Correction: Suriya et al. Integration of In Silico Strategies for Drug Repositioning towards P38α Mitogen-Activated Protein Kinase (MAPK) at the Allosteric Site. Pharmaceutics 2022, 14, 1461



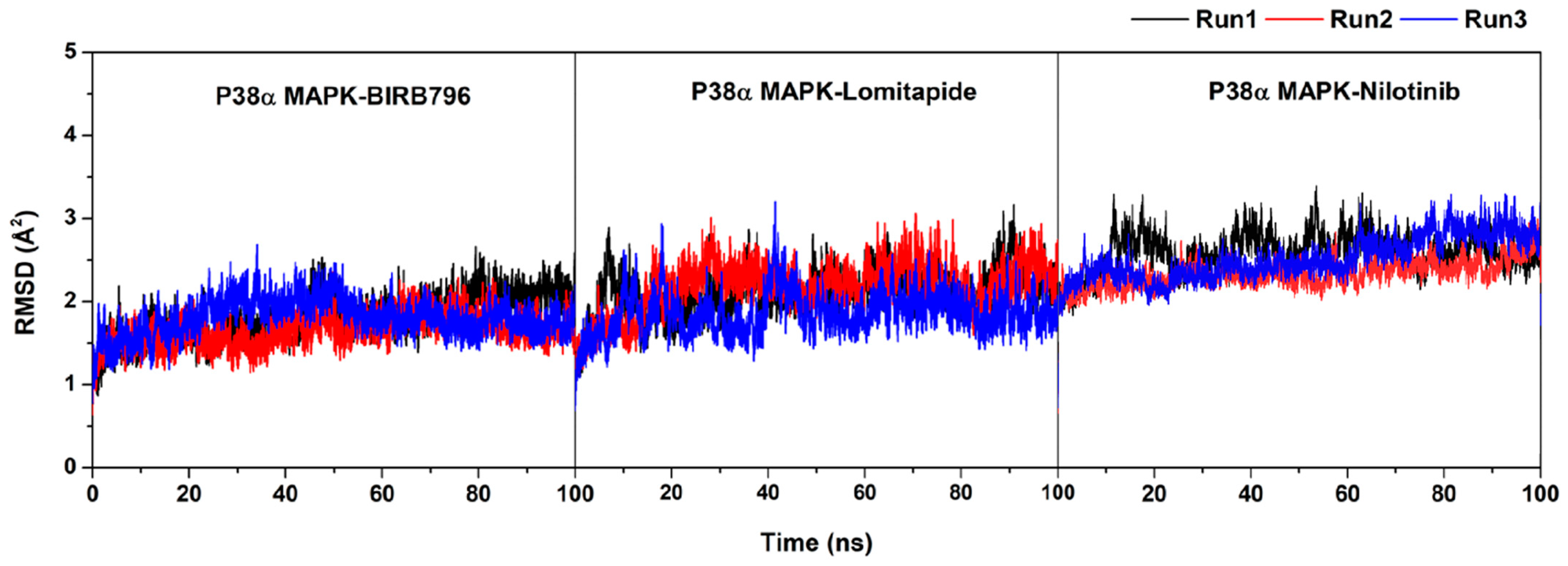
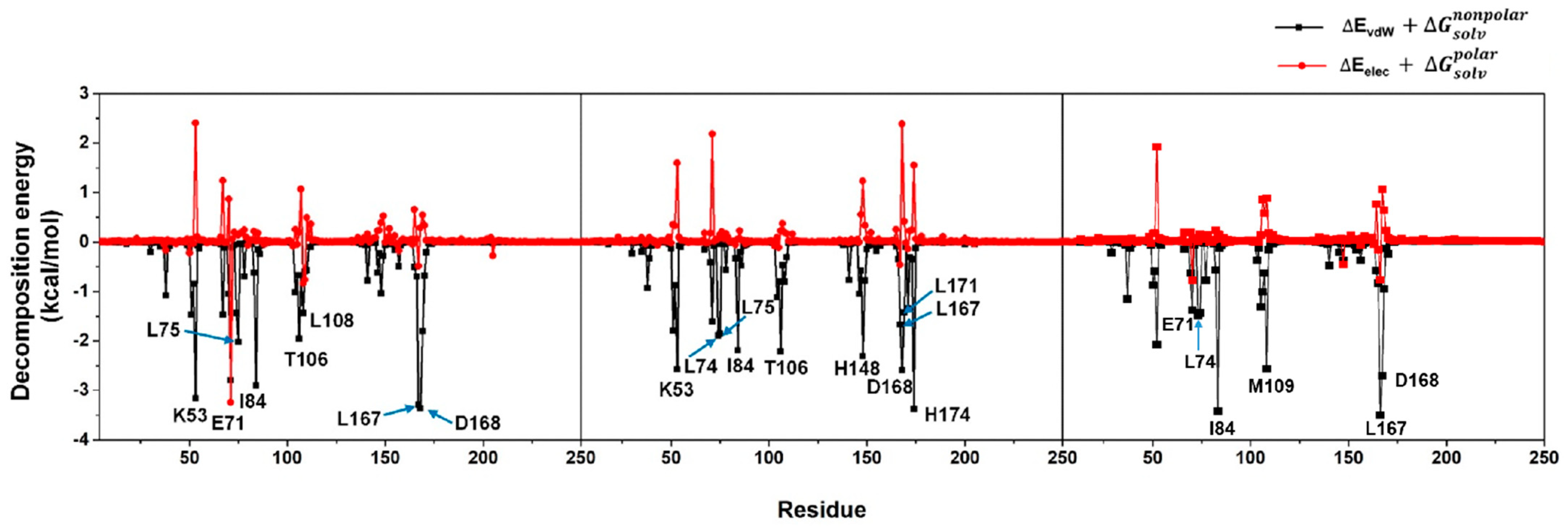
Error in Figure and Table
Text Correction
Reference
- Suriya, U.; Mahalapbutr, P.; Rungrotmongkol, T. Integration of In Silico Strategies for Drug Repositioning towards P38α Mitogen-Activated Protein Kinase (MAPK) at the Allosteric Site. Pharmaceutics 2022, 14, 1461. [Google Scholar] [CrossRef] [PubMed]
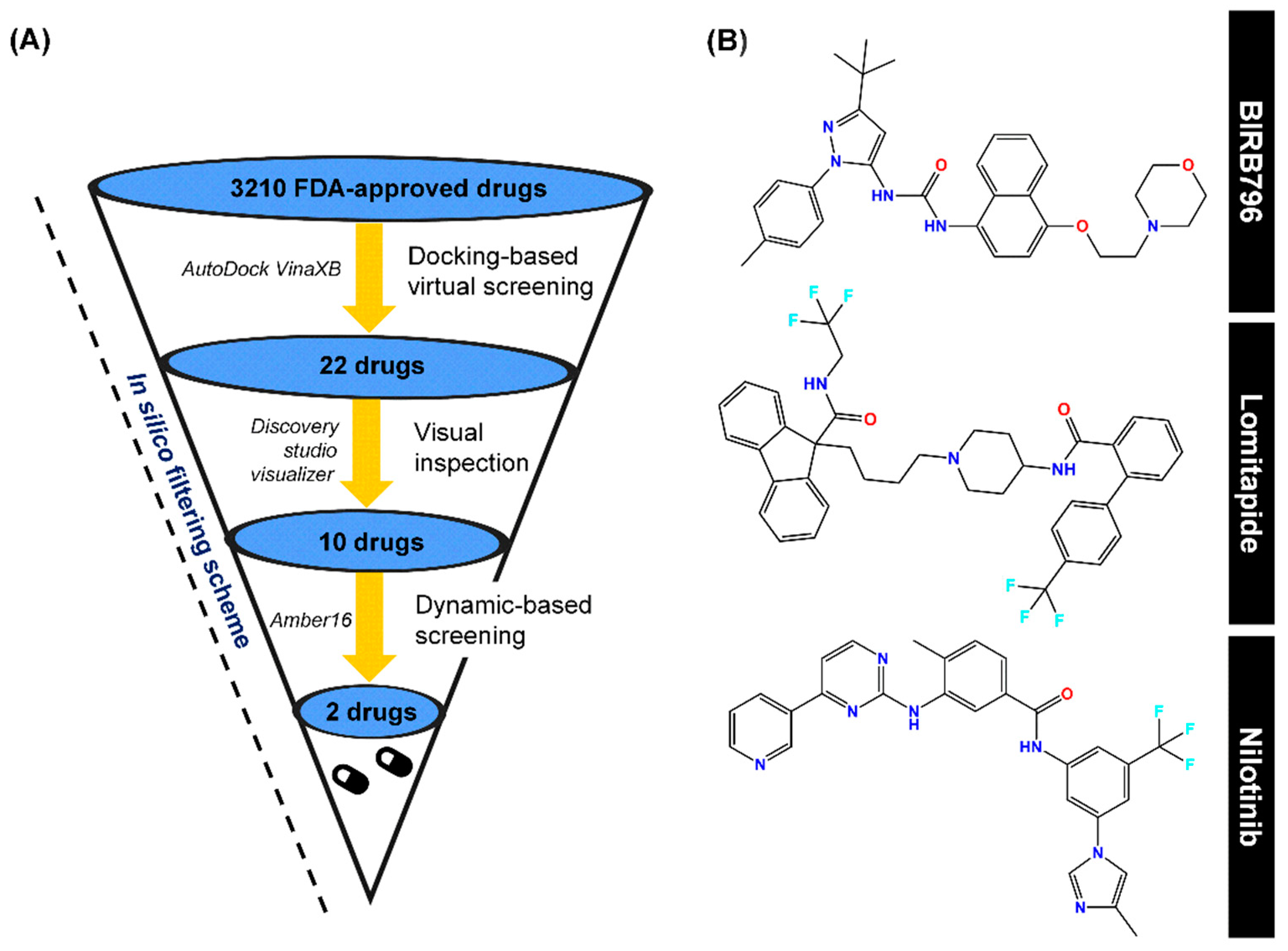

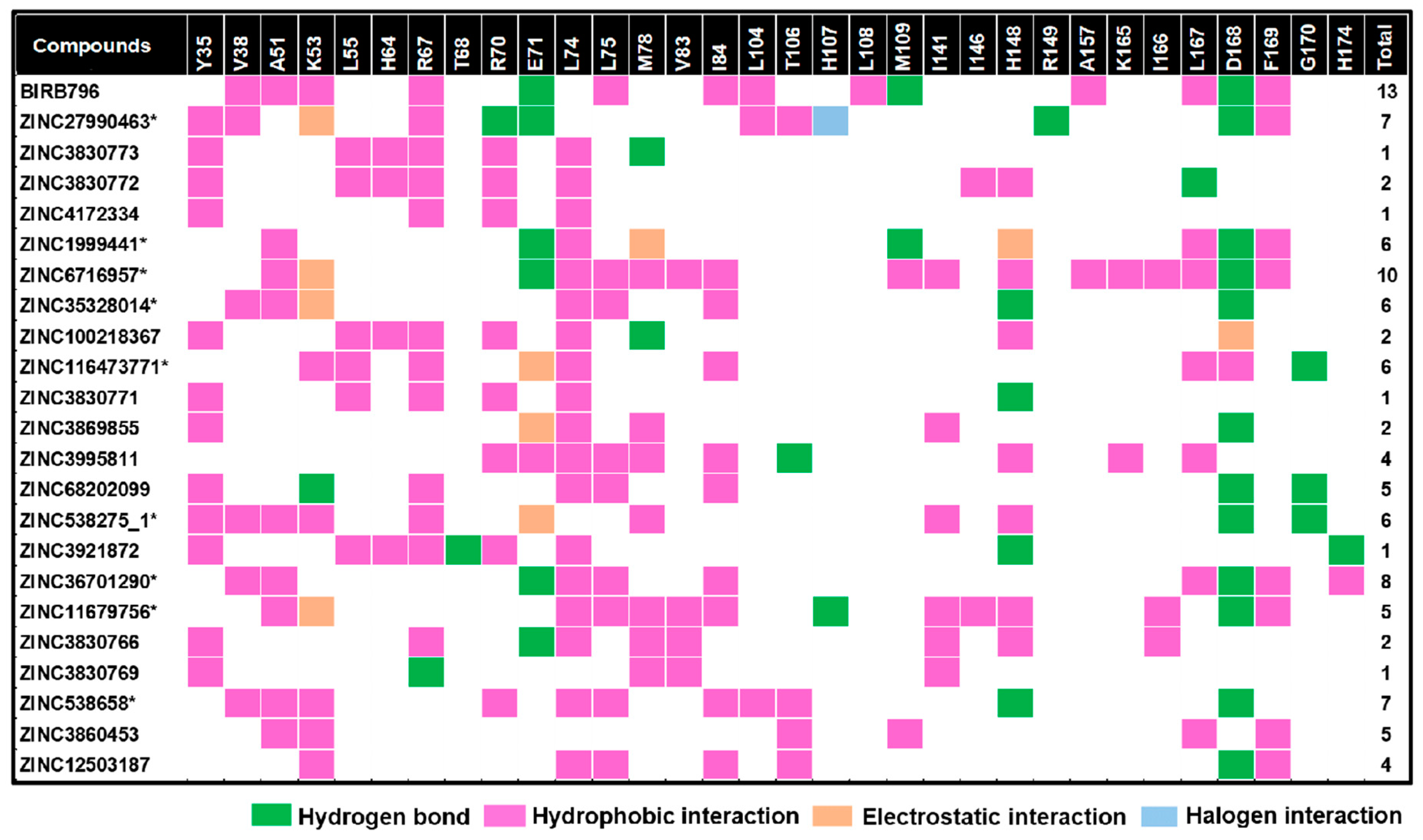
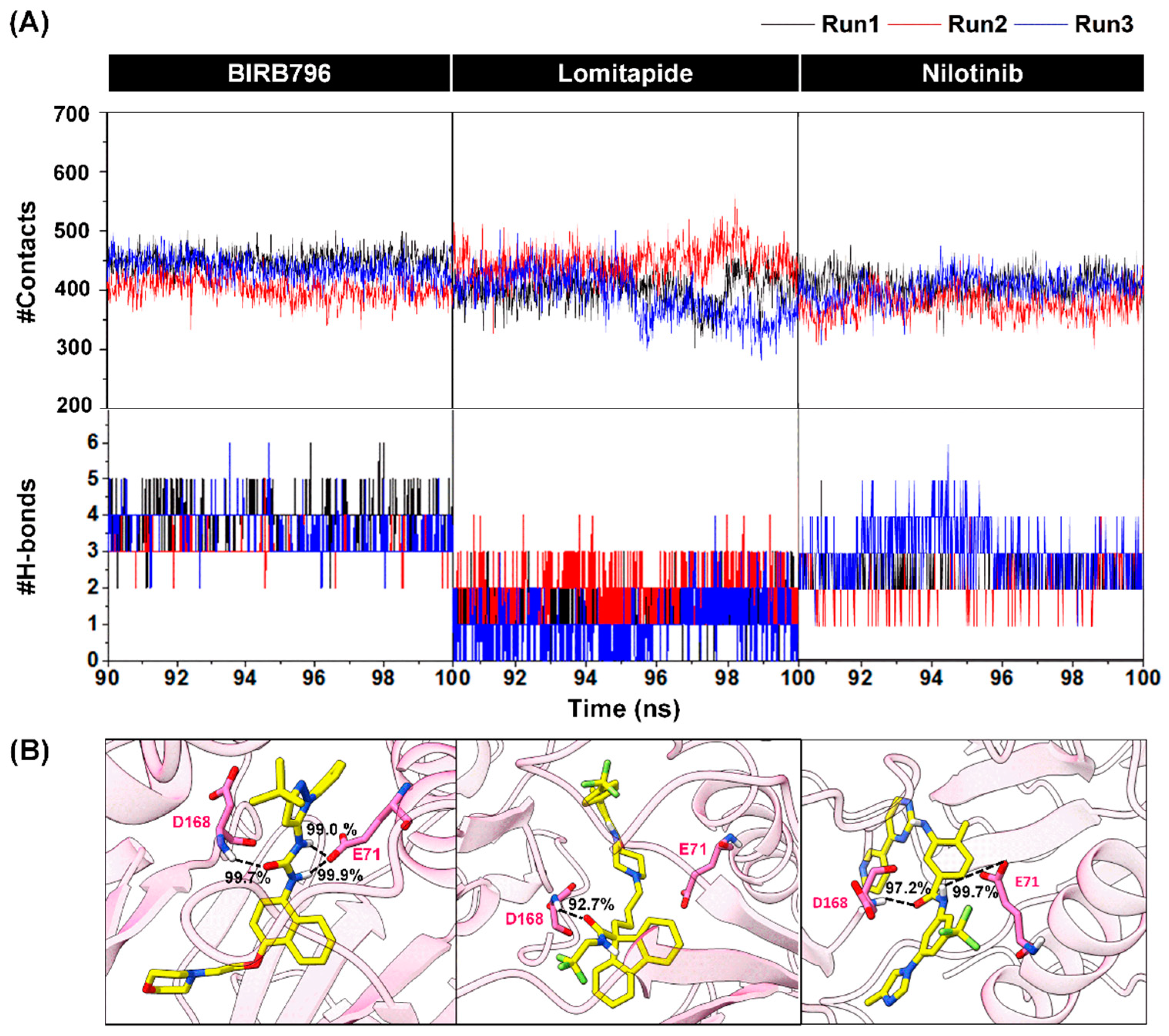
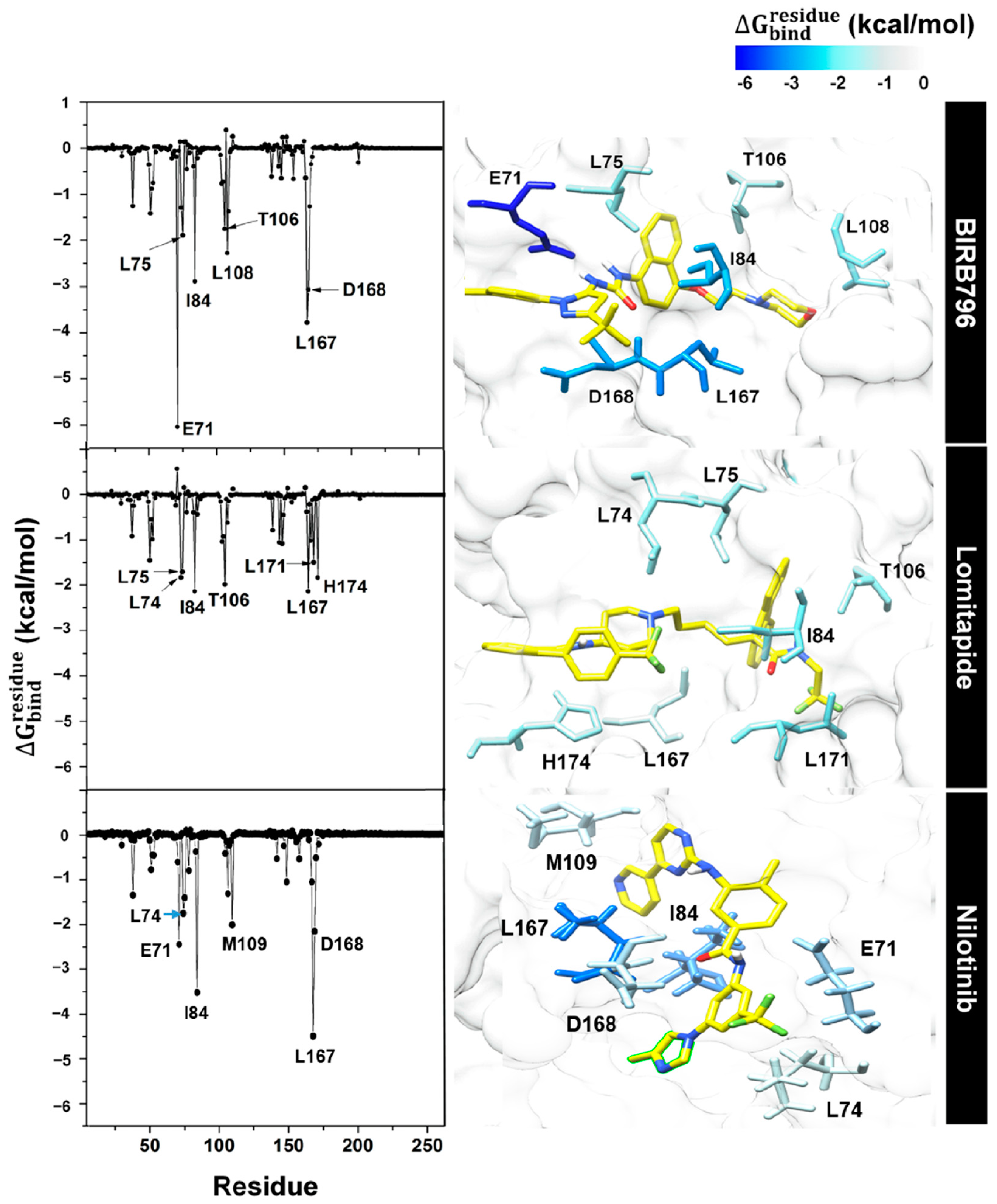

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs (ZINC ID) | Energy Components (kcal/mol) | ||||
|---|---|---|---|---|---|
| EVdW | Ecoul | ΔGRF | ΔGcavity | ΔGbind | |
| BIRB796 | experiment | −10.98 * | |||
| −78.53 ± 0.29 | −9.93 ± 0.15 | 15.60 ± 0.20 | −13.63 ± 0.04 | −11.95 ± 0.04 | |
| Lomitapide (ZINC27990463) | −77.77 ± 0.39 | −5.95 ± 0.18 | 17.01 ± 0.24 | −14.43 ± 0.04 | −11.39 ± 0.05 |
| Nebivolol (ZINC1999441) | −49.57 ± 0.29 | −12.66 ± 0.26 | 12.78 ± 0.21 | −10.19 ± 0.03 | −9.14 ± 0.03 |
| Nilotinib (ZINC6716957) | −71.93 ± 0.26 | −12.36 ± 0.19 | 17.15 ± 0.17 | −12.81 ± 0.04 | −11.27 ± 0.03 |
| Ibrutinib (ZINC35328014) | −61.87 ± 0.29 | −4.07 ± 0.17 | 13.21 ± 0.23 | −10.81 ± 0.04 | −9.55 ± 0.04 |
| Atovaquone (ZINC116473771) | −42.15 ± 0.27 | −2.43 ± 0.39 | 10.94 ± 0.23 | −7.90 ± 0.05 | −7.24 ± 0.03 |
| Dicumarol (ZINC3869855) | −39.68 ± 0.26 | −13.87 ± 0.40 | 20.55 ± 0.32 | −7.13 ± 0.03 | −7.09 ± 0.03 |
| Raloxifene (ZINC538275) | −51.19 ± 0.44 | −12.45 ± 0.50 | 18.50 ± 0.25 | −9.90 ± 0.05 | −8.66 ± 0.07 |
| Ponatinib (ZINC36701290) | −61.66 ± 0.25 | −4.92 ± 0.14 | 16.99 ± 0.24 | −12.08 ± 0.05 | −9.35 ± 0.03 |
| Eltrombopag (ZINC11679756) | −59.44 ± 0.33 | −5.39 ± 0.17 | 10.52 ± 0.17 | −10.97 ± 0.03 | −9.73 ± 0.04 |
| Samsca (ZINC538658) | −51.69 ± 0.26 | −16.63 ± 0.21 | 19.86 ± 0.18 | −10.38 ± 0.04 | −9.05 ± 0.03 |
| Compounds | Run | Energy Components (kcal/mol) | ||||
|---|---|---|---|---|---|---|
| EvdW | Ecoul | ΔGRF | ΔGcavity | ΔGbind | ||
| BIRB796 | 1 | −78.53 ± 0.29 | −9.93 ± 0.15 | 15.60 ± 0.20 | −13.63 ± 0.04 | −11.95 ± 0.04 |
| 2 | −82.37 ± 0.28 | −10.85 ± 0.14 | 15.07 ± 0.20 | −13.91 ± 0.04 | −11.64 ± 0.03 | |
| 3 | −82.01 ± 0.32 | −8.32 ± 0.16 | 11.41 ± 0.19 | −13.58 ± 0.04 | −11.69 ± 0.04 | |
| Average | −11.76 ± 0.04 | |||||
| Lomitapide | 1 | −77.77 ± 0.39 | −5.95 ± 0.18 | 17.01 ± 0.24 | −14.43 ± 0.04 | −11.39 ± 0.05 |
| 2 | −78.41 ± 0.34 | −13.12 ± 0.19 | 29.61 ± 0.35 | −14.58 ± 0.06 | −10.90 ± 0.04 | |
| 3 | −81.79 ± 0.34 | −6.11 ± 0.18 | 16.21 ± 0.24 | −13.16 ± 0.04 | −11.78 ± 0.04 | |
| Average | −11.35 ± 0.04 | |||||
| Nilotinib | 1 | −71.93 ± 0.26 | −12.36 ± 0.19 | 17.15 ± 0.17 | −12.81 ± 0.04 | −11.27 ± 0.03 |
| 2 | −65.37 ± 0.35 | −13.35 ± 0.24 | 17.40 ± 0.19 | −12.36 ± 0.05 | −10.61 ± 0.04 | |
| 3 | −69.27 ± 0.32 | −12.30 ± 0.21 | 20.18 ± 0.18 | −12.95 ± 0.04 | −10.68 ± 0.04 | |
| Average | −10.85 ± 0.04 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suriya, U.; Mahalapbutr, P.; Rungrotmongkol, T. Correction: Suriya et al. Integration of In Silico Strategies for Drug Repositioning towards P38α Mitogen-Activated Protein Kinase (MAPK) at the Allosteric Site. Pharmaceutics 2022, 14, 1461. Pharmaceutics 2025, 17, 419. https://doi.org/10.3390/pharmaceutics17040419
Suriya U, Mahalapbutr P, Rungrotmongkol T. Correction: Suriya et al. Integration of In Silico Strategies for Drug Repositioning towards P38α Mitogen-Activated Protein Kinase (MAPK) at the Allosteric Site. Pharmaceutics 2022, 14, 1461. Pharmaceutics. 2025; 17(4):419. https://doi.org/10.3390/pharmaceutics17040419
Chicago/Turabian StyleSuriya, Utid, Panupong Mahalapbutr, and Thanyada Rungrotmongkol. 2025. "Correction: Suriya et al. Integration of In Silico Strategies for Drug Repositioning towards P38α Mitogen-Activated Protein Kinase (MAPK) at the Allosteric Site. Pharmaceutics 2022, 14, 1461" Pharmaceutics 17, no. 4: 419. https://doi.org/10.3390/pharmaceutics17040419
APA StyleSuriya, U., Mahalapbutr, P., & Rungrotmongkol, T. (2025). Correction: Suriya et al. Integration of In Silico Strategies for Drug Repositioning towards P38α Mitogen-Activated Protein Kinase (MAPK) at the Allosteric Site. Pharmaceutics 2022, 14, 1461. Pharmaceutics, 17(4), 419. https://doi.org/10.3390/pharmaceutics17040419