


New Temporin A Analogues Modified in Positions 1 and 10—Synthesis and Biological Studies

 ,
,  ,
,  , , and
, , and 
Abstract
1. Introduction
- -
- Direct killing by means of damage to the integrity of the membrane;
- -
- Interference with the production of intracellular components, such as proteins and nucleic acids.
- -
- Phe1 was replaced with another proteinogenic aromatic amino acid Tyr or non-proteinogenic fluorinated phenylalanine (Phe(4-F));
- -
- Ser10 was substituted with other hydroxyl-containing natural amino acids (Tyr and Thr).
2. Materials and Methods
2.1. Synthesis and Analytical Data
2.2. Safety Testing and Antiproliferative Potential
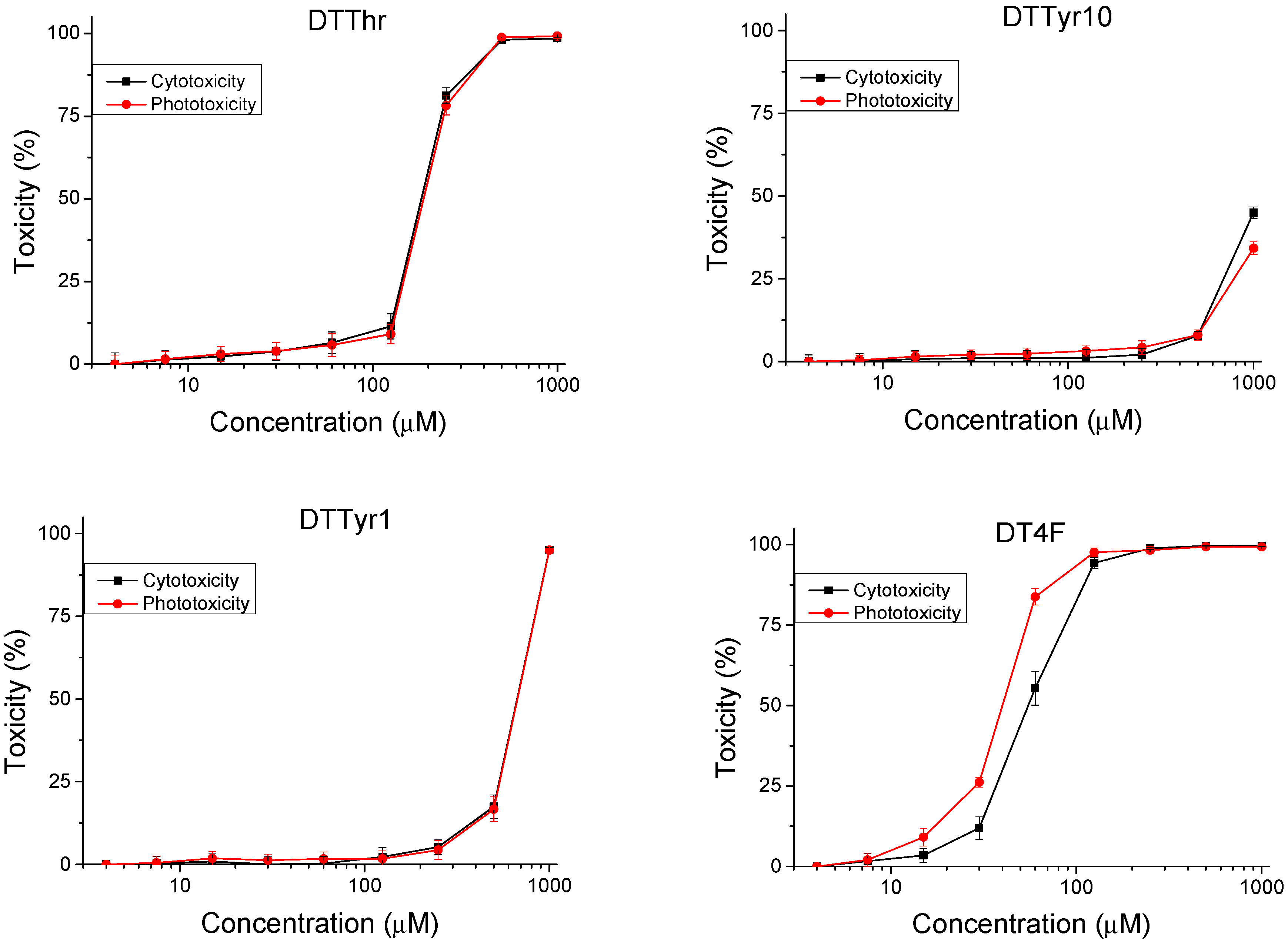
2.2.1. Safety Testing
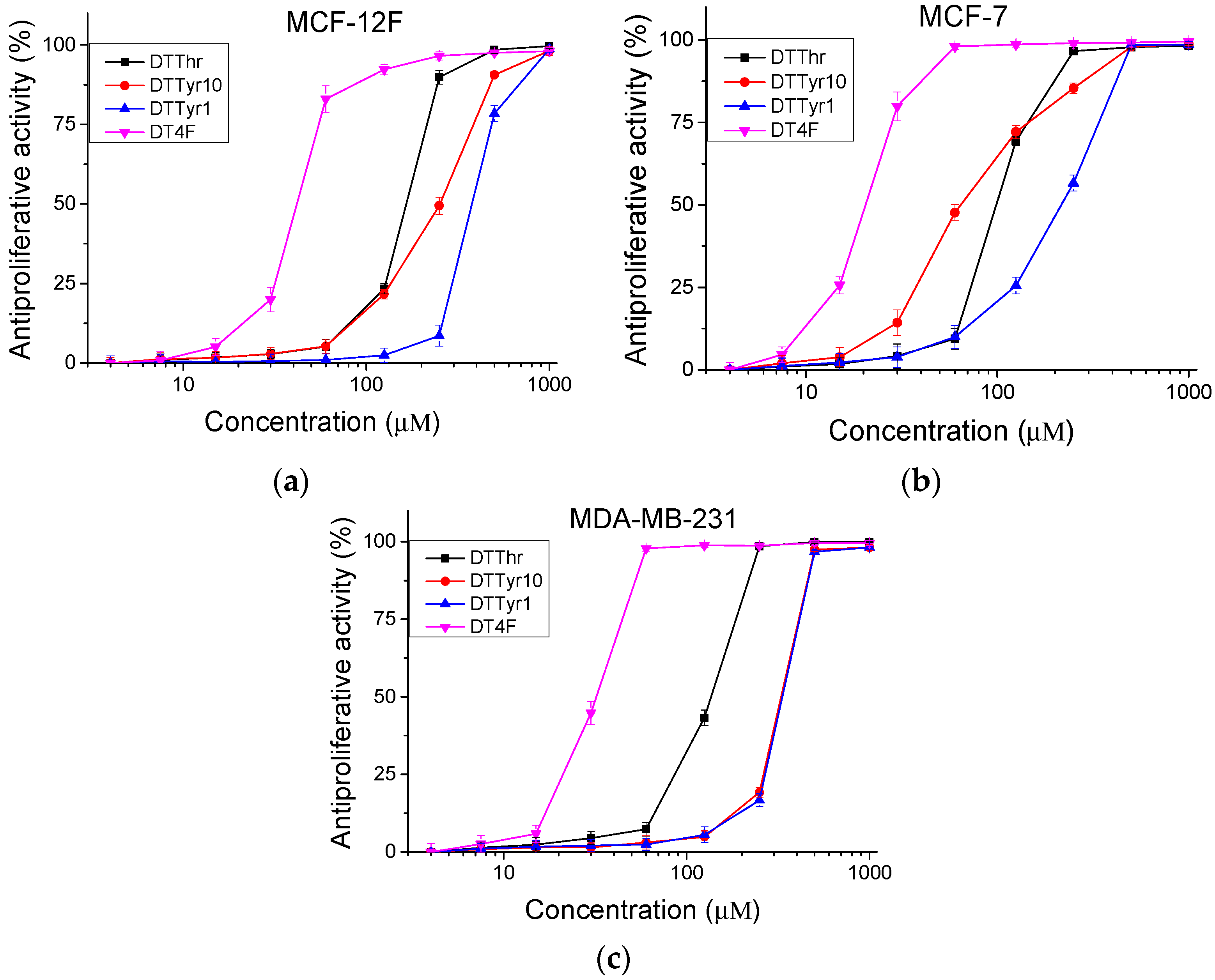
2.2.2. Antiproliferative Activity
2.3. Antimicrobial Assay
- E. coli 8785 in Luria–Bertani (LB, HiMedia, Mumbai, India) agar medium;
- B. subtilis 3562 in nutrient broth (NB, HiMedia, Mumbai, India) agar medium;
- P. aeruginosa 3700 and A. oxydans 9333 in Meat Peptone agar (MPA) medium;
- C. albicans 74 in Yeast Mold (YM) agar medium.
2.3.1. Disk Diffusion Method
2.3.2. Determination of Minimal Inhibitory Concentration (MIC)
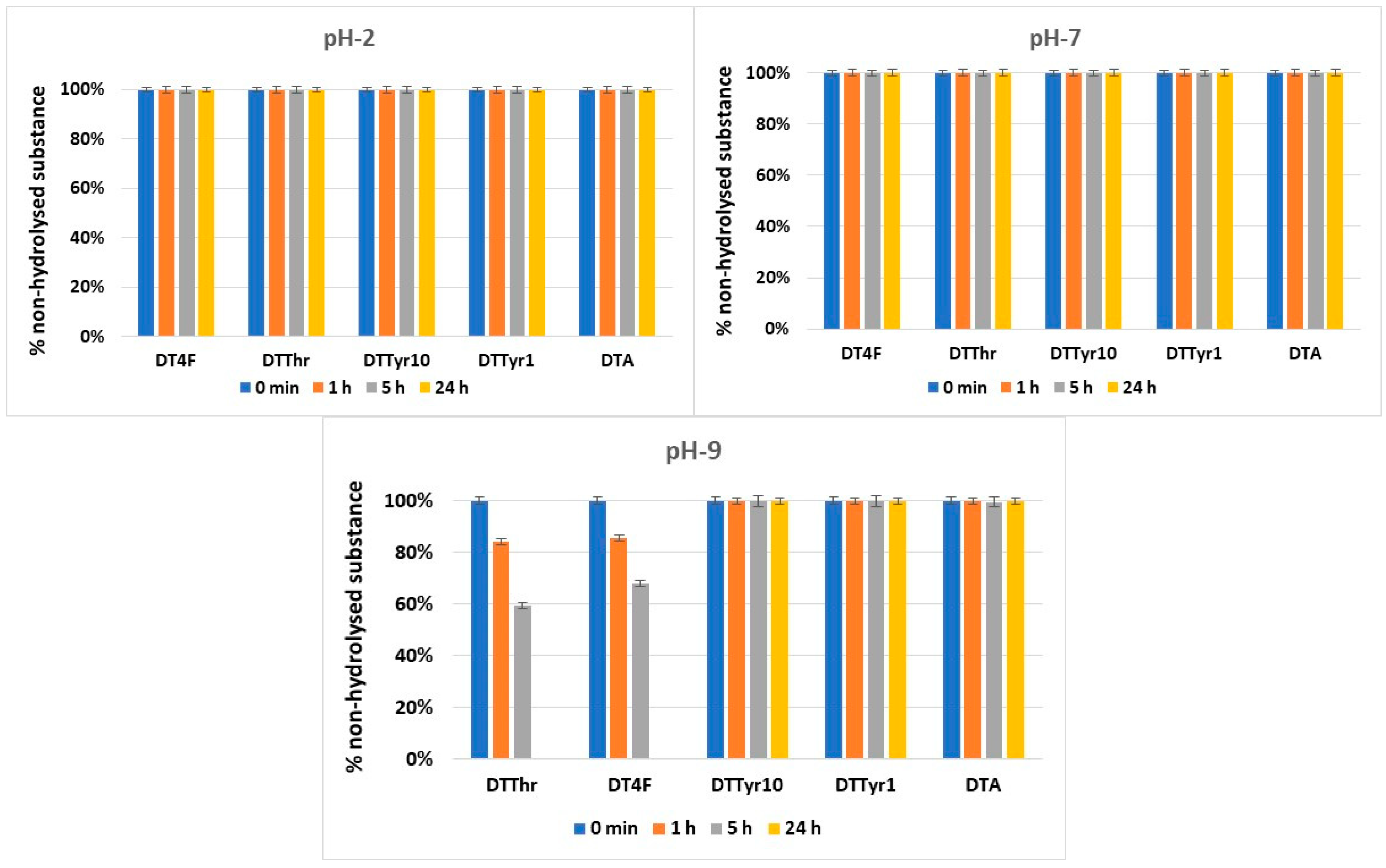
2.4. Hydrolytic Stability
- 1.
- System for pH 2.0:
- Solution A: In a volumetric flask, 119.0 mL of 0.1 mol/L HCl and 6.57 g of KCl in CO2-free water were combined, and the final solution was finished with 1000.0 mL of distilled water.
- Solution B: A volumetric flask containing 5 mg of pepsin was filled to the brim with solution A. Consequently, the final model solution had a pH of 2.0 and a pepsin concentration of 0.5 mg/mL.
- 2.
- System for pH 7.4: To achieve a final trypsin concentration of 0.1 mg/mL in a model system with pH 7.4, amounts of 0.1 g trypsin, 2.38 g Na2HPO4, 0.19 g KH2PO4, and 8.0 g NaCl were dissolved to a total of 1000.0 mL in distilled water. One milliliter of blood plasma (ACCUCLOTM Reference plasma, Normal, Sigma Diagnostics) was recovered using fifteen milliliters of the pH 7.4 buffer that was obtained.
- 3.
- System for pH 9.0: In total, 420.0 mL of a solution containing 0.1 mol/L NaOH in distilled water was combined with 1000 mL of a solution containing 6.18 g H3BO3 in 0.1 mol/L KCl in distilled water. To reach the final concentration of 0.1 mg/mL of trypsin, an additional 0.1 mg of trypsin was dissolved in the pH 9.0 solution and added to 10 mL in a volumetric flask.
3. Results
3.1. Peptide Synthesis and Characterization
3.2. Safety Testing and Antiproliferative Activity
3.2.1. Safety Testing
3.2.2. Antiproliferative Activity
3.3. Antimicrobial Activity
3.3.1. Disk Diffusion Method
3.3.2. Minimal Inhibitory Concentration (MIC)
3.4. Hydrolytic Stability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salam, M.A.; Al-Amin, M.Y.; Salam, M.T.; Pawar, J.S.; Akhter, N.; Rabaan, A.A.; Alqumber, M.A.A. Antimicrobial Resistance: A Growing Serious Threat for Global Public Health. Healthcare 2023, 11, 1946. [Google Scholar] [CrossRef] [PubMed]
- Durand, G.A.; Raoult, D.; Dubourg, G. Antibiotic Discovery: History, Methods and Perspectives. Int. J. Antimicrob. Agents 2019, 53, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yang, M. Antimicrobial Peptides: From Design to Clinical Application. Antibiotics 2022, 11, 349. [Google Scholar] [CrossRef] [PubMed]
- Thayer, A.M. IMPROVING PEPTIDES: Small Firms Develop Better PEPTIDE DRUG CANDIDATES to Expand This Pharmaceutical Class and Attract Big Pharma Partners. Chem. Eng. News Arch. 2011, 89, 13–20. [Google Scholar] [CrossRef]
- Borghouts, C.; Kunz, C.; Groner, B. Current Strategies for the Development of Peptide-based Anti-cancer Therapeutics. J. Pept. Sci. 2005, 11, 713–726. [Google Scholar] [CrossRef]
- Amblard, M.; Fehrentz, J.-A.; Martinez, J.; Subra, G. Methods and Protocols of Modern Solid Phase Peptide Synthesis. Mol. Biotechnol. 2006, 33, 239–254. [Google Scholar] [CrossRef]
- Deslouches, B.; Di, Y.P. Antimicrobial Peptides with Selective Antitumor Mechanisms: Prospect for Anticancer Applications. Oncotarget 2017, 8, 46635–46651. [Google Scholar] [CrossRef]
- Javadpour, M.M.; Juban, M.M.; Lo, W.-C.J.; Bishop, S.M.; Alberty, J.B.; Cowell, S.M.; Becker, C.L.; McLaughlin, M.L. De Novo Antimicrobial Peptides with Low Mammalian Cell Toxicity. J. Med. Chem. 1996, 39, 3107–3113. [Google Scholar] [CrossRef]
- Mai, J.C.; Mi, Z.; Kim, S.H.; Ng, B.; Robbins, P.D. A Proapoptotic Peptide for the Treatment of Solid Tumors. Cancer Res. 2001, 61, 7709–7712. [Google Scholar]
- Oelkrug, C.; Hartke, M.; Schubert, A. Mode of Action of Anticancer Peptides (ACPs) from Amphibian Origin. Anticancer Res. 2015, 35, 635–643. [Google Scholar]
- Avitabile, C.; Netti, F.; Orefice, G.; Palmieri, M.; Nocerino, N.; Malgieri, G.; D’Andrea, L.D.; Capparelli, R.; Fattorusso, R.; Romanelli, A. Design, Structural and Functional Characterization of a Temporin-1b Analog Active against Gram-Negative Bacteria. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2013, 1830, 3767–3775. [Google Scholar] [CrossRef]
- Dimitrova, D.; Nemska, V.; Foteva, T.; Iliev, I.; Georgieva, N.; Danalev, D. Synthesis and Biological Studies of New Temporin A Analogs Containing Unnatural Amino Acids in Position 7. Pharmaceutics 2024, 16, 716. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Muhldorfer, I.; Ziebuhr, W.; Hacker, J. Escherichia Coli in Urinary Tract Infections. In Molecular Medical Microbiology; Elsevier: Amsterdam, The Netherlands, 2002; Volume 2, pp. 1515–1540. ISBN 978-0-12-677530-3. [Google Scholar]
- Sharma, G.; Rao, S.; Bansal, A.; Dang, S.; Gupta, S.; Gabrani, R. Pseudomonas Aeruginosa Biofilm: Potential Therapeutic Targets. Biologicals 2014, 42, 1–7. [Google Scholar] [CrossRef]
- Kim, J.; Sudbery, P. Candida Albicans, a Major Human Fungal Pathogen. J. Microbiol. 2011, 49, 171–177. [Google Scholar] [CrossRef]
- Tsonis, I.; Karamani, L.; Xaplanteri, P.; Kolonitsiou, F.; Zampakis, P.; Gatzounis, G.; Marangos, M.; Assimakopoulos, S.F. Spontaneous Cerebral Abscess Due to Bacillus subtilis in an Immunocompetent Male Patient: A Case Report and Review of Literature. World J. Clin. Cases 2018, 6, 1169–1174. [Google Scholar] [CrossRef]
- La Jeon, Y.; Yang, J.J.; Kim, M.J.; Lim, G.; Cho, S.Y.; Park, T.S.; Suh, J.-T.; Park, Y.H.; Lee, M.S.; Kim, S.C.; et al. Combined Bacillus Licheniformis and Bacillus Subtilis Infection in a Patient with Oesophageal Perforation. J. Med. Microbiol. 2012, 61, 1766–1769. [Google Scholar] [CrossRef]
- Gobbetti, M.; Rizzello, C.G. Arthrobacter. In Encyclopedia of Food Microbiology; Elsevier: Amsterdam, The Netherlands, 2014; pp. 69–76. ISBN 978-0-12-384733-1. [Google Scholar]
- Mangoni, M.L.; Shai, Y. Temporins and Their Synergism against Gram-Negative Bacteria and in Lipopolysaccharide Detoxification. Biochim. Biophys. Acta (BBA)-Biomembr. 2009, 1788, 1610–1619. [Google Scholar] [CrossRef]
- Rosenfeld, Y.; Barra, D.; Simmaco, M.; Shai, Y.; Mangoni, M.L. A Synergism between Temporins toward Gram-Negative Bacteria Overcomes Resistance Imposed by the Lipopolysaccharide Protective Layer. J. Biol. Chem. 2006, 281, 28565–28574. [Google Scholar] [CrossRef]
- Zapadka, K.L.; Becher, F.J.; Gomes Dos Santos, A.L.; Jackson, S.E. Factors Affecting the Physical Stability (Aggregation) of Peptide Therapeutics. Interface Focus. 2017, 7, 20170030. [Google Scholar] [CrossRef]
- Mercer, D.K.; Torres, M.D.T.; Duay, S.S.; Lovie, E.; Simpson, L.; Von Köckritz-Blickwede, M.; De La Fuente-Nunez, C.; O’Neil, D.A.; Angeles-Boza, A.M. Antimicrobial Susceptibility Testing of Antimicrobial Peptides to Better Predict Efficacy. Front. Cell. Infect. Microbiol. 2020, 10, 326. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, F. Cationic Amphiphilic Peptides with Cancer-Selective Toxicity. Eur. J. Pharmacol. 2009, 625, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Mader, J.S.; Hoskin, D.W. Cationic Antimicrobial Peptides as Novel Cytotoxic Agents for Cancer Treatment. Expert Opin. Investig. Drugs 2006, 15, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Marqus, S.; Pirogova, E.; Piva, T.J. Evaluation of the Use of Therapeutic Peptides for Cancer Treatment. J. Biomed. Sci. 2017, 24, 21. [Google Scholar] [CrossRef]
- Mistry, N.; Drobni, P.; Näslund, J.; Sunkari, V.G.; Jenssen, H.; Evander, M. The Anti-Papillomavirus Activity of Human and Bovine Lactoferricin. Antivir. Res. 2007, 75, 258–265. [Google Scholar] [CrossRef]
- Giese, C.; Lepthien, S.; Metzner, L.; Brandsch, M.; Budisa, N.; Lilie, H. Intracellular Uptake and Inhibitory Activity of Aromatic Fluorinated Amino Acids in Human Breast Cancer Cells. ChemMedChem 2008, 3, 1449–1456. [Google Scholar] [CrossRef]
- Danalev, D.; Borisova, D.; Yaneva, S.; Georgieva, M.; Balacheva, A.; Dzimbova, T.; Iliev, I.; Pajpanova, T.; Zaharieva, Z.; Givechev, I.; et al. Synthesis, in Vitro Biological Activity, Hydrolytic Stability and Docking of New Analogs of BIM-23052 Containing Halogenated Amino Acids. Amino Acids 2020, 52, 1581–1592. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Code | Structure | Molecular Formula | MMexact g/mol | [M + H]+ Observed g/mol | [M + Na]+ Observed g/mol | RT min | αD20 [°] ** | M.p. [°C] |
|---|---|---|---|---|---|---|---|---|---|
| 1 | DTA * | FLPLIGRVL-S-GILNH2 | C68H117N17O14 | 1395.90 | 1397.00 | 1418.95 | 4.513 | −38 | 158 ± 2 |
| 2 | DTThr | FLPLIGRVL-T-GILNH2 | C69H119N17O14 | 1409.91 | 1410.75 | 1432.70 | 4.486 | −40 | 135 ± 1 |
| 3 | DTTyr10 | FLPLIGRVL-Y-GILNH2 | C74H121N17O14 | 1471.93 | 1472.70 | 1494.70 | 4.712 | −38 | 145 ± 1 |
| 4 | DTTyr1 | Y-LPLIGRVLSGILNH2 | C68H117N17O15 | 1411.89 | 1412.60 | - | 4.177 | −58 | 123 ± 2 |
| 5 | DT4F | Phe(4F)-LPLIGRVLSGILNH2 | C68H116FN17O14 | 1413.89 | 1415.05 | 1437.10 | 4.177 | −64 | 141 ± 1 |
| Compounds | Mean CC50 ± SD (µM) | PIF ** | |
|---|---|---|---|
| −Irr | +Irr * | ||
| DTA *** | 106.32 ± 4.18 | 105.40 ± 4.88 | 1.01 |
| DTThr | 183.36 ± 2.91 | 188.42 ± 3.55 | 1.0 |
| DTTyr10 | >1000 | >1000 | - |
| DTTyr1 | 668.98 ± 13.39 | 670.76 ± 12.2 | 1.0 |
| DT4F | 55.41 ± 4.64 | 39.99 ± 0.78 | 1.4 |
| Compounds | Mean IC50 ± SD (µM) | Selectivity Index (SI) ** | |||
|---|---|---|---|---|---|
| MCF-12F | MCF-7 | MDA-MB-231 | MCF-7 | MDA-MB-231 | |
| DTA * | 138.65 ± 8.36 | 73.15 ± 3.36 | 115.13 ± 4.04 | 1.90 | 1.20 |
| DTThr | 165.09 ± 2.21 | 98.57 ± 1.19 | 136.12 ± 3.61 | 1.67 | 1.21 |
| DTTyr10 | 251.44 ± 11.81 | 64.51 ± 3.93 | 328.47 ± 2.62 | 3.9 | 0.77 |
| DTTyr1 | 317.74 ± 9.79 | 216.37 ± 10.3 | 333.55 ± 3.72 | 1.75 | 1.13 |
| DT4F | 41.78 ± 1.64 | 20.33 ± 0.60 | 32.05 ± 1.43 | 2.06 | 1.3 |
| Code | Structure | B. subtilis 3562 | A. oxydans 9333 | ||||
|---|---|---|---|---|---|---|---|
| 1.4 mg/mL | 10 mg/mL | Chloramphenicol [30 µg/disk] | 1.4 mg/mL | 10 mg/mL | Gentamicin [10 µg/disk] | ||
| DTA * | FLPLIGRVLSGILNH2 | 8.8 ± 0.3 | 8.8 ± 0.8 | 26 | 6.8 ± 0.3 | 9.7 ± 0.6 | 18 |
| DTThr | FLPLIGRVLTGILNH2 | 7.2 ± 0.3 | 0 | 24 | 8 | 10.5 ± 0.5 | 21 |
| DTTyr10 | FLPLIGRVLYGILNH2 | 0 | 0 | 27 | 0 | 0 | 23 |
| DTTyr1 | YLPLIGRVLSGILNH2 | 0 | 0 | 28 | 10 | 10 | 22 |
| DT4F | Phe(4F)LPLIGRVLSGILNH2 | 8.2 ± 0.3 | 9.7 ± 0.3 | 24 | 10 | 12.8 ± 0.3 | 20 |
| Code | Structure | E. coli 8785 | P. aeruginosa 3700 | ||||
|---|---|---|---|---|---|---|---|
| 1.4 mg/mL | 10 mg/mL | Gentamicin [10 µg/disk] | 1.4 mg/mL | 10 mg/mL | Gentamicin [10 µg/disk] | ||
| DTA * | FLPLIGRVLSGILNH2 | 0 | 0 | 18 | 7.8 ± 0.3 | 9.3 ± 0.6 | 17 |
| DTThr | FLPLIGRVLTGILNH2 | 0 | 0 | 17.5 | 0 | 0 | 17 |
| DTTyr10 | FLPLIGRVLYGILNH2 | 0 | 0 | 16.5 | 0 | 0 | 18 |
| DTTyr1 | YLPLIGRVLSGILNH2 | 0 | 0 | 17 | 0 | 0 | 16 |
| DT4F | Phe(4F)LPLIGRVLSGILNH2 | 0 | 0 | 17 | 9.2 ± 0.3 | 12.3 ± 0.6 | 18 |
| Strain | B. subtilis 3562 | A. oxydans 9333 | P. aeruginosa 3700 | ||||
|---|---|---|---|---|---|---|---|
| Code | Concentration | 1.4 mg/mL | 10 mg/mL | 1.4 mg/mL | 10 mg/mL | 1.4 mg/mL | 10 mg/mL |
| 1 | DTA * | 33.8 | 33.8 | 37.8 | 53.9 | 45.9 | 54.7 |
| 2 | DTThr | 30.0 | 0.0 | 36.4 | 47.7 | 0.0 | 0.0 |
| 3 | DTTyr10 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 4 | DTTyr1 | 0.0 | 0.0 | 47.6 | 42.9 | 0.0 | 0.0 |
| 5 | DT4F | 32.8 | 48.5 | 50.0 | 64.0 | 51.1 | 68.3 |
| Code | Structure | B. subtilis 3562 | E. coli 8785 | A. oxydans 9333 | P. aeruginosa 3700 | C. albicans 74 | |
|---|---|---|---|---|---|---|---|
| 1 | DTA * | FLPLIGRVLSGILNH2 | 80 µg/mL | 320 µg/mL | 80 µg/mL | 320 µg/mL | 320 µg/mL |
| 2 | DTThr | FLPLIGRVLTGILNH2 | 160 µg/mL | NI ** | 320 µg/mL | 320 µg/mL | NI ** |
| 3 | DTTyr10 | FLPLIGRVLYGILNH2 | 320 µg/mL | NI ** | 320 µg/mL | 320 µg/mL | NI ** |
| 4 | DTTyr1 | YLPLIGRVLSGILNH2 | 160 µg/mL | NI ** | 320 µg/mL | 320 µg/mL | NI ** |
| 5 | DT4F | Phe(4F)LPLIGRVLSGILNH2 | 80 µg/mL | NI ** | 160 µg/mL | 80 µg/mL | NI ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dimitrova, D.; Nemska, V.; Iliev, I.; Petrin, S.; Georgieva, N.; Danalev, D. New Temporin A Analogues Modified in Positions 1 and 10—Synthesis and Biological Studies. Pharmaceutics 2025, 17, 396. https://doi.org/10.3390/pharmaceutics17040396
Dimitrova D, Nemska V, Iliev I, Petrin S, Georgieva N, Danalev D. New Temporin A Analogues Modified in Positions 1 and 10—Synthesis and Biological Studies. Pharmaceutics. 2025; 17(4):396. https://doi.org/10.3390/pharmaceutics17040396
Chicago/Turabian StyleDimitrova, Dilyana, Veronica Nemska, Ivan Iliev, Stoyko Petrin, Nelly Georgieva, and Dancho Danalev. 2025. "New Temporin A Analogues Modified in Positions 1 and 10—Synthesis and Biological Studies" Pharmaceutics 17, no. 4: 396. https://doi.org/10.3390/pharmaceutics17040396
APA StyleDimitrova, D., Nemska, V., Iliev, I., Petrin, S., Georgieva, N., & Danalev, D. (2025). New Temporin A Analogues Modified in Positions 1 and 10—Synthesis and Biological Studies. Pharmaceutics, 17(4), 396. https://doi.org/10.3390/pharmaceutics17040396