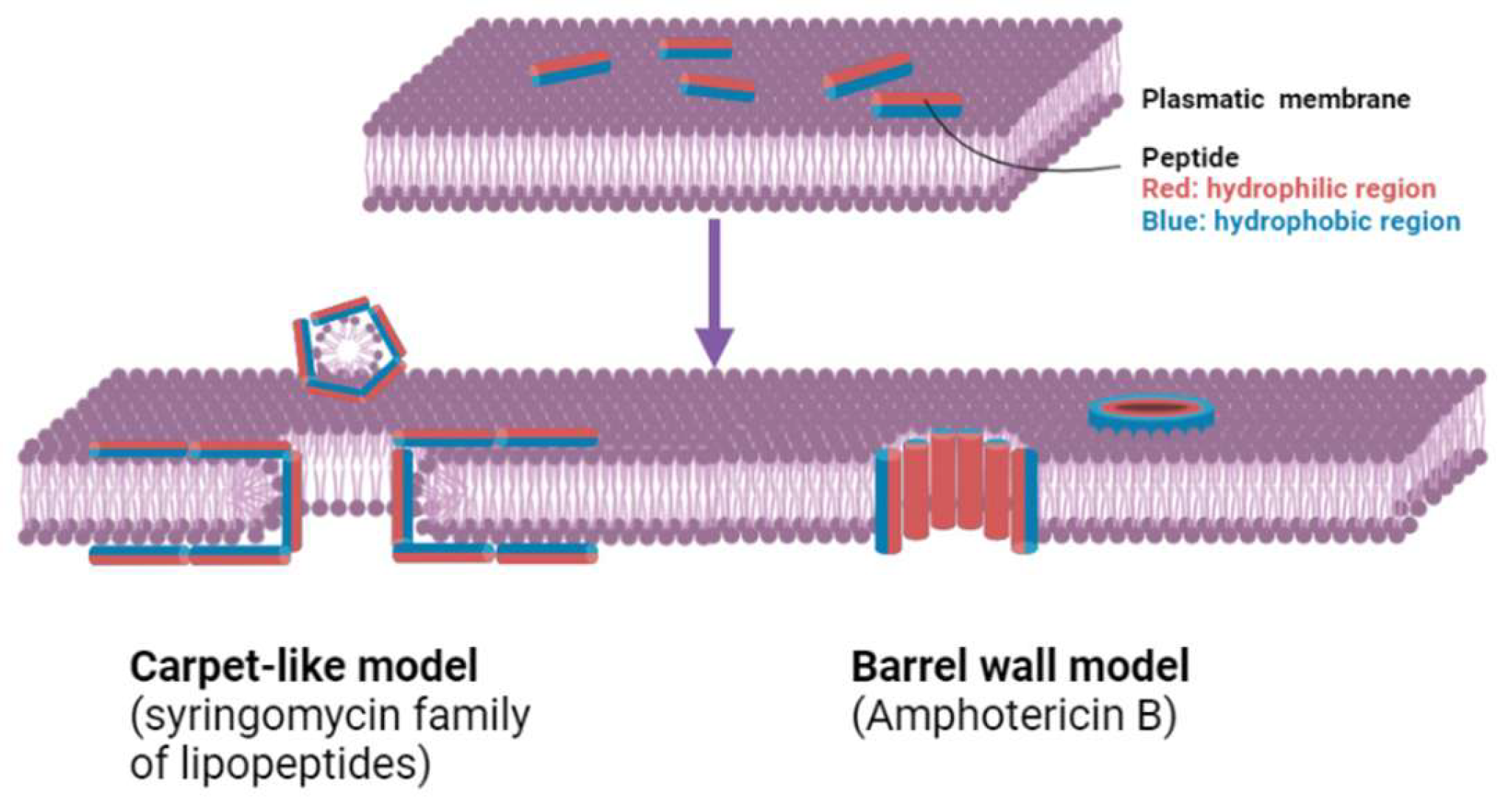
2.2. Antibacterial Therapeutic Proteins and Their Potential Biosimilars

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Examples | Mechanism of Action | Target Pathogen | Reference |
|---|---|---|---|---|
| Interferons: IFN-α, INF-γ | Stimulation of antiviral defenses | HBV, HCV, Ebola, fungal infections | [41,42] | |
| Antiviral therapeutic proteins | mAbs: Palivizumab, Casirivimab, Imdevimab | Neutralization of virions, block entry into host cells | RSV, SARS-CoV-2 | [33,34,35] |
| Fusion inhibitors: Enfuvirtide | Prevention of viral fusion with host cell membranes | HIV | [49] | |
| Enzybiotics: Cpl-1, Staphefekt SA.100 | Enzymatic degradation of bacterial cell walls | S. pneumoniae, S. aureus | [63,64,65] | |
| Antibacterial therapeutic proteins | AMPs: Defensins and cathelicidins | Disruption of bacterial membranes, cell lysis | Broad-spectrum antibacterial activity | [66,67] |
| mAbs: Bezlotoxumab, Tefibazumab | Target bacterial virulence factors, neutralization of toxins | C. difficile, S. aureus, | [58,60,61] | |
| Antifungal therapeutic proteins | mAbs: Mycograb | Neutralize fungal toxins and prevent cell invasion | Candida and Aspergillus species | [68] |
| AFPs: histatin 5 | Disruption of membrane integrity | Candida and Aspergillus species | [69] | |
| mAbs | Target specific parasite antigens | P. falciparum, P. vivax | [70,71] | |
| Anti-parasitic therapeutic proteins | Fusion proteins | Target parasite proteins and elicit a more robust immune response | L. amazonensis | [72] |
| Recombinant vaccines: Mosquirix | Emulate parasite antigens to stimulate immunity and ensure long-term protection | P. falciparum | [73] |
2.3. Antifungal Therapeutic Proteins
2.4. Antiparasitic Therapeutic Proteins and Their Potential Biosimilars
3. Challenges for Anti-Pathogen Biosimilars
3.1. Difficulties in Designing Appropriate In Vitro and In Vivo Models
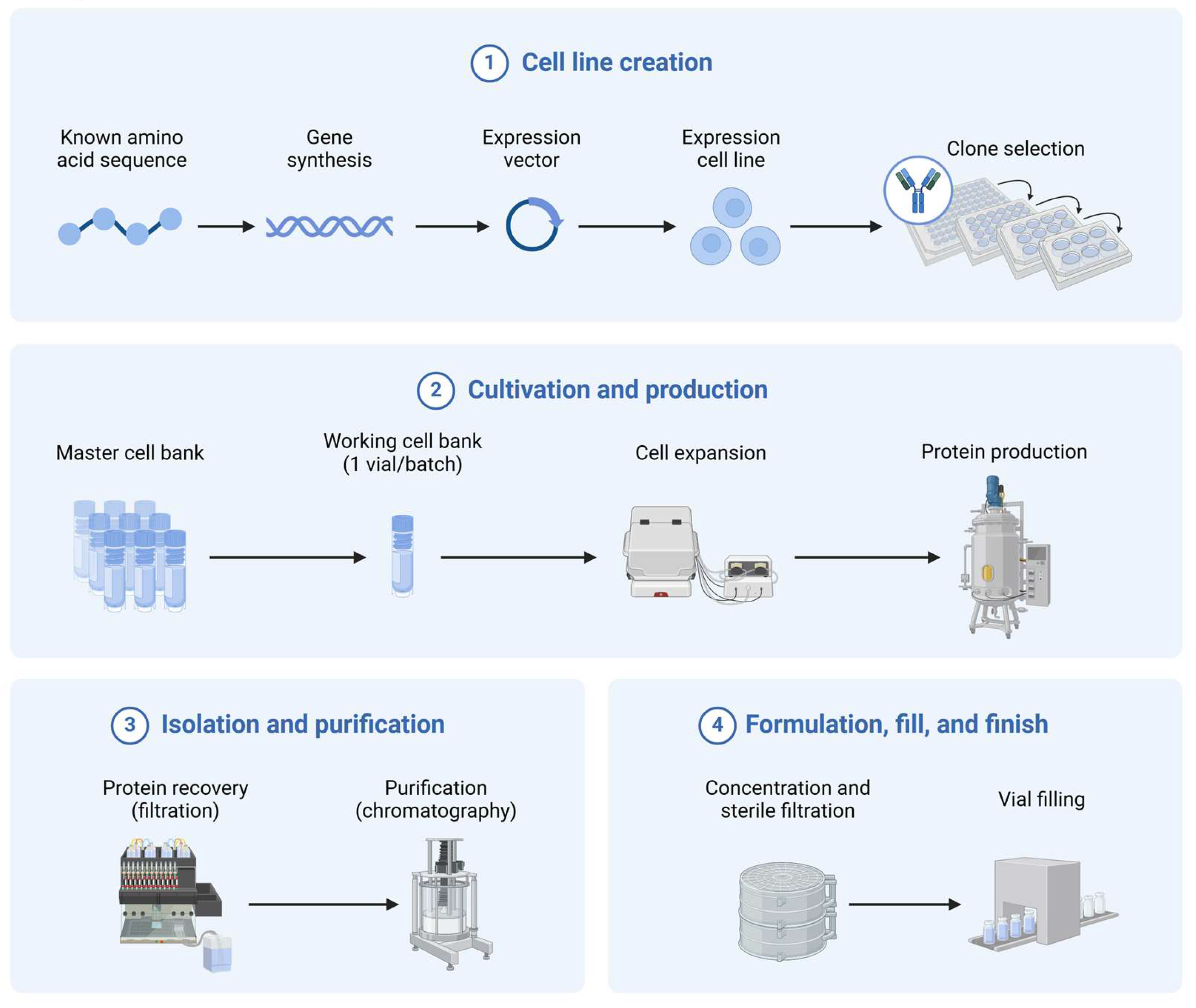
3.2. Choice of Expression Systems for Different Types of Anti-Pathogen Biologics
3.3. Scaling up Production and Regulatory Guidelines
3.4. Structural and Functional Complexity of Biosimilars
3.5. Cost and Time of Development
3.6. Stability and Storage Requirements
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial peptides: Classification, design, application and research progress in multiple fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Lin, X.; Gao, X.; Khan, R.U.; Liao, J.-Y.; Du, S.; Ge, J.; Zeng, S.; Yao, S.Q. The dawn of a new era: Targeting the “undruggables” with antibody-based therapeutics. Chem. Rev. 2023, 123, 7782–7853. [Google Scholar] [CrossRef] [PubMed]
- Jana, M.K.; Chiang, M.-H. Monoclonal antibodies: A promising weapon against the silent pandemic of multidrug resistant bacteria. In Microbes of Medical Importance; IIP Series: Mumbai, India, 2024; Volume 3, pp. 546–568. ISBN 978-93-6252-020-3. [Google Scholar]
- Drayton, M.; Kizhakkedathu, J.N.; Straus, S.K. Towards robust delivery of antimicrobial peptides to combat bacterial resistance. Molecules 2020, 25, 3048. [Google Scholar] [CrossRef] [PubMed]
- Ul Haq, I.; Maryam, S.; Shyntum, D.Y.; Khan, T.A.; Li, F. Exploring the frontiers of therapeutic breadth of antifungal peptides: A new avenue in antifungal drugs. J. Ind. Microbiol. Biotechnol. 2024, 51, kuae018. [Google Scholar] [CrossRef]
- Agarwal, G.; Gabrani, R. Antiviral peptides: Identification and validation. Int. J. Pept. Res. Ther. 2021, 27, 149–168. [Google Scholar] [CrossRef]
- Cao, N.; Cai, Y.; Huang, X.; Jiang, H.; Huang, Z.; Xing, L.; Lu, L.; Jiang, S.; Xu, W. Inhibition of influenza A virus and SARS-CoV-2 infection or co-infection by griffithsin and griffithsin-based bivalent entry inhibitor. Mbio 2024, 15, e00741-24. [Google Scholar] [CrossRef]
- Duan, Z.; Zhang, J.; Chen, X.; Liu, M.; Zhao, H.; Jin, L.; Zhang, Z.; Luan, N.; Meng, P.; Wang, J. Role of LL-37 in thrombotic complications in patients with COVID-19. Cell. Mol. Life Sci. 2022, 79, 309. [Google Scholar] [CrossRef]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef]
- Shi, M.; McHugh, K.J. Strategies for overcoming protein and peptide instability in biodegradable drug delivery systems. Adv. Drug Deliv. Rev. 2023, 199, 114904. [Google Scholar] [CrossRef]
- Lee, Y.J.; Shirkey, J.D.; Park, J.; Bisht, K.; Cowan, A.J. An overview of antiviral peptides and rational biodesign considerations. Biodes Res. 2022, 2022, 9898241. [Google Scholar] [CrossRef]
- Das, P.; Maity, A.; Prabhu, D.; Kumar, S.R. Biosimilars-New horizon of the generic drugs. Int. J. Pharm. Drug Anal. 2018, 6, 479–498. [Google Scholar]
- Niazi, S.K. Biosimilars and Interchangeable Biologics: Tactical Elements; CRC Press: Boca Raton, FL, USA; Taylor & Francis Group: New York, NY, USA, 2016. [Google Scholar]
- Ditani, A.S.; Mallick, P.P.; Anup, N.; Tambe, V.; Polaka, S.; Sengupta, P.; Rajpoot, K.; Tekade, R.K. Biosimilars accessible in the market for the treatment of cancer. J. Control. Release 2021, 336, 112–129. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Mody, R.; Malhotra, H. Global regulatory landscape of biosimilars: Emerging and established market perspectives. Biosimilars 2015, 2015, 19–32. [Google Scholar]
- Adegoke, B.O.; Odugbose, T.; Adeyemi, C. Data analytics for predicting disease outbreaks: A review of models and tools. Int. J. Life Sci. Res. Updates 2024, 2, 1–9. [Google Scholar] [CrossRef]
- El-Fakharany, E.M.; El-Gendi, H.; Saleh, A.K.; El-Sayed, M.H.; Alalawy, A.I.; Jame, R.; Abdelaziz, M.A.; Alshareef, S.A.; El-Maradny, Y.A. The use of proteins and peptides-based therapy in managing and preventing pathogenic viruses. Int. J. Biol. Macromol. 2024, 270, 132254. [Google Scholar] [CrossRef]
- Mancuso, G.; Midiri, A.; Gerace, E.; Biondo, C. Bacterial antibiotic resistance: The most critical pathogens. Pathogens 2021, 10, 1310. [Google Scholar] [CrossRef]
- Blackman, L.D.; Sutherland, T.D.; De Barro, P.J.; Thissen, H.; Locock, K.E. Addressing a future pandemic: How can non-biological complex drugs prepare us for antimicrobial resistance threats? Mater. Horiz. 2022, 9, 2076–2096. [Google Scholar] [CrossRef]
- Jarab, A.S.; Abu Heshmeh, S.R.; Al Meslamani, A.Z. Biosimilars as antivirals: Opportunities and challenges. Expert Rev. Anti-Infect. Ther. 2024, 22, 273–275. [Google Scholar] [CrossRef]
- Salam, M.A.; Al-Amin, M.Y.; Salam, M.T.; Pawar, J.S.; Akhter, N.; Rabaan, A.A.; Alqumber, M.A. Antimicrobial resistance: A growing serious threat for global public health. Healthcare 2023, 11, 1946. [Google Scholar] [CrossRef]
- Pelfrene, E.; Mura, M.; Sanches, A.C.; Cavaleri, M. Monoclonal antibodies as anti-infective products: A promising future? Clin. Microbiol. Infect. 2019, 25, 60–64. [Google Scholar] [CrossRef]
- Vachher, M.; Sen, A.; Kapila, R.; Nigam, A. Microbial therapeutic enzymes: A promising area of biopharmaceuticals. Curr. Res. Biotechnol. 2021, 3, 195–208. [Google Scholar] [CrossRef]
- Sumrall, E.T.; Hofstee, M.I.; Arens, D.; Röhrig, C.; Baertl, S.; Gehweiler, D.; Schmelcher, M.; Loessner, M.J.; Zeiter, S.; Richards, R.G.; et al. An enzybiotic regimen for the treatment of methicillin-resistant Staphylococcus aureus orthopaedic device-related infection. Antibiotics 2021, 10, 1186. [Google Scholar] [CrossRef] [PubMed]
- Manoharadas, S.; Witte, A.; Bläsi, U. Antimicrobial activity of a chimeric enzybiotic towards Staphylococcus aureus. J. Biotechnol. 2009, 139, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, V.; Mishra, S.; Chaudhuri, T.K. Enhanced production of recombinant serratiopeptidase in Escherichia coli and its characterization as a potential biosimilar to native biotherapeutic counterpart. Microb. Cell Fact. 2019, 18, 215. [Google Scholar] [CrossRef]
- Kabir, E.R.; Moreino, S.S.; Sharif Siam, M.K. The breakthrough of biosimilars: A twist in the narrative of biological therapy. Biomolecules 2019, 9, 410. [Google Scholar] [CrossRef]
- Tinsley, S.M.; Grande, C.; Olson, K.; Plato, L.; Jacobs, I. Potential of biosimilars to increase access to biologics: Considerations for advanced practice providers in oncology. J. Adv. Pract. Oncol. 2018, 9, 699–716. [Google Scholar]
- Jarab, A.S.; Abu Heshmeh, S.R.; Al Meslamani, A.Z. Bridging the gap: The future of biosimilars regulations. Hum. Vaccines Immunother. 2024, 20, 2362450. [Google Scholar] [CrossRef] [PubMed]
- Pelegrin, M.; Naranjo-Gomez, M.; Piechaczyk, M. Antiviral monoclonal antibodies: Can they be more than simple neutralizing agents? Trends Microbiol. 2015, 23, 653–665. [Google Scholar] [CrossRef]
- Pattnaik, G.P.; Chakraborty, H. Entry inhibitors: Efficient means to block viral infection. J. Membr. Biol. 2020, 253, 425–444. [Google Scholar] [CrossRef]
- He, Y.; Shen, M.; Wang, X.; Yin, A.; Liu, B.; Zhu, J.; Zhang, Z. Suppression of interferon response and antiviral strategies of Bunyaviruses. Trop. Med. Infect. Dis. 2024, 9, 205. [Google Scholar] [CrossRef]
- Caserta, M.T.; O’Leary, S.T.; Munoz, F.M.; Ralston, S.L. Palivizumab prophylaxis in infants and young children at increased risk of hospitalization for respiratory syncytial virus infection. Pediatrics 2023, 152, e2023061803. [Google Scholar] [CrossRef] [PubMed]
- Portal-Celhay, C.; Forleo-Neto, E.; Eagan, W.; Musser, B.J.; Davis, J.D.; Turner, K.C.; Norton, T.; Hooper, A.T.; Hamilton, J.D.; Pan, C.; et al. V Virologic efficacy of casirivimab and imdevimab COVID-19 antibody combination in outpatients with SARS-CoV-2 infection: A phase 2 dose-ranging randomized clinical trial. JAMA Netw. Open 2022, 5, e2225411. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.P.; Forleo-Neto, E.; Sarkar, N.; Isa, F.; Hou, P.; Chan, K.-C.; Musser, B.J.; Bar, K.J.; Barnabas, R.V.; Barouch, D.H. Effect of subcutaneous casirivimab and imdevimab antibody combination vs placebo on development of symptomatic COVID-19 in early asymptomatic SARS-CoV-2 infection: A randomized clinical trial. JAMA Netw. Open 2022, 327, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Teran, C.; Tiruthani, K.; McSweeney, M.; Ma, A.; Pickles, R.; Lai, S.K. Challenges and opportunities for antiviral monoclonal antibodies as COVID-19 therapy. Adv. Drug Del. Rev. 2021, 169, 100–117. [Google Scholar] [CrossRef]
- Carabetta, V.J. Available online: https://BioRender.com/5vu0bux (accessed on 20 March 2025).
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Nallar, S.C.; Kalvakolanu, D.V. Interferons, signal transduction pathways, and the central nervous system. J. Interferon Cytokine Res. 2014, 34, 559–576. [Google Scholar] [CrossRef]
- Wittling, M.C.; Cahalan, S.R.; Levenson, E.A.; Rabin, R.L. Shared and unique features of human interferon-beta and interferon-alpha subtypes. Front. Immunol. 2021, 11, 605673. [Google Scholar] [CrossRef]
- Gibbert, K.; Schlaak, J.F.; Yang, D.; Dittmer, U. IFN-α subtypes: Distinct biological activities in anti-viral therapy. Br. J. Pharmacol. 2013, 168, 1048–1058. [Google Scholar] [CrossRef]
- Rong, L.; Perelson, A.S. Treatment of hepatitis C virus infection with interferon and small molecule direct antivirals: Viral kinetics and modeling. Crit. Rev. Immunol. 2010, 30, 131–148. [Google Scholar] [CrossRef]
- Lai, J.Y.; Ho, J.X.; Kow, A.S.F.; Liang, G.; Tham, C.L.; Ho, Y.C.; Lee, M.T. Interferon therapy and its association with depressive disorders—A review. Front. Immunol. 2023, 14, 1048592. [Google Scholar] [CrossRef]
- Rhein, B.A.; Powers, L.S.; Rogers, K.; Anantpadma, M.; Singh, B.K.; Sakurai, Y.; Bair, T.; Miller-Hunt, C.; Sinn, P.; Davey, R.A.; et al. Interferon-γ inhibits Ebola virus infection. PLoS Pathog. 2015, 11, e1005263. [Google Scholar] [CrossRef] [PubMed]
- Delsing, C.E.; Gresnigt, M.S.; Leentjens, J.; Preijers, F.; Frager, F.A.; Kox, M.; Monneret, G.; Venet, F.; Bleeker-Rovers, C.P.; van de Veerdonk, F.L.; et al. Interferon-gamma as adjunctive immunotherapy for invasive fungal infections: A case series. BMC Infect. Dis. 2014, 14, 166. [Google Scholar] [CrossRef]
- Segal, B.H.; Walsh, T.J. Current approaches to diagnosis and treatment of invasive aspergillosis. Am. J. Respir. Crit. Care Med. 2006, 173, 707–717. [Google Scholar] [CrossRef]
- Kak, G.; Raza, M.; Tiwari, B.K. Interferon-gamma (IFN-γ): Exploring its implications in infectious diseases. Biomol. Concepts 2018, 9, 64–79. [Google Scholar] [CrossRef]
- Carabetta, V.J. Available online: https://BioRender.com/h40s893 (accessed on 20 March 2025).
- Louis D., S.; Cervia, J.S.; Smith, M.A. Enfuvirtide (T-20): A novel human immunodeficiency virus type 1 fusion inhibitor. Clin. Infect. Dis. 2003, 37, 1102–1106. [Google Scholar] [CrossRef] [PubMed]
- Lederman, M.M.; Penn-Nicholson, A.; Cho, M.; Mosier, D. Biology of CCR5 and its role in HIV infection and treatment. JAMA Netw. Open 2006, 296, 815–826. [Google Scholar] [CrossRef]
- Dawre, S.; Maru, S. Human Respiratory Viral Infections: Current Status and Future Prospects of Nanotechnology-Based Approaches for Prophylaxis and Treatment. Life Sci. 2021, 278, 119561. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.P.; Doms, R.W. The entry of entry inhibitors: A fusion of science and medicine. Proc. Natl. Acad. Sci. USA 2003, 100, 10598–10602. [Google Scholar] [CrossRef]
- Hoffmann, A.R.; Guha, S.; Wu, E.; Ghimire, J.; Wang, Y.; He, J.; Garry, R.F.; Wimley, W.C. Broad-spectrum antiviral entry inhibition by interfacially active peptides. J. Virol. 2020, 94, 10–1128. [Google Scholar] [CrossRef]
- Carabetta, V.J. Available online: https://BioRender.com/urvqk2k (accessed on 20 March 2025).
- Halawa, M.; Akantibila, M.; Reid, B.E.; Carabetta, V.J. Therapeutic proteins have the potential to become new weapons in the fight against antibiotic resistance. Front. Bacteriol. 2023, 2, 1304444. [Google Scholar] [CrossRef]
- Singh, R.; Chandley, P.; Rohatgi, S. Recent advances in the development of monoclonal antibodies and next-generation antibodies. Immunol. Horiz. 2023, 7, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Lavergne, V.; Skinner, A.M.; Gonzales-Luna, A.J.; Garey, K.W.; Kelly, C.P.; Wilcox, M.H. Clinical practice guideline by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA): 2021 focused update guidelines on management of Clostridioides difficile infection in adults. Clin. Infect. Dis. 2021, 73, e1029–e1044. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, M.H.; Gerding, D.N.; Poxton, I.R.; Kelly, C.; Nathan, R.; Birch, T.; Cornely, O.A.; Rahav, G.; Bouza, E.; Lee, C.; et al. Bezlotoxumab for prevention of recurrent Clostridium difficile infection. N. Engl. J. Med. 2017, 376, 305–317. [Google Scholar] [CrossRef]
- Speziale, P.; Pietrocola, G. Monoclonal antibodies targeting surface-exposed and secreted proteins from staphylococci. Vaccines 2021, 9, 459. [Google Scholar] [CrossRef]
- Hall, A.E.; Domanski, P.J.; Patel, P.R.; Vernachio, J.H.; Syribeys, P.J.; Gorovits, E.L.; Johnson, M.A.; Ross, J.M.; Hutchins, J.T.; Patti, J.M. Characterization of a protective monoclonal antibody recognizing Staphylococcus aureus MSCRAMM protein clumping factor A. Infect. Immun. 2003, 71, 6864–6870. [Google Scholar] [CrossRef]
- Patti, J.M. A humanized monoclonal antibody targeting Staphylococcus aureus. Vaccine 2004, 22 (Suppl. S1), S39–S43. [Google Scholar] [CrossRef]
- Weems, J.J., Jr.; Steinberg, J.P.; Filler, S.; Baddley, J.W.; Corey, G.R.; Sampathkumar, P.; Winston, L.; John, J.F.; Kubin, C.J.; Talwani, R.; et al. Phase II, randomized, double-blind, multicenter study comparing the safety and pharmacokinetics of tefibazumab to placebo for treatment of Staphylococcus aureus bacteremia. Antimicrob. Agents Chemother. 2006, 50, 2751–2755. [Google Scholar] [CrossRef]
- Entenza, J.M.; Loeffler, J.M.; Grandgirard, D.; Fischetti, V.A.; Moreillon, P. Therapeutic effects of bacteriophage Cpl-1 lysin against Streptococcus pneumoniae endocarditis in rats. Antimicrob. Agents Chemother. 2005, 49, 4789–4792. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, R.; García, E.; García, P. Phage lysins for fighting bacterial respiratory infections: A new generation of antimicrobials. Front. Immunol. 2018, 9, 2252. [Google Scholar] [CrossRef]
- Totté, J.E.E.; van Doorn, M.B.; Pasmans, S. Successful treatment of chronic Staphylococcus aureus-related dermatoses with the topical endolysin Staphefekt SA.100: A report of 3 cases. Case Rep. Dermatol. 2017, 9, 19–25. [Google Scholar] [CrossRef]
- Li, C.; Cai, Y.; Luo, L.; Tian, G.; Wang, X.; Yan, A.; Wang, L.; Wu, S.; Wu, Z.; Zhang, T.; et al. TC-14, a cathelicidin-derived antimicrobial peptide with broad-spectrum antibacterial activity and high safety profile. iScience 2024, 27, 110404. [Google Scholar] [CrossRef]
- Sathoff, A.E.; Lewenza, S.; Samac, D.A. Plant defensin antibacterial mode of action against Pseudomonas species. BMC Microbiol. 2020, 20, 173. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.C.; Rigg, G.; Hodgetts, S.; Carter, T.; Chapman, C.; Gregory, C.; Illidge, C.; Burnie, J. Preclinical assessment of the efficacy of mycograb, a human recombinant antibody against fungal HSP90. Antimicrob. Agents Chemother. 2003, 47, 2208–2216. [Google Scholar] [CrossRef]
- Zolin, G.V.S.; da Fonseca, F.H.; Zambom, C.R.; Garrido, S.S. Histatin 5 metallopeptides and their potential against Candida albicans pathogenicity and drug resistance. Biomolecules 2021, 11, 1209. [Google Scholar] [CrossRef] [PubMed]
- Kisalu, N.K.; Idris, A.H.; Weidle, C.; Flores-Garcia, Y.; Flynn, B.J.; Sack, B.K.; Murphy, S.; Schön, A.; Freire, E.; Francica, J.R.; et al. A human monoclonal antibody prevents malaria infection by targeting a new site of vulnerability on the parasite. Nat. Med. 2018, 24, 408–416. [Google Scholar] [CrossRef]
- Han, J.-H.; Cheng, Y.; Muh, F.; Ahmed, M.A.; Cho, J.-S.; Nyunt, M.H.; Jeon, H.-Y.; Ha, K.-S.; Na, S.; Park, W.S.; et al. Inhibition of parasite invasion by monoclonal antibody against epidermal growth factor-like domain of Plasmodium vivax merozoite surface protein 1 paralog. Sci. Rep. 2019, 9, 3906. [Google Scholar] [CrossRef] [PubMed]
- Martins, V.T.; Lage, D.P.; Duarte, M.C.; Carvalho, A.M.R.S.; Costa, L.E.; Mendes, T.A.O.; Vale, D.L.; Menezes-Souza, D.; Roatt, B.M.; Tavares, C.A.P.; et al. A recombinant fusion protein displaying murine and human MHC class I- and II-specific epitopes protects against Leishmania amazonensis infection. Cell Immunol. 2017, 313, 32–42. [Google Scholar] [CrossRef]
- Laurens, M.B. RTS, S/AS01 vaccine (Mosquirix™): An overview. Hum. Vaccines Immunother. 2020, 16, 480–489. [Google Scholar] [CrossRef]
- Fruitwala, S.; El-Naccache, D.W.; Chang, T.L. Multifaceted immune functions of human defensins and underlying mechanisms. Semin. Cell Dev. Biol. 2019, 88, 163–172. [Google Scholar] [CrossRef]
- Van Harten, R.M.; Van Woudenbergh, E.; Van Dijk, A.; Haagsman, H.P. Cathelicidins: Immunomodulatory antimicrobials. Vaccines 2018, 6, 63. [Google Scholar] [CrossRef]
- Wei, L.; Yang, J.; He, X.; Mo, G.; Hong, J.; Yan, X.; Lin, D.; Lai, R. Structure and function of a potent lipopolysaccharide-binding antimicrobial and anti-inflammatory peptide. J. Med. Chem. 2013, 56, 3546–3556. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yan, Z.; Meng, Y.; Hong, X.; Shao, G.; Ma, J.; Cheng, R.; Liu, J.; Kang, J.; Fu, C. Antimicrobial peptides: Mechanism of action, activity and clinical potential. Mil. Med. Res. 2021, 8, 48. [Google Scholar] [CrossRef]
- Ma, X.; Wang, Q.; Ren, K.; Xu, T.; Zhang, Z.; Xu, M.; Rao, Z.; Zhang, X. A review of antimicrobial peptides: Structure, mechanism of action, and molecular optimization strategies. Fermentation 2024, 10, 540. [Google Scholar] [CrossRef]
- Seyfi, R.; Kahaki, F.A.; Ebrahimi, T.; Montazersaheb, S.; Eyvazi, S.; Babaeipour, V.; Tarhriz, V. Antimicrobial peptides (AMPs): Roles, functions and mechanism of action. Int. J. Pept. Res. Ther. 2020, 26, 1451–1463. [Google Scholar] [CrossRef]
- Dijksteel, G.S.; Ulrich, M.M.W.; Middelkoop, E.; Boekema, B.K.H.L. Review: Lessons learned from clinical trials using antimicrobial peptides (AMPs). Front. Microbiol. 2021, 12, 616979. [Google Scholar] [CrossRef]
- Cresti, L.; Cappello, G.; Pini, A. Antimicrobial peptides towards clinical application-A long history to be concluded. Int. J. Mol. Sci. 2024, 25, 4870. [Google Scholar] [CrossRef] [PubMed]
- Carabetta, V.J. Available online: https://BioRender.com/u06x820 (accessed on 20 March 2025).
- São-José, C. Engineering of phage-derived lytic enzymes: Improving their potential as antimicrobials. Antibiotics 2018, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wang, Q.; Jiang, J.; Gao, L. Nanozybiotics: Nanozyme-based antibacterials against bacterial resistance. Antibiotics 2022, 11, 390. [Google Scholar] [CrossRef]
- Elfadadny, A.; Ragab, R.F.; AlHarbi, M.; Badshah, F.; Ibáñez-Arancibia, E.; Farag, A.; Hendawy, A.O.; De Ríos-Escalante, P.R.; Aboubakr, M.; Zakai, S.A. Antimicrobial resistance of Pseudomonas aeruginosa: Navigating clinical impacts, current resistance trends, and innovations in breaking therapies. Front. Microbiol. 2024, 15, 1374466. [Google Scholar] [CrossRef]
- Dams, D.; Briers, Y. Enzybiotics: Enzyme-based antibacterials as therapeutics. In Therapeutic Enzymes: Function and Clinical Implications; Labrou, N., Ed.; Springer: Singapore, 2019; pp. 233–253. [Google Scholar]
- Moretta, A.; Scieuzo, C.; Petrone, A.M.; Salvia, R.; Manniello, M.D.; Franco, A.; Lucchetti, D.; Vassallo, A.; Vogel, H.; Sgambato, A. Antimicrobial peptides: A new hope in biomedical and pharmaceutical fields. Front. Cell. Infect. Microbiol. 2021, 11, 668632. [Google Scholar] [CrossRef]
- Fernández de Ullivarri, M.; Arbulu, S.; Garcia-Gutierrez, E.; Cotter, P.D. Antifungal peptides as therapeutic agents. Front. Cell. Infect. MicroBiol. 2020, 10, 105. [Google Scholar] [CrossRef] [PubMed]
- Boniche, C.; Rossi, S.A.; Kischkel, B.; Vieira Barbalho, F.; Nogueira D’Aurea Moura, Á.; Nosanchuk, J.D.; Travassos, L.R.; Pelleschi Taborda, C. Immunotherapy against systemic fungal infections based on monoclonal antibodies. J. Fungi 2020, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Prusty, J.S.; Kumar, A.; Kumar, A. Anti-Fungal Peptides: An Emerging Category with Enthralling Therapeutic Prospects in the Treatment of Candidiasis. Crit. Rev. Microbiol. 2024, 1–37. [Google Scholar] [CrossRef]
- Rabaan, A.A.; Alfaraj, A.H.; Alshengeti, A.; Alawfi, A.; Alwarthan, S.; Alhajri, M.; Al-Najjar, A.H.; Al Fares, M.A.; Najim, M.A.; Almuthree, S.A. Antibodies to combat fungal infections: Development strategies and progress. Microorganisms 2023, 11, 671. [Google Scholar] [CrossRef]
- Matthews, R.C.; Burnie, J.P. Recombinant antibodies: A natural partner in combinatorial antifungal therapy. Vaccine 2004, 22, 865–871. [Google Scholar] [CrossRef]
- Feng, Z.; Lu, H.; Jiang, Y. Promising immunotherapeutic targets for treating candidiasis. Front. Cell. Infect. Microbiol. 2024, 14, 1339501. [Google Scholar] [CrossRef]
- Pachl, J.; Svoboda, P.; Jacobs, F.; Vandewoude, K.; van der Hoven, B.; Spronk, P.; Masterson, G.; Malbrain, M.; Aoun, M.; Garbino, J.; et al. A randomized, blinded, multicenter trial of lipid-associated amphotericin B alone versus in combination with an antibody-based inhibitor of heat shock protein 90 in patients with invasive candidiasis. Clin. Infect. Dis. 2006, 42, 1404–1413. [Google Scholar] [CrossRef]
- Louie, A.; Stein, D.S.; Zack, J.Z.; Liu, W.; Conde, H.; Fregeau, C.; Vanscoy, B.D.; Drusano, G.L. Dose range evaluation of Mycograb C28Y variant, a human recombinant antibody fragment to heat shock protein 90, in combination with amphotericin B-desoxycholate for treatment of murine systemic candidiasis. Antimicrob. Agents Chemother. 2011, 55, 3295–3304. [Google Scholar] [CrossRef]
- Konakbayeva, D.; Karlsson, A.J. Strategies and opportunities for engineering antifungal peptides for therapeutic applications. Curr. Opin. Biotechnol. 2023, 81, 102926. [Google Scholar] [CrossRef]
- Gu, J.; Isozumi, N.; Yuan, S.; Jin, L.; Gao, B.; Ohki, S.; Zhu, S. Evolution-based protein engineering for antifungal peptide improvement. Mol. Biol. Evol. 2021, 38, 5175–5189. [Google Scholar] [CrossRef]
- Van der Weerden, N.L.; Bleackley, M.R.; Anderson, M.A. Properties and mechanisms of action of naturally occurring antifungal peptides. Cell Molecular. Life Sci. 2013, 70, 3545–3570. [Google Scholar] [CrossRef] [PubMed]
- Struyfs, C.; Cammue, B.P.A.; Thevissen, K. Membrane-interacting antifungal peptides. Front. Cell. Dev. Biol. 2021, 9, 649875. [Google Scholar] [CrossRef]
- Tong, S.; Li, M.; Keyhani, N.O.; Liu, Y.; Yuan, M.; Lin, D.; Jin, D.; Li, X.; Pei, Y.; Fan, Y. Characterization of a fungal competition factor: Production of a conidial cell-wall associated antifungal peptide. PLoS Pathog. 2020, 16, e1008518. [Google Scholar] [CrossRef] [PubMed]
- Kadri, S.S. Key Takeaways From the U.S. CDC’s 2019 Antibiotic Resistance Threats Report for Frontline Providers. Crit. Care Med. 2020, 48, 939–945. [Google Scholar] [CrossRef]
- Li, T.; Li, L.; Du, F.; Sun, L.; Shi, J.; Long, M.; Chen, Z. Activity and mechanism of action of antifungal peptides from microorganisms: A review. Molecules 2021, 26, 3438. [Google Scholar] [CrossRef]
- Longoni, S.S.; Tiberti, N.; Bisoffi, Z.; Piubelli, C. Monoclonal antibodies for protozoan infections: A future reality or a utopic idea? Front. Med. 2021, 8, 745665. [Google Scholar] [CrossRef]
- Stutzer, C.; Richards, S.A.; Ferreira, M.; Baron, S.; Maritz-Olivier, C. Metazoan parasite vaccines: Present status and future prospects. Front. Cell. Infect. Microbiol. 2018, 8, 67. [Google Scholar] [CrossRef]
- WHO. WHO Recommends Groundbreaking Malaria Vaccine for Children at Risk. Available online: https://www.who.int/news/item/06-10-2021-who-recommends-groundbreaking-malaria-vaccine-for-children-at-risk (accessed on 20 March 2025).
- Niazi, S.K. Biosimilars: A futuristic fast-to-market advice to developers. Expert. Opin. Biolog. Ther. 2022, 22, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.J.; Choudhary, M.C.; Deo, R.; Yousuf, F.; Gomez, A.N.; Edelstein, G.E.; Boucau, J.; Glover, O.T.; Barry, M.; Gilbert, R.F. Emerging SARS-CoV-2 resistance after antiviral treatment. JAMA Netw. Open 2024, 7, e2435431. [Google Scholar] [CrossRef]
- Ferreira, G.S.; Veening-Griffioen, D.H.; Boon, W.P.; Moors, E.H.; Gispen-de Wied, C.C.; Schellekens, H.; van Meer, P.J. A standardised framework to identify optimal animal models for efficacy assessment in drug development. PLoS ONE 2019, 14, e0218014. [Google Scholar] [CrossRef]
- Singh, D.; Mathur, A.; Arora, S.; Roy, S.; Mahindroo, N. Journey of organ on a chip technology and its role in future healthcare scenario. Appl. Surf. Sci. Adv. 2022, 9, 100246. [Google Scholar] [CrossRef]
- Wang, L.; Hu, D.; Xu, J.; Hu, J.; Wang, Y. Complex in vitro model: A transformative model in drug development and precision medicine. Clin. Transl. Sci. 2023, 17, e13695. [Google Scholar] [CrossRef]
- Yan, H.; Semple, K.M.; Gonzaléz, C.M.; Howard, K.E. Bone marrow-liver-thymus (BLT) immune humanized mice as a model to predict cytokine release syndrome. Transl. Res. 2019, 210, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Kachhawaha, K.; Singh, S.K. Comprehensive approaches to preclinical evaluation of monoclonal antibodies and their next-generation derivatives. Biochem. Pharmacol. 2024, 225, 116303. [Google Scholar] [CrossRef]
- Garg, R.K.; Chhabra, D.; Yadav, D.; Adlakha, S. Quest for dexterous prospects in AI regulated arena: Opportunities and challenges in healthcare. Int. J. Healthc. Technol. Manag. 2020, 18, 22–50. [Google Scholar] [CrossRef]
- Mascarenhas-Melo, F.; Diaz, M.; Gonçalves, M.B.S.; Vieira, P.; Bell, V.; Viana, S.; Nunes, S.; Paiva-Santos, A.C.; Veiga, F. An Overview of Biosimilars-Development, Quality, Regulatory Issues, and Management in Healthcare. Pharmaceuticals 2024, 17, 235. [Google Scholar] [CrossRef]
- Markus, R.; Liu, J.; Ramchandani, M.; Landa, D.; Born, T.; Kaur, P. Developing the totality of evidence for biosimilars: Regulatory considerations and building confidence for the healthcare community. BioDrugs 2017, 31, 175–187. [Google Scholar] [CrossRef]
- Lee, S.J.; Chow, S.-C. Methodologies in Biosimilar Product Development, 1st ed.; Lee, S.J., Chow, S.-C., Eds.; Chapman and Hall/CRC: New York, NY, USA, 2021. [Google Scholar]
- Barbier, L.; Ebbers, H.C.; Declerck, P.; Simoens, S.; Vulto, A.G.; Huys, I. The efficacy, safety, and immunogenicity of switching between reference biopharmaceuticals and biosimilars: A systematic review. Clin. Pharmacol. Ther. 2020, 108, 734–755. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.; Joshi, S.; Guttman, A.; Rathore, A.S. N-Glycosylation of monoclonal antibody therapeutics: A comprehensive review on significance and characterization. Anal. Chim. Acta 2022, 1209, 339828. [Google Scholar] [CrossRef]
- Tobin, P.H.H.; Richards, D.A.; Callender, R.J.; Wilson, C. Protein engineering: A new frontier for biological therapeutics. Curr. Drug Metab. 2014, 15, 743–756. [Google Scholar] [CrossRef]
- Nosaki, S.; Hoshikawa, K.; Ezura, H.; Miura, K. Transient protein expression systems in plants and their applications. Plant Biotechnol. 2021, 38, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Garvey, M. Non-mammalian eukaryotic expression systems yeast and fungi in the production of biologics. J. Fungi 2022, 8, 1179. [Google Scholar] [CrossRef] [PubMed]
- Chong, S. Overview of cell-free protein synthesis: Historic landmarks, commercial systems, and expanding applications. Curr. Protoc. Mol. Biol. 2014, 108, 16.30.1–16.30.11. [Google Scholar] [CrossRef]
- Niazi, S.K.; Magoola, M. Advances in Escherichia coli-based therapeutic protein expression: Conversion, continuous manufacturing, and cell-free production. Biologics 2023, 3, 380–401. [Google Scholar] [CrossRef]
- Dondapati, S.K.; Stech, M.; Zemella, A.; Kubick, S. Cell-free protein synthesis: A promising option for future drug development. BioDrugs 2020, 34, 327–348. [Google Scholar] [CrossRef]
- Garenne, D.; Haines, M.C.; Romantseva, E.F.; Freemont, P.; Strychalski, E.A.; Noireaux, V. Cell-free gene expression. Nat. Rev. Methods Primers 2021, 1, 49. [Google Scholar] [CrossRef]
- Vulto, A.G.; Jaquez, O.A. The process defines the product: What really matters in biosimilar design and production? Rheumatology 2017, 56, iv14–iv29. [Google Scholar] [CrossRef]
- Shin, Y.D.; Landauer, K. Manufacturing of recombinant proteins using quality by design (QbD) methodology: Current Trend and Challenges. In Biopharmaceutical Manufacturing: Progress, Trends and Challenges; Pörtner, R., Ed.; Springer Nature: Cham, Switzerland, 2023. [Google Scholar]
- Carabetta, V.J. Available online: https://BioRender.com/dotfvw1. (accessed on 20 March 2025).
- Cordeiro, M.A.; Vitorino, C.; Sinogas, C.; Sousa, J.J. A regulatory perspective on biosimilar medicines. Pharmaceutics 2024, 16, 321. [Google Scholar] [CrossRef]
- Amaral, C.; Rodrigues, A.R.; Veiga, F.; Bell, V. Biosimilar medicines: From development process to marketing authorization by the EMA and the FDA. Appl. Sci. 2024, 14, 7529. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Wang, M.; Zhai, S.; Hou, C.; Sun, F.; Jian, L. Evaluating Biosimilars: Safety, Efficacy, and Regulatory Considerations in Clinical Studies. Int. J. Clin. Pharm. 2024, 47, 232–236. [Google Scholar] [CrossRef]
- Nupur, N.; Joshi, S.; Gulliarme, D.; Rathore, A.S. Analytical similarity assessment of biosimilars: Global regulatory landscape, recent studies and major advancements in orthogonal platforms. Front. Bioeng. Biotechnol. 2022, 10, 832059. [Google Scholar] [CrossRef] [PubMed]
- Alsenaidy, M.A.; Jain, N.K.; Kim, J.H.; Middaugh, C.R.; Volkin, D.B. Protein comparability assessments and potential applicability of high throughput biophysical methods and data visualization tools to compare physical stability profiles. Front. Pharmacol. 2014, 5, 39. [Google Scholar] [CrossRef] [PubMed]
- Blackstone, E.A.; Joseph, P.F. The economics of biosimilars. Am. Health Drug Benefits 2013, 6, 469–478. [Google Scholar]
- Jayeoba, D. The US pharmaceutical market maze: How exploitative practices and legal loopholes trap competition and delay affordable medicines. Soc. Sci. Res. Netw. 2025. [Google Scholar] [CrossRef]
- Kabir, E.; Moreino, S.; Sharif Siam, M.; Kabir, E.R.; Moreino, S.S.; Sharif Siam, M.K. An Empirical Analysis of the Perceived Challenges and Benefits of Introducing Biosimilars in Bangladesh: A Paradigm Shift. Biomolecules 2018, 8, 89. [Google Scholar] [CrossRef]
- Yu, Y.B.; Briggs, K.T.; Taraban, M.B.; Brinson, R.G.; Marino, J.P. Grand challenges in pharmaceutical research series: Ridding the cold chain forbiologics. Pharm. Res. 2021, 38, 3–7. [Google Scholar] [CrossRef]
- Ndochinwa, G.O.; Wang, Q.Y.; Okoro, N.O.; Amadi, O.C.; Nwagu, T.N.; Nnamchi, C.I.; Moneke, A.N.; Odiba, A.S. New advances in protein engineering for industrial applications: Key takeaways. Open Life Sci. 2024, 19, 20220856. [Google Scholar] [CrossRef] [PubMed]
- Butreddy, A.; Janga, K.Y.; Ajjarapu, S.; Sarabu, S.; Dudhipala, N. Instability of therapeutic proteins—An overview of stresses, stabilization mechanisms and analytical techniques involved in lyophilized proteins. Int. J. Biol. Macromol. 2021, 167, 309–325. [Google Scholar] [CrossRef]
- Kim, H.; Alten, R.; Avedano, L.; Dignass, A.; Gomollón, F.; Greveson, K.; Halfvarson, J.; Irving, P.M.; Jahnsen, J.; Lakatos, P.L.; et al. The future of biosimilars: Maximizing benefits across immune-mediated inflammatory diseases. Drugs 2020, 80, 99–113. [Google Scholar] [CrossRef]
- Evangelatos, G.; Bamias, G.; Kitas, G.D.; Kollias, G.; Sfikakis, P.P. The second decade of anti-TNF-a therapy in clinical practice: New lessons and future directions in the COVID-19 era. Rheumatol. Int. 2022, 42, 1493–1511. [Google Scholar] [CrossRef]
- Messner, K.; Eickhoff, C.; Schulz, M.; Allemann, S.S.; Arnet, I. Knowledge and attitudes of German and Swiss community pharmacists towards biologicals and biosimilars–a prospective survey before and after the COVID-19 pandemic. BMC Health Serv. Res. 2023, 23, 1432. [Google Scholar] [CrossRef]
- Wong, G.; Zhang, L.; Majeed, H.; Razvi, Y.; DeAngelis, C.; Lam, E.; McKenzie, E.; Wang, K.; Pasetka, M. A retrospective review of the real-world experience of the Pegfilgrastim biosimilar (Lapelga®) to the reference biologic (Neulasta®). J. Oncol. Pharm. Pract. 2022, 28, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, R.N.; Bhushan, B.; Singh, K.; Chopra, H.; Kumar, S.; Agrawal, M.; Pathak, D.; Chanchal, D.K.; Laxmikant. Recent advances in pharmaceutical design: Unleashing the potential of novel therapeutics. Curr. Pharm. Biotechnol. 2024, 25, 2060–2077. [Google Scholar] [CrossRef] [PubMed]
- Yaroson, E.V.; Breen, L.; Hou, J.; Sowter, J. Examining the impact of resilience strategies in mitigating medicine shortages in the United Kingdom’s (UK) pharmaceutical supply chain (PSC). Benchmarking Int. J. 2024, 31, 683–706. [Google Scholar] [CrossRef]
- Canter, B.; Sussman, S.; Colvill, S.; Arad, N.; Staton, E.; Rai, A. Introducing biosimilar competition for cell and gene therapy products. J. Law Biosci. 2024, 11, lsae015. [Google Scholar] [CrossRef]
- Godman, B.; Bishop, I.; Finlayson, A.E.; Campbell, S.; Kwon, H.Y.; Bennie, M. Reforms and initiatives in Scotland in recent years to encourage the prescribing of generic drugs, their influence and implications for other countries. Expert Rev. Pharmacoecon. Outcomes Res. 2013, 13, 469–482. [Google Scholar] [CrossRef]
- Oskouei, S.T.; Kusmierczyk, A.R. Biosimilar uptake: The importance of healthcare provider education. Pharm. Med. 2021, 35, 215–224. [Google Scholar] [CrossRef]
- Rathore, A.S.; Thakur, G.; Kateja, N. Continuous integrated manufacturing for biopharmaceuticals: A new paradigm or an empty promise? Biotechnol. Bioeng. 2023, 120, 333–351. [Google Scholar] [CrossRef]
- Monga, A.; Gagan, P.; Jamwal, P.; Sharma, S.; Kaur, A. Biosimilars: A critical review of development, regulatory landscape, and clinical implications. AAPS PharmSciTech 2025, 26, 46. [Google Scholar] [CrossRef]
- Moreno, S.G.; Epstein, D. The price of innovation—The role of drug pricing in financing pharmaceutical innovation. A conceptual framework. J. Mark. Access Health Policy 2019, 7, 1583536. [Google Scholar] [CrossRef]
- Feagan, B.G.; Marabani, M.; Wu, J.J.; Faccin, F.; Spronk, C.; Castañeda-Hernández, G. The challenges of switching therapies in an evolving multiple biosimilars landscape: A narrative review of current evidence. Adv. Ther. 2020, 37, 4491–4518. [Google Scholar] [CrossRef] [PubMed]
- Kandari, D.; Bhatnagar, R. Antibody engineering and its therapeutic applications. Int. Rev. Immunol. 2023, 42, 156–183. [Google Scholar] [CrossRef] [PubMed]
- Chiba, C.H.; Knirsch, M.C.; Azzoni, A.R.; Moreira, A.R.; Stephano, M.A. Cell-free protein synthesis: Advances on production process for biopharmaceuticals and immunobiological products. BioTechniques 2021, 70, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Stamatis, C.; Farid, S.S. Process economics evaluation of cell-free synthesis for the commercial manufacture of antibody drug conjugates. Biotechnol. J. 2021, 16, e2000238. [Google Scholar] [CrossRef]

| Conventional Therapies | Therapeutic Proteins/Biosimilars | |
|---|---|---|
| MOA | Direct pathogen inhibition (e.g., cell wall disruption, essential enzyme inhibition, etc.) | Targeted immune modulation, neutralization of toxins/virulence factors, direct pathogen inhibition |
| Chemistry | Chemical (natural or synthetic) | Proteins (mAbs, peptides, enzybiotics) |
| Safety | Toxicity concerns (e.g., nephrotoxicity, hepatotoxicity, GI issues) | Risk of immunogenicity and hypersensitivity |
| Spectrum | Broad spectrum, harm to native microbiota | Narrow spectrum, highly specific, often spares native microbiota |
| Resistance | Rapid resistance development | Reduced resistance development |
| Cost | Low for generics, high for new drugs | Higher initial cost, but long-term savings |
| Challenge | Description | Solutions |
|---|---|---|
| Difficulties in designing appropriate in vitro and in vivo models | The development of models that accurately simulate human–pathogen interactions presents considerable complexity. In vitro systems often fail to completely replicate in vivo environments, whereas animal models may not consistently reflect human immune responses. | Development and improvement of humanized animal models and organ-on-a-chip models, optimization of cell culture conditions, and standardization of models. |
| Choice of appropriate and optimal expression systems | The selection of an appropriate host system, e.g., bacterial, yeast, or mammalian, is paramount to ensuring adequate protein folding, glycosylation, and biological activity, all of which significantly influence both efficacy and safety. | Optimization of production conditions, creating host cells for enhanced PTM addition, and the use of CFPS. Standardization would be beneficial for reproducibility. |
| Scaling up production and regulatory guidelines | The process of large-scale production necessitates the maintenance of consistency, stability, and potency. Regulatory agencies enforce stringent guidelines for the approval of biosimilars, necessitating comprehensive comparability studies. | Advancement of bioreactor technologies, and implementation of rigorous quality control. Prompt regulatory participation is required for faster approval. |
| Structural and functional complexity of biosimilars | Unlike small-molecule pharmaceuticals, biosimilars consist of large, intricate molecules that demand meticulous replication of their structural and functional attributes, a task that poses significant challenges during the manufacturing process. | Application of modern biophysical and biochemical techniques. Multidisciplinary approach requiring scientific collaboration. |
| Immunogenicity and safety concerns | Biosimilars have the potential to elicit immune responses that could diminish efficacy or result in adverse effects. The prediction and mitigation of immunogenicity continue to represent a substantial obstacle in the realm of clinical development. | Creation of risk assessment methods, possibly using AI methodologies. Performance of rigorous immunogenicity testing, and clinical monitoring for side effects. |
| Cost and time of development | The process of obtaining biosimilar approval is protracted and financially burdensome, largely due to the necessity for extensive characterization, clinical trials, and regulatory endorsements, which frequently restrict competition and accessibility. | Optimized, adaptive clinical trial formats that make use of scientific data in real time. Government support by providing pricing incentives, tax advantages, or reimbursement frameworks. |
| Stability and storage requirements | Biologics, including biosimilars, exhibit heightened sensitivity to temperature fluctuations and storage conditions, necessitating specialized handling protocols to preserve stability and avert degradation. | Development of modern packaging technologies, enhanced formulation methods, and cold chain systems for management. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halawa, M.; ElSayed, R.M.R.; Aderibigbe, T.; Newman, P.M.; Reid, B.E.; Carabetta, V.J. Biosimilars Targeting Pathogens: A Comprehensive Review of Their Role in Bacterial, Fungal, Parasitic, and Viral Infections. Pharmaceutics 2025, 17, 581. https://doi.org/10.3390/pharmaceutics17050581
Halawa M, ElSayed RMR, Aderibigbe T, Newman PM, Reid BE, Carabetta VJ. Biosimilars Targeting Pathogens: A Comprehensive Review of Their Role in Bacterial, Fungal, Parasitic, and Viral Infections. Pharmaceutics. 2025; 17(5):581. https://doi.org/10.3390/pharmaceutics17050581
Chicago/Turabian StyleHalawa, Mohamed, Ramez M. Rashad ElSayed, Tope Aderibigbe, Precious M. Newman, Briana E. Reid, and Valerie J. Carabetta. 2025. "Biosimilars Targeting Pathogens: A Comprehensive Review of Their Role in Bacterial, Fungal, Parasitic, and Viral Infections" Pharmaceutics 17, no. 5: 581. https://doi.org/10.3390/pharmaceutics17050581
APA StyleHalawa, M., ElSayed, R. M. R., Aderibigbe, T., Newman, P. M., Reid, B. E., & Carabetta, V. J. (2025). Biosimilars Targeting Pathogens: A Comprehensive Review of Their Role in Bacterial, Fungal, Parasitic, and Viral Infections. Pharmaceutics, 17(5), 581. https://doi.org/10.3390/pharmaceutics17050581