
Matrix Approach Assessment of Cabotegravir Drug–Drug Interactions with OAT1/OAT3 Substrates and UGT1A1/UGT1A9 Inhibitors Using Physiologically-Based Pharmacokinetic Modeling

 ,
,
Abstract
1. Introduction
2. Materials and Methods
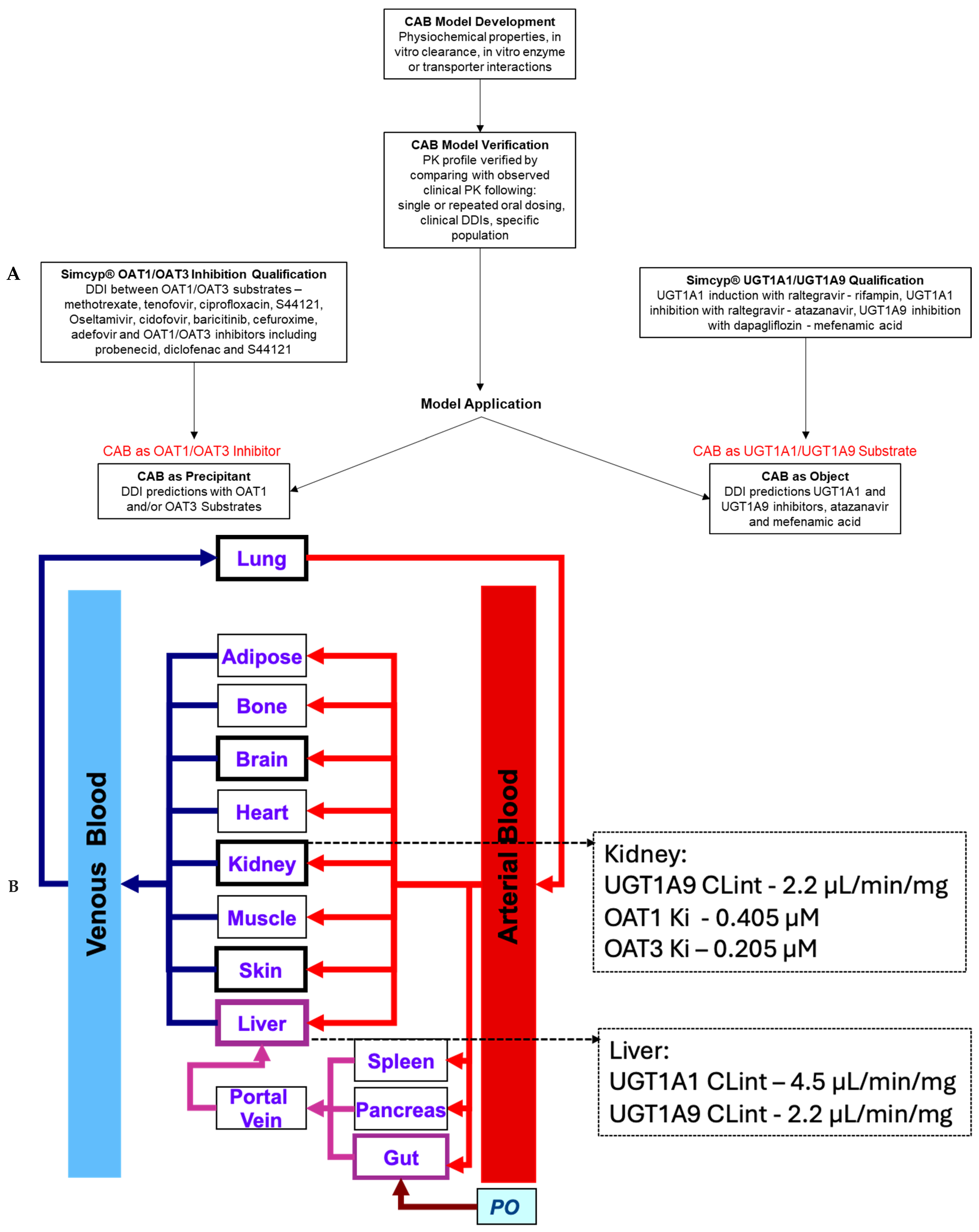
2.1. PBPK Modeling Strategy
2.2. Simcyp® v17.1 Software Qualification
2.2.1. Qualification for Prediction of OAT1 and/or OAT3 DDIs
2.2.2. Qualification for the Prediction of UGT1A1 and/or UGT1A9 DDIs
2.3. Cabotegravir Model Development
2.4. Cabotegravir Model Verification
2.5. CAB DDI Simulation Trial Design
2.6. Model Applications
2.6.1. OAT1 and OAT3 Inhibition
2.6.2. Sensitivity Analysis
2.6.3. UGT1A1 and UGT1A9 Object Interaction
2.7. Data Analysis and Statistics
3. Results
3.1. Cabotegravir Model Development
3.2. Qualification for Prediction of OAT1 and/or OAT3 DDIs
3.3. Qualification for Prediction of UGT1A1 and/or UGT1A9 DDIs
3.4. Cabotegravir Model Verification
3.5. Model Applications
3.5.1. OAT1 and OAT3 Inhibition
3.5.2. Sensitivity Analysis
3.5.3. UGT1A1 and UGT1A9 Object Interaction
4. Discussion
5. Conclusions
6. Study Highlights
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bowers, G.D.; Culp, A.; Reese, M.J.; Tabolt, G.; Moss, L.; Piscitelli, S.; Huynh, P.; Wagner, D.; Ford, S.L.; Gould, E.P.; et al. Drug interaction profile of the HIV integrase inhibitor cabotegravir: Assessment from in vitro studies and a clinical investigation with midazolam. Xenobiotica 2016, 46, 445–456. [Google Scholar] [CrossRef]
- Bowers, G.D.; Culp, A.; Reese, M.J.; Tabolt, G.; Moss, L.; Piscitelli, S.; Huynh, P.; Wagner, D.; Ford, S.L.; Gould, E.P.; et al. Disposition and metabolism of cabotegravir: A comparison of biotransformation and excretion between different species and routes of administration in humans. Xenobiotica 2016, 46, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention Diagnoses of HIV Infection in the United States and dependent areas, 2022. In HIV Surveillance Report, 2022; National Center for HIV, Viral Hepatitis, STD, and Tuberculosis Prevention: Atlanta, GA, USA, 2024; Volume 35.
- Deeks, S.G.; Phillips, A.N. HIV infection, antiretroviral treatment, ageing, and non-AIDS related morbidity. BMJ 2009, 338, a3172. [Google Scholar] [CrossRef]
- Guaraldi, G.; Orlando, G.; Zona, S.; Menozzi, M.; Carli, F.; Garlassi, E.; Berti, A.; Rossi, E.; Roverato, A.; Palella, F. Premature Age-Related Comorbidities Among HIV-Infected Persons Compared with the General Population. Clin. Infect. Dis. 2011, 53, 1120–1126. [Google Scholar] [CrossRef]
- Nachega, J.B.; Hsu, A.J.; Uthman, O.A.; Spinewine, A.; Pham, P.A. Antiretroviral therapy adherence and drug–drug interactions in the aging HIV population. AIDS 2012, 26, S39–S53. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.D.M.; Mateo, M.G.; Corbacho, N.; Vidal, F.; Domingo, P. Drug-drug interactions when treating HIV-related metabolic disorders. Expert Opin. Drug Metab. Toxicol. 2019, 15, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Ambrosioni, J.; Díaz, N.A.; Marzolini, C.; Dragovic, G.; Imaz, A.; Calcagno, A.; Luque, S.; Curran, A.; Troya, J.; Tuset, M.; et al. Outcomes of Drug Interactions Between Antiretrovirals and Co-Medications, Including Over-the-Counter Drugs: A Real-World Study. Infect. Dis. Ther. 2024, 13, 609–617. [Google Scholar] [CrossRef]
- Brooks, K.M.; George, J.M.; Kumar, P. Drug interactions in HIV treatment: Complementary & alternative medicines and over-the-counter products. Expert Rev. Clin. Pharmacol. 2017, 10, 59–79. [Google Scholar] [CrossRef]
- Tetart, M.; Passecountrin, P.; Lesourd, A.; Sanderink, D.; Le Moal, G.; Surgers, L.; Beraud, G.; Katlama, C.; Robineau, O.; Parienti, J.-J.; et al. Are unknown co-medications, over-the-counter and off-label drug use still problems among people living with HIV? Results from a transversal survey in 23 centres in France. J. Antimicrob. Chemother. 2023, 78, 2731–2734. [Google Scholar] [CrossRef]
- Cihlar, T.; Lin, D.C.; Pritchard, J.B.; Fuller, M.D.; Mendel, D.B.; Sweet, D.H. The Antiviral Nucleotide Analogs Cidofovir and Adefovir Are Novel Substrates for Human and Rat Renal Organic Anion Transporter 1. Mol. Pharmacol. 1999, 56, 570–580. [Google Scholar] [CrossRef]
- Cha, S.H.; Sekine, T.; Fukushima, J.-I.; Kanai, Y.; Kobayashi, Y.; Goya, T.; Endou, H. Identification and Characterization of Human Organic Anion Transporter 3 Expressing Predominantly in the Kidney. Mol. Pharmacol. 2001, 59, 1277–1286. [Google Scholar] [CrossRef]
- Kohler, J.J.; Hosseini, S.H.; Green, E.; Abuin, A.; Ludaway, T.; Russ, R.; Santoianni, R.; Lewis, W. Tenofovir renal proximal tubular toxicity is regulated By OAT1 and MRP4 transporters. Lab. Investig. 2011, 91, 852–858. [Google Scholar] [CrossRef] [PubMed]
- VanWert, A.L.; Srimaroeng, C.; Sweet, D.H. Organic Anion Transporter 3 (Oat3/Slc22a8) Interacts with Carboxyfluoroquinolones, and Deletion Increases Systemic Exposure to Ciprofloxacin. Mol. Pharmacol. 2008, 74, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Ose, A.; Ito, M.; Kusuhara, H.; Yamatsugu, K.; Kanai, M.; Shibasaki, M.; Hosokawa, M.; Schuetz, J.D.; Sugiyama, Y. Limited Brain Distribution of [3R,4R,5S]-4-Acetamido-5-amino-3-(1-ethylpropoxy)-1-cyclohexene-1-carboxylate Phosphate (Ro 64-0802), a Pharmacologically Active Form of Oseltamivir, by Active Efflux Across the Blood-Brain Barrier Mediated by Organic Anion Transporter 3 (Oat3/Slc22a8<) and Multidrug Resistance-Associated Protein 4 (Mrp4/Abcc4). Drug Metab. Dispos. 2009, 37, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Apiwattanakul, N.; Sekine, T.; Chairoungdua, A.; Kanai, Y.; Nakajima, N.; Sophasan, S.; Endou, H. Transport Properties of Nonsteroidal Anti-Inflammatory Drugs by Organic Anion Transporter 1 Expressed in Xenopus laevis Oocytes. Mol. Pharmacol. 1999, 55, 847–854. [Google Scholar] [CrossRef]
- Food and Drug Administration. In Vitro Drug Interaction Studies–Cytochrome P450 Enzyme-and Transporter-Mediated Drug Interactions; US Department of Health and Human Services: Silver Spring, MD, USA, 2020.
- European Medicines Agency. Guideline on the Investigation of Drug Interactions; European Medicines Agency: London, UK, 2012. [Google Scholar]
- Harbourt, D.E.; Fallon, J.K.; Ito, S.; Baba, T.; Ritter, J.K.; Glish, G.L.; Smith, P.C. Quantification of Human Uridine-Diphosphate Glucuronosyl Transferase 1A Isoforms in Liver, Intestine, and Kidney Using Nanobore Liquid Chromatography–Tandem Mass Spectrometry. Anal. Chem. 2012, 84, 98–105. [Google Scholar] [CrossRef]
- Sato, Y.; Nagata, M.; Tetsuka, K.; Tamura, K.; Miyashita, A.; Kawamura, A.; Usui, T. Optimized Methods for Targeted Peptide-Based Quantification of Human Uridine 5′-Diphosphate-Glucuronosyltransferases in Biological Specimens Using Liquid Chromatography–Tandem Mass Spectrometry. Drug Metab. Dispos. 2014, 42, 885–889. [Google Scholar] [CrossRef]
- Ohno, S.; Nakajin, S. Determination of mRNA Expression of Human UDP-Glucuronosyltransferases and Application for Localization in Various Human Tissues by Real-Time Reverse Transcriptase-Polymerase Chain Reaction. Drug Metab. Dispos. 2009, 37, 32–40. [Google Scholar] [CrossRef]
- Strassburg, C.P.; Nguyen, N.; Manns, M.P.; Tukey, R.H. Polymorphic Expression of the UDP-Glucuronosyltransferase UGT1A Gene Locus in Human Gastric Epithelium. Mol. Pharmacol. 1998, 54, 647–654. [Google Scholar] [CrossRef]
- Ford, S.L.; Sutton, K.; Lou, Y.; Zhang, Z.; Tenorio, A.; Trezza, C.; Patel, P.; Spreen, W. Effect of Rifampin on the Single-Dose Pharmacokinetics of Oral Cabotegravir in Healthy Subjects. Antimicrob. Agents Chemother. 2017, 61, e00487-17. [Google Scholar] [CrossRef]
- Miners, J.O.; Polasek, T.M.; Hulin, J.-A.; Rowland, A.; Meech, R. Drug-drug interactions that alter the exposure of glucuronidated drugs: Scope, UDP-glucuronosyltransferase (UGT) enzyme selectivity, mechanisms (inhibition and induction), and clinical significance. Pharmacol. Ther. 2023, 248, 108459. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Saka, H.; Ando, M.; Sawa, T.; Muro, K.; Ueoka, H.; Yokoyama, A.; Saitoh, S.; Shimokata, K.; Hasegawa, Y. Polymorphisms of UDP-Glucuronosyltransferase Gene and Irinotecan Toxicity: A Pharmacogenetic Analysis1. Cancer Res. 2000, 60, 6921–6926. [Google Scholar] [PubMed]
- Posner, J.; Cohen, A.; Land, G.; Winton, C.; Peck, A. The pharmacokinetics of lamotrigine (BW430C) in healthy subjects with unconjugated hyperbilirubinaemia (Gilbert’s syndrome). Br. J. Clin. Pharmacol. 1989, 28, 117–120. [Google Scholar] [CrossRef]
- Goey, A.K.L.; Sissung, T.M.; Peer, C.J.; Trepel, J.B.; Lee, M.-J.; Tomita, Y.; Ehrlich, S.; Bryla, C.; Balasubramaniam, S.; Piekarz, R.; et al. Effects of UGT1A1 genotype on the pharmacokinetics, pharmacodynamics, and toxicities of belinostat administered by 48-hour continuous infusion in patients with cancer. J. Clin. Pharmacol. 2016, 56, 461–473. [Google Scholar] [CrossRef]
- Patel, P.; Xue, Z.; King, K.S.; Parham, L.; Ford, S.; Lou, Y.; Bakshi, K.K.; Sutton, K.; Margolis, D.; Hughes, A.R.; et al. Evaluation of the effect of UGT1A1 polymorphisms on the pharmacokinetics of oral and long-acting injectable cabotegravir. J. Antimicrob. Chemother. 2020, 75, 2240–2248. [Google Scholar] [CrossRef]
- Johnson, T.N.; Rostami-Hodjegan, A. Resurgence in the use of physiologically based pharmacokinetic models in pediatric clinical pharmacology: Parallel shift in incorporating the knowledge of biological elements and increased applicability to drug development and clinical practice. Pediatr. Anesth. 2011, 21, 291–301. [Google Scholar] [CrossRef]
- Chetty, M.; Johnson, T.N.; Polak, S.; Salem, F.; Doki, K.; Rostami-Hodjegan, A. Physiologically based pharmacokinetic modelling to guide drug delivery in older people. Adv. Drug Deliv. Rev. 2018, 135, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Ke, A.B.; Greupink, R.; Abduljalil, K. Drug Dosing in Pregnant Women: Challenges and Opportunities in Using Physiologically Based Pharmacokinetic Modeling and Simulations. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 103–110. [Google Scholar] [CrossRef]
- Hsueh, C.-H.; Hsu, V.; Zhao, P.; Zhang, L.; Giacomini, K.; Huang, S.-M. PBPK Modeling of the Effect of Reduced Kidney Function on the Pharmacokinetics of Drugs Excreted Renally by Organic Anion Transporters. Clin. Pharmacol. Ther. 2018, 103, 485–492. [Google Scholar] [CrossRef]
- Heimbach, T.; Chen, Y.; Chen, J.; Dixit, V.; Parrott, N.; Peters, S.A.; Poggesi, I.; Sharma, P.; Snoeys, J.; Shebley, M.; et al. Physiologically-Based Pharmacokinetic Modeling in Renal and Hepatic Impairment Populations: A Pharmaceutical Industry Perspective. Clin. Pharmacol. Ther. 2021, 110, 297–310. [Google Scholar] [CrossRef]
- Cheeti, S.; Budha, N.R.; Rajan, S.; Dresser, M.J.; Jin, J.Y. A physiologically based pharmacokinetic (PBPK) approach to evaluate pharmacokinetics in patients with cancer. Biopharm. Drug Dispos. 2013, 34, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Budha, N.; Jin, J. Impact of Physiologically Based Pharmacokinetic Models on Regulatory Reviews and Product Labels: Frequent Utilization in the Field of Oncology. Clin. Pharmacol. Ther. 2017, 101, 597–602. [Google Scholar] [CrossRef]
- Jamei, M. Recent Advances in Development and Application of Physiologically-Based Pharmacokinetic (PBPK) Models: A Transition from Academic Curiosity to Regulatory Acceptance. Curr. Pharmacol. Rep. 2016, 2, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Chen, Y.; Unadkat, J.D.; Zhang, X.; Wu, D.; Heimbach, T. Applications, Challenges, and Outlook for PBPK Modeling and Simulation: A Regulatory, Industrial and Academic Perspective. Pharm. Res. 2022, 39, 1701–1731. [Google Scholar] [CrossRef]
- Aherne, G.W.; Piall, E.; Marks, V.; Mould, G.; White, W.F. Prolongation and enhancement of serum methotrexate concentrations by probenecid. Br. Med. J. 1978, 1, 1097–1099. [Google Scholar] [CrossRef] [PubMed]
- Ball, K.; Jamier, T.; Parmentier, Y.; Denizot, C.; Mallier, A.; Chenel, M. Prediction of renal transporter-mediated drug-drug interactions for a drug which is an OAT substrate and inhibitor using PBPK modelling. Eur. J. Pharm. Sci. 2017, 106, 122–132. [Google Scholar] [CrossRef]
- Posada, M.M.; Cannady, E.A.; Payne, C.D.; Zhang, X.; Bacon, J.A.; Pak, Y.A.; Higgins, J.W.; Shahri, N.; Hall, S.D.; Hillgren, K.M. Prediction of Transporter-Mediated Drug-Drug Interactions for Baricitinib. Clin. Transl. Sci. 2017, 10, 509–519. [Google Scholar] [CrossRef]
- Hsu, V.; de LT Vieira, M.; Zhao, P.; Zhang, L.; Zheng, J.H.; Nordmark, A.; Berglund, E.G.; Giacomini, K.M.; Huang, S.-M. Towards Quantitation of the Effects of Renal Impairment and Probenecid Inhibition on Kidney Uptake and Efflux Transporters, Using Physiologically Based Pharmacokinetic Modelling and Simulations. Clin. Pharmacokinet. 2014, 53, 283–293. [Google Scholar] [CrossRef]
- Tahara, H.; Kusuhara, H.; Endou, H.; Koepsell, H.; Imaoka, T.; Fuse, E.; Sugiyama, Y. A Species Difference in the Transport Activities of H2 Receptor Antagonists by Rat and Human Renal Organic Anion and Cation Transporters. J. Pharmacol. Exp. Ther. 2005, 315, 337–345. [Google Scholar] [CrossRef]
- Posada, M.M.; Bacon, J.A.; Schneck, K.B.; Tirona, R.G.; Kim, R.B.; Higgins, J.W.; Pak, Y.A.; Hall, S.D.; Hillgren, K.M. Prediction of Renal Transporter Mediated Drug-Drug Interactions for Pemetrexed Using Physiologically Based Pharmacokinetic Modeling. Drug Metab. Dispos. 2015, 43, 325–334. [Google Scholar] [CrossRef]
- Wenning Larissa, A.; Hanley William, D.; Brainard Diana, M.; Petry Amelia, S.; Ghosh, K.; Jin, B.; Mangin, E.; Marbury Thomas, C.; Berg Jolene, K.; Chodakewitz Jeffrey, A.; et al. Effect of Rifampin, a Potent Inducer of Drug-Metabolizing Enzymes, on the Pharmacokinetics of Raltegravir. Antimicrob. Agents Chemother. 2009, 53, 2852–2856. [Google Scholar] [CrossRef] [PubMed]
- Krishna, R.; East, L.; Larson, P.; Valiathan, C.; Deschamps, K.; Luk, J.A.; Bethel-Brown, C.; Manthos, H.; Brejda, J.; Gartner, M. Atazanavir increases the plasma concentrations of 1200 mg raltegravir dose. Biopharm. Drug Dispos. 2016, 37, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Kasichayanula, S.; Liu, X.; Griffen, S.C.; LaCreta, F.P.; Boulton, D.W. Effects of rifampin and mefenamic acid on the pharmacokinetics and pharmacodynamics of dapagliflozin. Diabetes Obes. Metab. 2013, 15, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Callegari, E.; Lin, J.; Tse, S.; Goosen, T.C.; Sahasrabudhe, V. Physiologically-Based Pharmacokinetic Modeling of the Drug–Drug Interaction of the UGT Substrate Ertugliflozin Following Co-Administration with the UGT Inhibitor Mefenamic Acid. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 127–136. [Google Scholar] [CrossRef]
- PONSTEL® (Mefenamic Acid Capsules, USP) FDA Label. Effective Date: 02/19/2008. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/015034s040lbl.pdf (accessed on 8 April 2025).
- Parasrampuria, R.; Ford, S.L.; Lou, Y.; Fu, C.; Bakshi, K.K.; Tenorio, A.R.; Trezza, C.; Spreen, W.R.; Patel, P. A Phase I Study to Evaluate the Pharmacokinetics and Safety of Cabotegravir in Adults with Severe Renal Impairment and Healthy Matched Control Participants. Clin. Pharmacol. Drug Dev. 2019, 8, 674–681. [Google Scholar] [CrossRef]
- Shaik, J.S.B.; Ford, S.L.; Lou, Y.; Zhang, Z.; Bakshi, K.K.; Tenorio, A.R.; Trezza, C.; Spreen, W.R.; Patel, P. A Phase 1 Study to Evaluate the Pharmacokinetics and Safety of Cabotegravir in Patients With Hepatic Impairment and Healthy Matched Controls. Clin. Pharmacol. Drug Dev. 2019, 8, 664–673. [Google Scholar] [CrossRef]
- Ford, S.L.; Lou, Y.; Lewis, N.; Kostapanos, M.; D’Amico, R.; Spreen, W.; Patel, P. Effect of Rifabutin on the Pharmacokinetics of Oral Cabotegravir in Healthy Subjects. Antivir. Ther. 2019, 24, 301–308. [Google Scholar] [CrossRef]
- Jaehde, U.; Sörgel, F.; Reiter, A.; Sigl, G.; Naber, K.G.; Schunack, W. Effect of probenecid on the distribution and elimination of ciprofloxacin in humans. Clin. Pharmacol. Ther. 1995, 58, 532–541. [Google Scholar] [CrossRef]
- Hill, G.; Cihlar, T.; Oo, C.; Ho, E.S.; Prior, K.; Wiltshire, H.; Barrett, J.; Liu, B.; Ward, P. The Anti-Influenza Drug Oseltamivir Exhibits Low Potential to Induce Pharmacokinetic Drug Interactions via Renal Secretion—Correlation of in Vivo and in Vitro Studies. Drug Metab. Dispos. 2002, 30, 13–19. [Google Scholar] [CrossRef]
- Cundy, K.C.; Petty, B.G.; Flaherty, J.; Fisher, P.E.; Polis, M.A.; Wachsman, M.; Lietman, P.S.; Lalezari, J.P.; Hitchcock, M.J.; Jaffe, H.S. Clinical pharmacokinetics of cidofovir in human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 1995, 39, 1247–1252. [Google Scholar] [CrossRef]
- Iqbal, M.P.; Baig, J.A.; Ali, A.A.; Niazi, S.K.; Mehboobali, N.; Hussain, M.A. The effects of non-steroidal anti-inflammatory drugs on the disposition of methotrexate in patients with rheumatoid arthritis. Biopharm. Drug Dispos. 1998, 19, 163–167. [Google Scholar] [CrossRef]
- Garton, A.M.; Rennie, R.P.; Gilpin, J.; Marrelli, M.; Shafran, S.D. Comparison of dose doubling with probenecid for sustaining serum cefuroxime levels. J. Antimicrob. Chemother. 1997, 40, 903–906. [Google Scholar] [CrossRef]
- Maeda, K.; Tian, Y.; Fujita, T.; Ikeda, Y.; Kumagai, Y.; Kondo, T.; Tanabe, K.; Nakayama, H.; Horita, S.; Kusuhara, H.; et al. Inhibitory effects of p-aminohippurate and probenecid on the renal clearance of adefovir and benzylpenicillin as probe drugs for organic anion transporter (OAT) 1 and OAT3 in humans. Eur. J. Pharm. Sci. 2014, 59, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; Buchanan, A.M.; Chen, S.; Ford, S.L.; Gould, E.; Margolis, D.; Spreen, W.R.; Patel, P. Effect of Cabotegravir on Cardiac Repolarization in Healthy Subjects. Clin. Pharmacol. Drug Dev. 2016, 5, 509–516. [Google Scholar] [CrossRef] [PubMed]
- CABENUVA (Cabotegravir Extended-Release Injectable Suspension; Rilpivirine Extended-Release Injectable Suspension). In Co-Packaged for Intramuscular Use [Package Insert]; Viiv Healthcare: Durham, NC, USA, 2024.
- Bentz, J.; O’Connor, M.P.; Bednarczyk, D.; Coleman, J.; Lee, C.; Palm, J.; Pak, Y.A.; Perloff, E.S.; Reyner, E.; Balimane, P.; et al. Variability in P-Glycoprotein Inhibitory Potency (IC50) Using Various in Vitro Experimental Systems: Implications for Universal Digoxin Drug-Drug Interaction Risk Assessment Decision Criteria. Drug Metab. Dispos. 2013, 41, 1347–1366. [Google Scholar] [CrossRef]
- McFeely, S.J.; Ritchie, T.K.; Ragueneau-Majlessi, I. Variability in In Vitro OATP1B1/1B3 Inhibition Data: Impact of Incubation Conditions on Variability and Subsequent Drug Interaction Predictions. Clin. Transl. Sci. 2020, 13, 47–52. [Google Scholar] [CrossRef]
- Margolis, D.A.; Gonzalez-Garcia, J.; Stellbrink, H.-J.; Eron, J.J.; Yazdanpanah, Y.; Podzamczer, D.; Lutz, T.; Angel, J.B.; Richmond, G.J.; Clotet, B.; et al. Long-acting intramuscular cabotegravir and rilpivirine in adults with HIV-1 infection (LATTE-2): 96-week results of a randomised, open-label, phase 2b, non-inferiority trial. Lancet 2017, 390, 1499–1510. [Google Scholar] [CrossRef]
- Trezza, C.; Ford, S.L.; Gould, E.; Lou, Y.; Huang, C.; Ritter, J.M.; Buchanan, A.M.; Spreen, W.; Patel, P. Lack of effect of oral cabotegravir on the pharmacokinetics of a levonorgestrel/ethinyl oestradiol-containing oral contraceptive in healthy adult women. Br. J. Clin. Pharmacol. 2017, 83, 1499–1505. [Google Scholar] [CrossRef]
- Mori, K.; Saito, R.; Nakamaru, Y.; Shimizu, M.; Yamazaki, H. Physiologically based pharmacokinetic–pharmacodynamic modeling to predict concentrations and actions of sodium-dependent glucose transporter 2 inhibitor canagliflozin in human intestines and renal tubules. Biopharm. Drug Dispos. 2016, 37, 491–506. [Google Scholar] [CrossRef]
- Obermeier, M.; Yao, M.; Khanna, A.; Koplowitz, B.; Zhu, M.; Li, W.; Komoroski, B.; Kasichayanula, S.; Discenza, L.; Washburn, W.; et al. In Vitro Characterization and Pharmacokinetics of Dapagliflozin (BMS-512148), a Potent Sodium-Glucose Cotransporter Type II Inhibitor, in Animals and Humans. Drug Metab. Dispos. 2010, 38, 405–414. [Google Scholar] [CrossRef]
- FDA Final Risk Evaluation and Mitigation Strategy (REMS). Review Dapagliflozin Application Number 202293 Orig1s000. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/202293orig1s000riskr.pdf (accessed on 8 April 2025).
- Komoroski, B.; Vachharajani, N.; Boulton, D.; Kornhauser, D.; Geraldes, M.; Li, L.; Pfister, M. Dapagliflozin, a Novel SGLT2 Inhibitor, Induces Dose-Dependent Glucosuria in Healthy Subjects. Clin. Pharmacol. Ther. 2009, 85, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Djajadisastra, J.; Purnama, H.D.; Yanuar, A. In Silico Binding Interaction Study Of Mefenamic Acid and Piroxicam on HumanAlbumin. Int. J. App. Pharm. 2017, 9 (Suppl. 1), 102–106. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, I.; Gyéresi, Á.; Kiss, L.; Deli, M.A.; Croitoru, M.D.; Szabó-Révész, P.; Aigner, Z. Preparation and investigation of mefenamic acid—polyethylene glycol—sucrose ester solid dispersions. Acta Pharm. 2015, 65, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Mistilis, S.P. Studies on Ponstan (mefenamic acid): I. Gastro-intestinal blood loss; II. Absorbtion and excretion of a new formulation. J. Int. Med. Res. 1975, 3, 176–182. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Weight (g/mol) | 405.4 | Calculated |
|---|---|---|
| Log P | 1.58 | Based on compound structure and physicochemical properties; Log P measured using a potentiometric assay; Pka1 measured using a UV metric assay |
| Compound type | Monoprotic Acid | |
| pKa1 | 7.71 | |
| B/P ratio | 0.54 | |
| fu,p | 0.006 | Measured value using equilibrium dialysis method |
| Absorption | ADAM | |
| Papp MDCK (10−6 cm/s) | 25.6 | Measured value using monolayer MDCKII cells in presence of 5 uM elacridar |
| Dissolution profile | For micronized tablet formulation | Measured value from an in vitro dissolution assay |
| Distribution | ||
| Distribution model | Minimal PBPK model | |
| SAC kin (1/h) | 0.03 | Estimated using Simcyp Parameter Estimate function |
| SAC kout (1/h) | 0.07 | |
| Vss (L/kg) | (Predicted) 0.12 | Vss predicted by Simcyp® using Method 2 |
| Scalar | 1.5 | |
| Elimination | Enzymatic | |
| UGT1A1 CLint (µL/min/mg) | 4.5 | [2] |
| UGT1A9 CLint (µL/min/mg) | 2.2 | |
| fu,mic | 0.5 | Optimized |
| Interaction | ||
| OAT1 Ki (µM) | 0.405 | [1] |
| OAT3 Ki (µM) | 0.205 | |
| UGT1A1 Ki (µM) | >50 | |
| UGT1A9 Ki (µM) | 23.0 | |
| Substrate-Inhibitor Pair | No. of Subjects in Trial | Age Range (Mean) | % of Females | Dose Regimen | Reference |
|---|---|---|---|---|---|
| OAT1/OAT3 substrates 1-CAB | 10 | 20–50 | 0.5 | 30 mg CAB was administered orally once a day for 14 days and a single dose of a specific OAT1/OAT3 substrate was administered 2 h after CAB dose on day 10. The OAT1 or OAT3 substrate was administered either orally or IV at doses based on clinical studies. | [38,39,40,52,53,54,55,56,57] |
| CAB-Atazanavir | 14 | 19–55 | 0.5 | Atazanavir was administered orally at 400 mg once daily for 9 days and CAB 30 mg single oral dose was co-administered on day 7. | [45] |
| CAB-Mefenamic Acid | 16 | 25–55 | 0.44 | Mefenamic acid was orally administered starting with a loading dose 0f 500 mg and then 250 mg Q6h for 4 days, Single dose of 30 mg oral CAB was administered on day 2. | [46] |
| DDI Drugs (Substrate – Precipitant) | AUC Ratio | Cmax Ratio | Mechanism of DDI | Reference | ||||
|---|---|---|---|---|---|---|---|---|
| Observed | Simulated | Observed | Simulated | |||||
| S44121 – Probenecid 1 | 2.2 (1.9–2.7) | 2.1 (1.2–4.8) | NR | 1.2 (1.1–1.5) | OAT1/OAT3 inhibition | [39] | ||
| Ciprofloxacin – Probenecid 1 | 1.7 | 1.6 (1.2–2.3) | 1.2 | 1.2 (1.1–1.3) | OAT3 inhibition | [52] | ||
| Tenofovir – S44121 1 | 0.89 (0.56–1.1) | 1.0 (1.0–1.1) | NR | 1.0 (1.0–1.1) | OAT1 inhibition | [39] | ||
| Baricitinib – Probenecid 2 | 2.0 (1.9, 2.2) 4 | 1.8 (1.3, 2.8) | 1.0 (0.94, 1.1) 4 | 1.1 (1.0, 1.2) | OAT1 inhibition | [40] | ||
| Oseltamivir carboxylate – Probenecid 2 | 2.5 (2.3, 2.8) 4 | 2.2 (1.5, 3.0) | 1.9 (1.7, 2.0)4 | 1.8 (1.4, 2.3) | OAT3 inhibition | [53] | ||
| Cidofovir – Probenecid 2 | 1.0 | 1.3 (1.1, 1.7) | 1.1 | 1.1 (1.0, 1.1) | OAT1 inhibition | [54] | ||
| Methotrexate – Diclofenac 3 | 1.3 | 1.0 (1.0–1.0) | NR | 1.0 (1.0–1.0) | OAT3 inhibition | [55] | ||
| Cefuroxime – Probenecid 3 | 1.4 | 1.8 (1.3–2.4) | 1.1 | 1.1 (1.0–1.2) | OAT1 inhibition | [56] | ||
| Adefovir – Probenecid | 1.8 | 1.7 (1.3–2.0) | NR | 1.5 (1.2–1.7) | OAT1 inhibition | [57] | ||
| Raltegravir – Rifampin | 0.6 5 | 0.06–0.48 7 | 0.62 5 | 0.09–0.55 7 | UGT1A1 induction | [44] | ||
| Raltegravir – Atazanavir | 1.67 (1.34, 2.10) 5 | 1.48 (1.24, 1.73) 2 | 1.16 (1.01, 1.33) 5 | 1.49 (1.17, 1.85) 2 | UGT1A1 inhibition | [45] | ||
| Dapagliflozin – Mefenamic acid | 1.51 (1.49, 1.53) 5 | 1.51 (1.44, 1.58) 2 | 1.13 (1.03, 2.10) 5 | 1.24 (1.23, 1.25) 2 | UGT1A9 inhibition | [46] | ||
| Half-Life Mean Ratio | Plasma Clearance Mean Ratio | C24 Mean Ratio 6 | ||||||
| Observed | Predicted | Observed | Predicted | Observed | Predicted | [38] | ||
| Methotrexate – Probenecid | 1.5 | 1.3 | 0.64 | 0.64 | 4.4 | 3.8 | ||
| CAB AUC0-inf Ratio | CAB Cmax Ratio | CAB CL Ratio | Reference | ||||
|---|---|---|---|---|---|---|---|
| Observed | Simulated | Observed | Simulated | Observed | Simulated | ||
| CAB + Rifampin vs. CAB | 0.41 1 | 0.51 (0.21–0.66) 2 | 0.94 1 | 0.93 (0.83–0.97) 2 | 2.4 1 | 2.3 (1.5–4.5) 2 | [23] |
| UGT1A1 poor metabolizers vs. normal metabolizers | 1.41 3 | 1.41 4 | 1.28 3 | 1.03 4 | NR | NR | [28] |
| Precipitant | Object (Transporter) | Predicted AUC Ratio 1 | Predicted Cmax Ratio 1 |
|---|---|---|---|
| CAB | S44121 (OAT1 and OAT3) | 1.18 (1.07–1.34) | 1.05 (1.02–1.10) |
| Methotrexate (OAT3) | 1.11 (1.04–1.23) | 1.01 (1.00–1.03) | |
| Ciprofloxacin (OAT3) | 1.11 (1.06–1.17) | 1.04 (1.02–1.05) | |
| Tenofovir (OAT1) | 1.04 (1.01–1.07) | 1.01 (1.00–1.02) | |
| Baricitinib (OAT1) | 1.08 (1.04–1.16) | 1.02 (1.00–1.04) | |
| Oseltamivir carboxylate (OAT3) | 1.14 (1.07–1.23) | 1.12 (1.07–1.20) | |
| Cidofovir (OAT3) | 1.05 (1.02–1.10) | 1.01 (1.00–1.03) | |
| Cefuroxime (OAT1) | 1.09 (1.04–1.17) | 1.02 (1.01–1.04) | |
| Adefovir (OAT1) | 1.07 (1.03–1.14) | 1.05 (1.03–1.10) | |
| Atazanavir (UGT1A1) | CAB–Atazanavir (UGT1A1) | 1.11 (1.04–1.20) | 1.02 (1.01–1.04) |
| Atazanavir (UGT1A1) 2 | CAB | 1.06 (1.03–1.12) | 1.03 (1.01–1.02) |
| Mefenamic Acid (UGT1A9) | CAB | 1.10 (1.04, 1.18) | 1.02 (1.01–1.02) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tracey, H.; Bate, S.T.; Ford, S.; Patel, P.; Bloomer, J.; Patel, A.; Taskar, K.S. Matrix Approach Assessment of Cabotegravir Drug–Drug Interactions with OAT1/OAT3 Substrates and UGT1A1/UGT1A9 Inhibitors Using Physiologically-Based Pharmacokinetic Modeling. Pharmaceutics 2025, 17, 531. https://doi.org/10.3390/pharmaceutics17040531
Tracey H, Bate ST, Ford S, Patel P, Bloomer J, Patel A, Taskar KS. Matrix Approach Assessment of Cabotegravir Drug–Drug Interactions with OAT1/OAT3 Substrates and UGT1A1/UGT1A9 Inhibitors Using Physiologically-Based Pharmacokinetic Modeling. Pharmaceutics. 2025; 17(4):531. https://doi.org/10.3390/pharmaceutics17040531
Chicago/Turabian StyleTracey, Helen, Simon T. Bate, Susan Ford, Parul Patel, Jackie Bloomer, Aarti Patel, and Kunal S. Taskar. 2025. "Matrix Approach Assessment of Cabotegravir Drug–Drug Interactions with OAT1/OAT3 Substrates and UGT1A1/UGT1A9 Inhibitors Using Physiologically-Based Pharmacokinetic Modeling" Pharmaceutics 17, no. 4: 531. https://doi.org/10.3390/pharmaceutics17040531
APA StyleTracey, H., Bate, S. T., Ford, S., Patel, P., Bloomer, J., Patel, A., & Taskar, K. S. (2025). Matrix Approach Assessment of Cabotegravir Drug–Drug Interactions with OAT1/OAT3 Substrates and UGT1A1/UGT1A9 Inhibitors Using Physiologically-Based Pharmacokinetic Modeling. Pharmaceutics, 17(4), 531. https://doi.org/10.3390/pharmaceutics17040531