

Design of Experiment Approach for Enhancing the Dissolution Profile and Robustness of Loratadine Tablet Using D-α-Tocopheryl Polyethylene Glycol 1000 Succinate
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Design of Experiment and Statistical Data Analysis
2.3. Preparation of Granules and Tablets
2.4. Evaluation of Granules
2.4.1. Granules Size Distribution
2.4.2. Flowability of Granules
2.4.3. Bulk and Taped Density
2.5. Evaluation of Tablets
2.5.1. Weight Variation
2.5.2. Uniformity of Drug Content
2.5.3. Hardness and Friability
2.5.4. Disintegration Time
2.5.5. In Vitro Dissolution
3. Results
4. Discussion
4.1. Influence of Key Variables on Average Granules Size (D50)
4.2. Influence of Key Variables on Granules’ Bulk Density
4.3. Influence of Key Variables on Granules’ Flowability
4.4. Influence of Key Variables on Tablet Strength
4.5. Influence of Key Variables on Disintegration Time
4.6. Influence of Key Variables on Tablet Dissolution
4.7. Lack-of-Fit Test
4.8. Optimization and Validation of Design
4.9. Scale-Up Considerations and Commercial Viability
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, S.; Xu, X.; Sun, W.; Zhang, Z.; Pan, B.; Hu, Q. Enteric and hydrophilic polymers enhance dissolution and absorption of poorly soluble acidic drugs based on micro-environmental pH-modifying solid dispersion. Eur. J. Pharm. Sci. 2022, 168, 106074. [Google Scholar] [CrossRef] [PubMed]
- Islam, N.; Khan, N.U.; Razzaq, A.; Menaa, F.; Khan, Z.U.; Hussain, A.; Rehman, S.F.; Iqbal, H.; Ni, J. Loratadine oral bioavailability enhancement via solid dispersion loaded oro-dispersible films: Formulation, characterization and pharmacokinetics. Colloids Surf. B. Biointerfaces 2023, 230, 113526. [Google Scholar] [CrossRef] [PubMed]
- Weerapol, Y.; Limmatvapirat, S.; Takeuchi, H.; Sriamornsak, P. Fabrication of spontaneous emulsifying powders for improved dissolution of poorly water-soluble drugs. Powder Technol. 2015, 271, 100–108. [Google Scholar] [CrossRef]
- Sotthivirat, S.; Ramesh, R.; Wasylaschuk, W.; Bottone, C.; Xia, B.; Stellabott, J.; McNevin, M.; Skomski, D.; Brown, C. Effect of TPGS surfactant on dissolution sensitivity of a poorly water-soluble drug using high-shear wet granulation. Powder Technol. 2020, 375, 302–309. [Google Scholar] [CrossRef]
- Cao, J.; Yang, B.; Wang, Y.; Wei, C.; Wang, H.; Li, S. Polymer brush hexadecyltrimethylammonium bromide (CTAB) modified poly (propylene-g-styrene sulphonic acid) fiber (ZB-1): CTAB/ZB-1 as a promising strategy for improving the dissolution and physical stability of poorly water-soluble drugs. Mater. Sci. Eng. C. 2017, 80, 282–295. [Google Scholar] [CrossRef]
- Goddeeris, C.; Willems, T.; Mooter, G.V.D. Formulation of fast disintegrating tablets of ternary solid dispersions consisting of TPGS 1000 and HPMC 2910 or PVPVA 64 to improve the dissolution of the anti-HIV drug UC 781. Eur. J. Pharm. Sci. 2008, 34, 293–302. [Google Scholar] [CrossRef]
- Benabbas, R.; Sanchez-Ballester, N.M.; Bataille, B.; Sharkawi, T.; Soulairol, I. Development and pharmaceutical performance of a novel co-processed excipient of alginic acid and microcrystalline cellulose. Powder Technol. 2021, 378, 576–584. [Google Scholar] [CrossRef]
- Jin, F.; Tatavarti, A. Tabletability assessment of conventional formulations containing Vitamin E tocopheryl polyethylene glycol succinate. Int. J. Pharm. 2010, 389, 58–65. [Google Scholar] [CrossRef]
- Uchiyama, H.; Tozuka, Y.; Nishikawa, M.; Takeuchi, H. Nanocomposite formation between alpha-glucosyl stevia and surfactant improves the dissolution profile of poorly water-soluble drug. Int. J. Pharm. 2012, 428, 183–186. [Google Scholar] [CrossRef]
- Guo, Y.; Luo, J.; Tan, S.; Otieno, B.O.; Zhang, Z. The applications of Vitamin E TPGS in drug delivery. Eur. J. Pharm. Sci. 2013, 49, 175–186. [Google Scholar] [CrossRef]
- Sotthivirat, S.; Ren, J.; Wasylaschuk, W.; Afanador, N.; Rosenberg, K.; Taggart, R.V.; Skomski, D.; Brown, C. An Integrated Approach for High-Shear Wet Granulation (HSWG) Processing of TPGS-Based Formulations: Demonstration of Process Robustness through Experimental Design Conditions. J. Pharm. Sci. 2021, 110, 2934–2945. [Google Scholar] [CrossRef] [PubMed]
- Kumbhar, P.S.; Nadaf, S.; Manjappa, A.S.; Jha, N.K.; Shinde, S.S.; Chopade, S.S.; Shete, A.S.; Disouza, J.I.; Sambamoorthy, U.; Kumar, S.A. D-ɑ-tocopheryl polyethylene glycol succinate: A review of multifarious applications in nanomedicines. OpenNano 2022, 6, 100036. [Google Scholar] [CrossRef]
- Cho, H.-J.; Lee, J.-Y.; Kang, W.-S.; Piao, J.; Yoon, I.-S.; Kim, D.-D. Soluplus®/TPGS-based solid dispersions prepared by hot-melt extrusion equipped with twin-screw systems for enhancing oral bioavailability of valsartan. Drug Des. Dev. Ther. 2015, 9, 2745–2756. [Google Scholar] [CrossRef] [PubMed]
- Arregui, J.R.; Kovvasu, S.P.; Kunamaneni, P.; Betageri, G.V. Carvedilol solid dispersion for enhanced oral bioavailability using rat model. J. Appl. Pharm. Sci. 2019, 9, 42–50. [Google Scholar]
- Leskinen, J.T.T.; Simonaho, S.-P.; Hakulinen, M.; Ketolainen, J. In-line ultrasound measurement system for detecting tablet integrity. Int. J. Pharm. 2010, 400, 104–113. [Google Scholar] [CrossRef]
- Frizon, F.; Eloy, J.d.O.; Donaduzzi, C.M.; Mitsui, M.L.; Marchetti, J.M. Dissolution rate enhancement of loratadine in polyvinylpyrrolidone K-30 solid dispersions by solvent methods. Powder Technol. 2013, 235, 532–539. [Google Scholar] [CrossRef]
- Omari, S.; Ashour, E.A.; Elkanayati, R.; Alyahya, M.; Almutairi, M.; Repka, M.A. Formulation development of loratadine immediate- release tablets using hot-melt extrusion and 3D printing technology. J. Drug. Deliver. Sci. Technol. 2022, 74, 103505. [Google Scholar] [CrossRef]
- Emami, M.S.; Haghshenasfard, M.; Zarghami, R.; Sadeghi, R.; Esfahany, M.N. Experimental study on the reduction of loratadine particle size through confined liquid impinging jets. Int. J. Pharm. 2020, 587, 119668. [Google Scholar] [CrossRef]
- AlAli, A.S.; Aldawsari, M.F.; Alalaiwe, A.; Almutairy, B.K.; Al-Shdefat, R.; Walbi, I.S.; Fayed, M.H. Exploitation of Design-of-Experiment Approach for Design and Optimization of Fast-Disintegrating Tablets for Sublingual Delivery of Sildenafil Citrate with Enhanced Bioavailability Using Fluid-Bed Granulation Technique. Pharmaceutics 2021, 13, 870. [Google Scholar] [CrossRef]
- Khafagy, E.-S.; Fayed, M.H.; Alrabahi, S.H.; Gad, S.; Alshahrani, S.M.; Aldawsari, M. Defining design space for optimization of escitalopram ultra-fast melting tablet using suspension spray-coating technique: In-vitro and in-vivo evaluation. J. Drug. Deliver. Sci. Technol. 2020, 57, 101631. [Google Scholar] [CrossRef]
- Aodah, A.H.; Fayed, M.H.; Alalaiwe, A.; Alsulays, B.B.; Aldawsari, M.F.; Khafagy, E.S. Design, Optimization, and Correlation of In Vitro/In Vivo Disintegration of Novel Fast Orally Disintegrating Tablet of High Dose Metformin Hydrochloride Using Moisture Activated Dry Granulation Process and Quality by Design Approach. Pharmaceutics 2020, 12, 598. [Google Scholar] [CrossRef] [PubMed]
- Alalaiwe, A.; Fayed, M.H.; Alshahrani, S.M.; Alsulays, B.B.; Alshetaili, A.S.; Tawfeek, H.M.; Khafagy, E.-S. Application of design of experiment approach for investigating the effect of partially pre-gelatinized starch on critical quality attributes of rapid orally disintegrating tablets. J. Drug. Deliver. Sci. Technol. 2019, 49, 227–234. [Google Scholar] [CrossRef]
- Rampado, R.; Peer, D. Design of experiments in the optimization of nanoparticle-based drug delivery systems. J. Control. Release 2023, 358, 398–419. [Google Scholar] [CrossRef] [PubMed]
- Putra, O.N.; Musfiroh, I.; Elisa, S.; Musa, M.; Ikram, E.H.K.; Chaidir, C.; Muchtaridi, M. Sodium Starch Glycolate (SSG) from Sago Starch (Metroxylon sago) as a Superdisintegrant: Synthesis and Characterization. Molecules 2023, 29, 151. [Google Scholar] [CrossRef]
- Fayed, M.H.; Aldawsari, M.F.; AlAli, A.S.; Alsaqr, A.; Almutairy, B.K.; Aodah, A.H.; Tawfeek, H.M.; Khafagy, E.-S.; Helal, D.A. Design-of-experiment approach to quantify the effect of nano-sized silica on tableting properties of microcrystalline cellulose to facilitate direct compression tableting of binary blend containing a low-dose drug. J. Drug. Deliver. Sci. Technol. 2022, 68, 103127. [Google Scholar] [CrossRef]
- USP 38-NF-33; United States Pharmacopeial. United States Pharmacopeial Convention: Rockville, MD, USA, 2015.
- El-Sherbiny, D.T.; El-Enany, N.; Belal, F.F.; Hansen, S.H. Simultaneous determination of loratadine and desloratadine in pharmaceutical preparations using liquid chromatography with a microemulsion as eluent. J. Pharm. Biomed. Anal. 2007, 43, 1236–1242. [Google Scholar] [CrossRef]
- Suknuntha, K.; Khumpirapang, N.; Tantishaiyakul, V.; Okonogi, S. Solubility and Physical Stability Enhancement of Loratadine by Preparation of Co-Amorphous Solid Dispersion with Chlorpheniramine and Polyvinylpyrrolidone. Pharmaceutics 2023, 15, 2558. [Google Scholar] [CrossRef]
- Li, J.; Zhou, Y.; Aisha, M.; Wu, J.; Wang, H.; Huang, F.; Sun, M. Preparation of loratadine nanocrystal tablets to improve the solubility and dissolution for enhanced oral bioavailability. J. Pharm. Pharmacol. 2021, 73, 937–946. [Google Scholar] [CrossRef]
- Galata, D.L.; Zsiros, B.; Knyihár, G.; Péterfi, O.; Mészáros, L.A.; Ronkay, F.; Nagy, B.; Szabó, E.; Nagy, Z.K.; Farkas, A. Convolutional neural network-based evaluation of chemical maps obtained by fast Raman imaging for prediction of tablet dissolution profiles. Int. J. Pharm. 2023, 640, 123001. [Google Scholar] [CrossRef]
- Pandey, P.; Sinko, P.D.; Bindra, D.S.; Hamey, R.; Gour, S.; Vema-Varapu, C. Processing challenges with solid dosage formulations containing vitamin E TPGS. Pharm. Dev. Technol. 2013, 18, 296–304. [Google Scholar] [CrossRef]
- Macho, O.; Gabrišová, Ľ.; Guštafík, A.; Jezso, K.; Juriga, M.; Kabát, J.; Blaško, J. The Influence of Wet Granulation Parameters on the Compaction Behavior and Tablet Strength of a Hydralazine Powder Mixture. Pharmaceutics 2023, 15, 2148. [Google Scholar] [CrossRef] [PubMed]
- Arndt, O.-R.; Baggio, R.; Adam, A.K.; Harting, J.; Franceschinis, E.; Kleinebudde, P. Impact of Different Dry and Wet Granulation Techniques on Granule and Tablet Properties: A Comparative Study. J. Pharm. Sci. 2018, 107, 3143–3152. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Tao, J.; Chaudhury, A.; Ramachandran, R.; Gao, J.Z.; Bindra, D.S. A combined experimental and modeling approach to study the effects of high-shear wet granulation process parameters on granule characteristics. Pharm. Dev. Technol. 2013, 18, 210–224. [Google Scholar] [CrossRef] [PubMed]
- Ke, W.-T.; Lin, S.-Y.; Ho, H.-O.; Sheu, M.-T. Physical characterizations of microemulsion systems using tocopheryl polyethylene glycol 1000 succinate (TPGS) as a surfactant for the oral delivery of protein drugs. J. Control. Release 2005, 102, 489–507. [Google Scholar] [CrossRef]
- Kalam, M.A.; Alhowyan, A.A.; Alhudaithi, S.S.; Khan, M.S.; Alshememry, A.K.; Alkholief, M. Solid Dispersions of Gefitinib with D-α-Tocopherol Polyethylene Glycol-1000 Succinate and 2-Hydroxypropyl β-Cyclodextrin Complex Improved Their Solubility, Dissolution and Apoptosis against A549 Cells. Appl. Sciences 2023, 13, 10859. [Google Scholar] [CrossRef]
- Zhang, Z.; Tan, S.; Feng, S.-S. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials 2012, 33, 4889–4906. [Google Scholar] [CrossRef]
- Mishra, S.M.; Rohera, B.D. An integrated, quality by design (QbD) approach for design, development and optimization of orally disintegrating tablet formulation of carbamazepine. Pharm. Dev. Technol. 2017, 22, 889–903. [Google Scholar] [CrossRef]
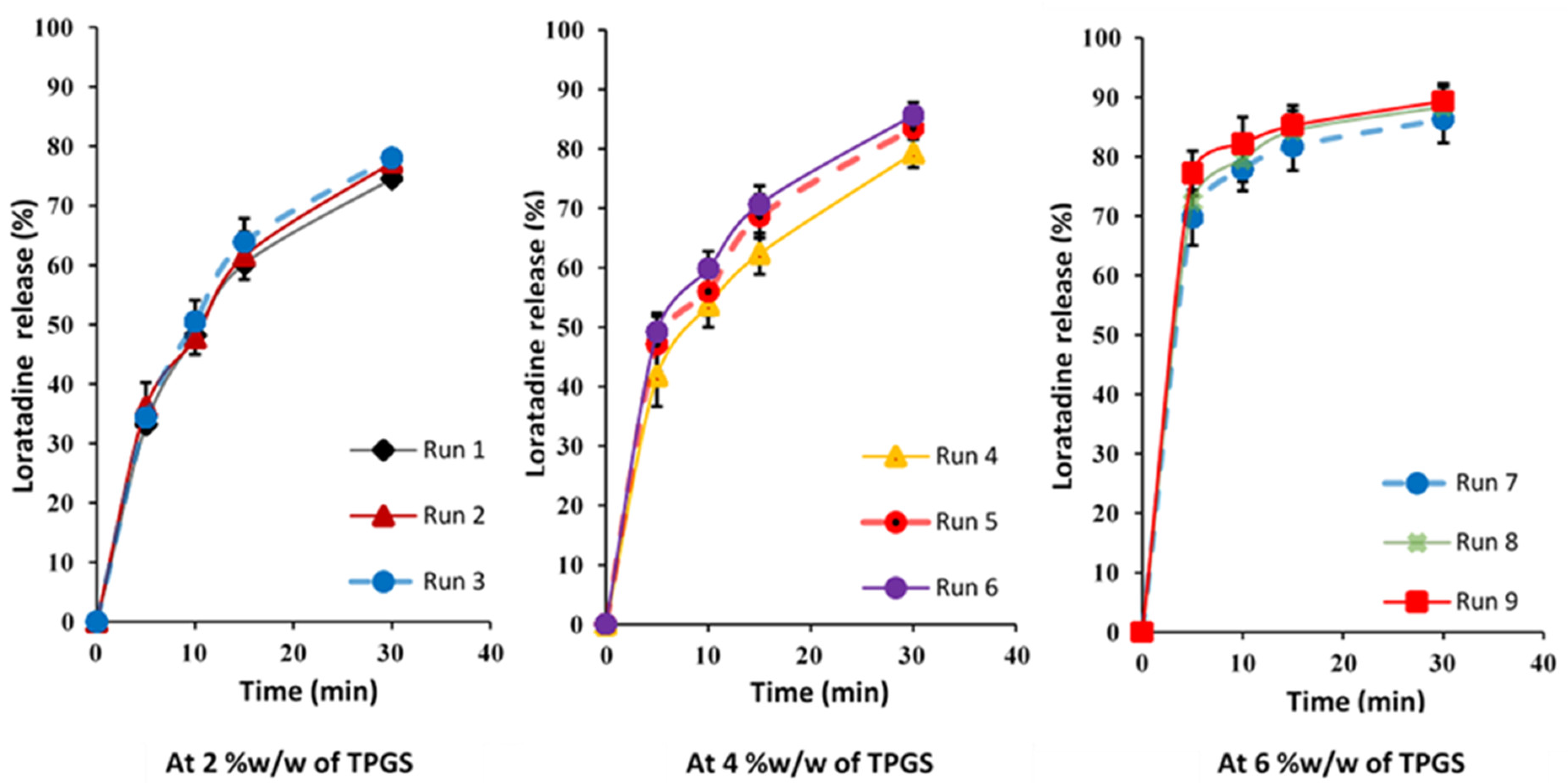
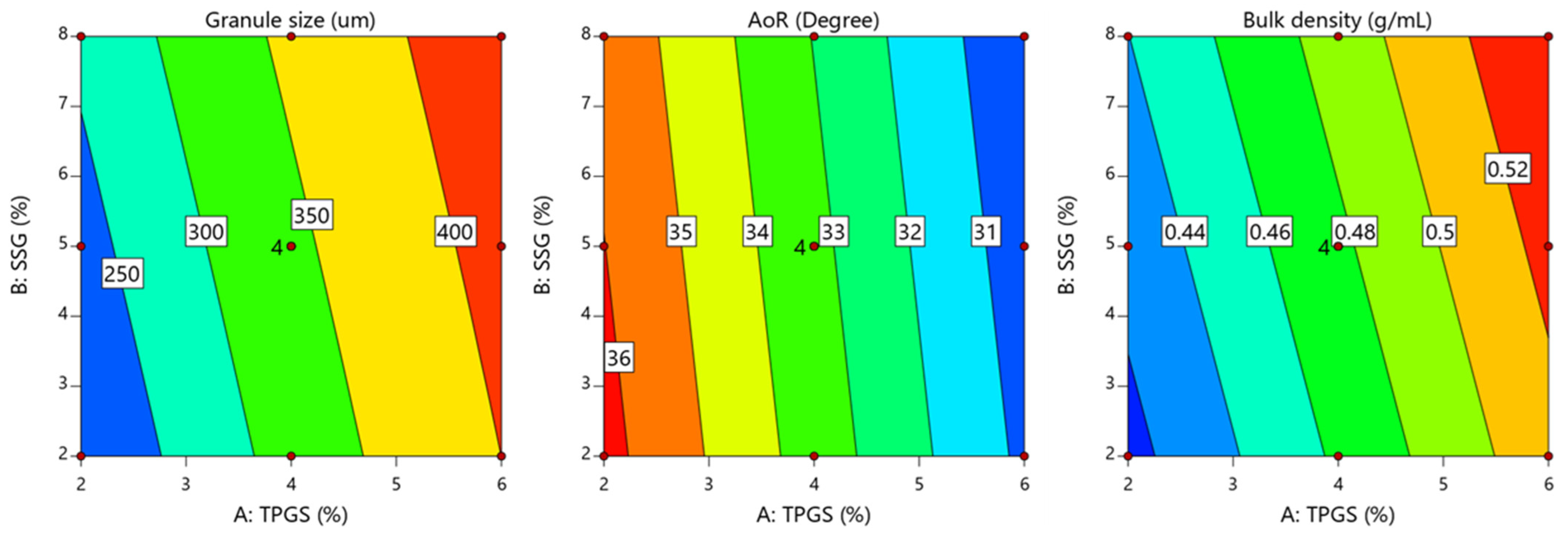



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run | TPGS (% w/w) | SSG (% w/w) |
|---|---|---|
| 1 | 2 | 2 |
| 2 | 2 | 5 |
| 3 | 2 | 8 |
| 4 | 4 | 2 |
| 5 | 4 | 5 |
| 5 * | 4 | 5 |
| 5 * | 4 | 5 |
| 5 * | 4 | 5 |
| 6 | 4 | 8 |
| 7 | 6 | 2 |
| 8 | 6 | 5 |
| 9 | 6 | 8 |
| Ingredients | Function | % w/w |
|---|---|---|
| Loratadine | Drug | 5 |
| TPGS | Surfactant | 2–6 |
| Sodium starch glycolate | Superdisintegrant | 2–8 |
| * Microcrystalline cellulose PH-102 | Filler | Up to 100 |
| * Lactose monohydrate | Filler | Up to 100 |
| Magnesium stearate | Hydrophobic lubricant | 1 |
| Total | - | 100 |
| Responses | Suggested Model | p-Value | R2 | Adjusted R2 | Predicted R2 | Adequate Precision |
|---|---|---|---|---|---|---|
| Y1: * D50 | Quadratic | <0.0001 | 0.9950 | 0.9908 | 0.9759 | 47.31 |
| Y2: Bulk density | Linear | <0.0001 | 0.8979 | 0.8753 | 0.8172 | 17.74 |
| Y3: Angle of repose | Linear | <0.0001 | 0.9846 | 0.9812 | 0.9779 | 43.16 |
| Y4: Crushing strength | Quadratic | <0.0001 | 0.9920 | 0.9853 | 0.9336 | 28.52 |
| Y5: Disintegration time | Quadratic | <0.0001 | 0.9892 | 0.9803 | 0.9715 | 30.58 |
| Y6: Percent release after 30 min | Linear | <0.0001 | 0.9331 | 0.9183 | 0.8529 | 25.05 |
| Formula | Mean Granule Size (µm ± SD) | Bulk Density (g/mL ± SD) | Tapped Density (g/mL ± SD) | Angle of Repose (Degree ± SD) | Carr’s Index | Hausner’s Ratio |
|---|---|---|---|---|---|---|
| 1 | 206.13 ± 0.35 | 0.413 ± 0.014 | 0.510 ± 0.005 | 36.21 ± 0.321 | 19 | 1.23 |
| 2 | 224.71 ± 0.32 | 0.431 ± 0.006 | 0.502 ± 0.009 | 35.89 ± 0.423 | 14.1 | 1.16 |
| 3 | 262.15 ± 0.21 | 0.452 ± 0.032 | 0.518 ± 0.019 | 35.57 ± 0.127 | 12.7 | 1.14 |
| 4 | 311.56 ± 0.26 | 0.463 ± 0.034 | 0.542 ± 0.007 | 33.46 ± 0.615 | 14.5 | 1.17 |
| 5 | 331.25 ± 0.21 | 0.461 ± 0.007 | 0.533 ± 0.015 | 33.54 ± 0.247 | 13.5 | 1.15 |
| 5 * | 346.11 ± 0.65 | 0.458 ± 0.033 | 0.521 ± 0.0024 | 33.11 ± 0.442 | 12.1 | 1.13 |
| 5 * | 344.21 ± 0.24 | 0.456 ± 0.008 | 0.513 ± 0.017 | 33.78 ± 0.361 | 11.1 | 1.12 |
| 5 * | 343.54 ± 0.32 | 0.486 ± 0.047 | 0.561 ± 0.031 | 33.61 ± 0.431 | 13.3 | 1.15 |
| 6 | 359.11 ± 0.45 | 0.501 ± 0.019 | 0.568 ± 0.003 | 32.84 ± 0.392 | 11.8 | 1.13 |
| 7 | 401.75 ± 0.64 | 0.532 ± 0.020 | 0.587 ± 0.012 | 30.77 ± 0.331 | 9.3 | 1.10 |
| 8 | 412.55 ± 0.25 | 0.528 ± 0.022 | 0.579 ± 0.043 | 30.11 ± 0.214 | 8.8 | 1.09 |
| 9 | 423.64 ± 0.81 | 0.533 ± 0.110 | 0.591 ± 0.027 | 30.23 ± 0.325 | 9.8 | 1.11 |
| Run | Weight (mg ± SD) | Drug Content (% ± SD) | Crushing Strength (N ± SD) | Friability (% ± SD) | Disintegration Time (S ± SD) | %Release at 30 min (% ± SD) |
|---|---|---|---|---|---|---|
| 1 | 200.6 ± 0.951 | 100.12 ± 1.31 | 68.352 ± 0.215 | 0.09 ± 0.03 | 7.11 ± 1.13 | 74.53 ± 1.15 |
| 2 | 201.03 ± 0.624 | 96.70 ± 0.13 | 68.058 ± 0.133 | 0.02 ± 0.06 | 4.15 ± 1.33 | 77.24 ± 1.14 |
| 3 | 200.59 ± 0.943 | 101.23 ± 1.12 | 61.095 ± 0.139 | 0.03 ± 0.02 | 2.35 ± 1.28 | 78.04 ± 1.13 |
| 4 | 201.0 5 ± 0.701 | 100.66 ± 1.6 | 68.254 ± 0.235 | 0.03 ± 0.01 | 7.5 ± 1.63 | 79.25 ± 2.36 |
| 5 | 200.96 ± 0.829 | 98.3 ± 1.61 | 69.431 ± 0.211 | 0.17 ± 0.05 | 5.38 ± 1.61 | 83.47 ± 1.82 |
| 5 * | 200.81 ± 0.853 | 101.54 ± 0.88 | 68.745 ± 0.210 | 0.04 ± 0.03 | 4.25 ± 1.32 | 82.79 ± 2.41 |
| 5 * | 200.47 ± 0.683 | 100.88 ± 1.63 | 67.568 ± 0.237 | 0.09 ± 0.02 | 5.33 ± 2.18 | 83.14 ± 2.84 |
| 5 * | 201.08 ± 0.755 | 98.76 ± 2.01 | 67.764 ± 0.209 | 0.06 ± 0.01 | 4.35 ± 0.57 | 82.91 ± 1.21 |
| 6 | 200.77 ± 1.06 | 100.93 ± 0.76 | 68.745 ± 0.342 | 0.15 ± 0.03 | 4.14 ± 0.81 | 85.63 ± 2.17 |
| 7 | 200.79 ± 0.871 | 100.4 ± 0.8 | 41.286 ± 0.233 | 0.08 ± 0.01 | 12.31 ± 0.36 | 83.52 ± 2.33 |
| 8 | 200.84 ± 0.739 | 101.3 ± 1.7 | 41.776 ± 0.257 | 0.16 ± 0.03 | 11.02 ± 0.84 | 85.11 ± 3.11 |
| 9 | 200.61 ± 0.828 | 100.1 ± 1.5 | 43.836 ± 0.353 | 0.12 ± 0.01 | 11.45 ± 0.68 | 91.52 ± 1.98 |
| Variables | Coefficient Estimate | Sum of Squares | Standard Error | * F-Value | p-Value |
|---|---|---|---|---|---|
| Granule size (Quadratic model) | |||||
| Intercept | 339.49 | - | 2.05 | - | - |
| X1 | 90.82 | 49,495.08 | 2.73 | 1109.18 | <0.0001 |
| X2 | 20.91 | 2623.37 | 2.73 | 58.79 | 0.0003 |
| X1X2 | –8.53 | 291.21 | 3.34 | 6.53 | 0.0432 |
| X12 | –17.28 | 796.49 | –17.28 | 17.85 | 0.0055 |
| X22 | –0.5775 | 0.8893 | –0.5775 | 0.0199 | 0.8924 |
| Granules bulk density (Linear model) | |||||
| Intercept | 0.4762 | – | 0.0041 | – | – |
| X1 | 0.0495 | 0.0147 | 0.0058 | 74.08 | <0.0001 |
| X2 | 0.013 | 0.0010 | 0.0058 | 5.11 | 0.0501 |
| Granule flowability (Linear model) | |||||
| Intercept | 33.26 | - | 0.0819 | - | - |
| X1 | –2.76 | 45.71 | 0.1158 | 568.48 | <0.0001 |
| X2 | –0.30 | 0.54 | 0.1158 | 6.72 | 0.0291 |
| Variables | Coefficient Estimate | Sum of Squares | Standard Error | * F-Value | p-Value |
|---|---|---|---|---|---|
| Crushing strength (Quadratic model) | |||||
| Intercept | 7.0 | - | 0.06 | - | - |
| X1 | –1.2 | 8.64 | 0.05 | 417.91 | <0.0001 |
| X2 | –0.071 | 0.03 | 0.05 | 1.49 | 0.267 |
| X1X2 | 0.25 | 0.25 | 0.07 | 12.09 | 0.013 |
| X12 | –1.44 | 5.56 | 0.0881 | 268.86 | <0.0001 |
| X22 | –0.058 | 0.0092 | 0.0881 | 0.4452 | 0.529 |
| Disintegration time (Quadratic model) | |||||
| Intercept | 4.87 | - | 0.212 | - | - |
| X1 | 3.53 | 74.69 | 0.189 | 345.88 | <0.0001 |
| X2 | –1.5 | 13.44 | 0.189 | 62.23 | 0.0002 |
| X1 X2 | 0.975 | 3.8 | 0.232 | 17.61 | 0.0057 |
| X12 | 2.62 | 18.32 | 0.2846 | 84.84 | <0.0001 |
| X22 | 0.856 | 1.96 | 0.2846 | 9.05 | 0.0237 |
| Percent release at 30 min (Linear model) | |||||
| Intercept | 82.26 | - | 0.37 | - | - |
| X1 | 5.06 | 153.42 | 0.52 | 93.19 | <0.0001 |
| X2 | 2.98 | 53.34 | 0.52 | 32.4 | 0.0003 |
| Response | * F-Value | p-Value | Comment |
|---|---|---|---|
| d50 | 0.945 | 0.517 | Not significant |
| bulk density | 1.02 | 0.537 | Not significant |
| Flowability | 0.984 | 0.551 | Not significant |
| Crushing strength | 4.24 | 0.133 | Not significant |
| Disintegration time | 0.157 | 0.918 | Not significant |
| Percent release at 30 min | 27.07 | 0.01 | Significant |
| Variables | Target | Range | Weight | Importance Coefficient |
|---|---|---|---|---|
| Input | ||||
| TPGS | In range | 2–6% w/w | 1 | NA |
| SSG | In range | 2–8% w/w | 1 | NA |
| Output | ||||
| Crushing strength (N) | 58.840 | 41.286–69.431 | +++ | |
| Disintegration time (Min) | 5 | 2.35–12.31 | 1 | +++ |
| Percent release at 30 min (%) | 85 | 74.53–91.52 | 1 | +++ |
| Variables | Value | ||
|---|---|---|---|
| TPGS (2–6% w/w) | 5.02 | ||
| SSG (2–8% w/w) | 5.13 | ||
| Overall desirability = 0.880 | |||
| Responses | Predicted values | Experimental values * | Prediction error (%) |
| Crushing strength (N) | 58.84 | 56.094 ± 0.32 | 4.66 |
| Disintegration time (Min.) | 7.33 | 7.11 ± 1.08 | 3.001 |
| Percent release at 30 min (%) | 85.0 | 86.21 ± 1.61 | –1.42 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jabbar, A.A.; Al-Ani, I.; Al-Shdefat, R.I.; Ghazal, N.; Jaffal, A.; Fayed, M.H. Design of Experiment Approach for Enhancing the Dissolution Profile and Robustness of Loratadine Tablet Using D-α-Tocopheryl Polyethylene Glycol 1000 Succinate. Pharmaceutics 2025, 17, 380. https://doi.org/10.3390/pharmaceutics17030380
Jabbar AA, Al-Ani I, Al-Shdefat RI, Ghazal N, Jaffal A, Fayed MH. Design of Experiment Approach for Enhancing the Dissolution Profile and Robustness of Loratadine Tablet Using D-α-Tocopheryl Polyethylene Glycol 1000 Succinate. Pharmaceutics. 2025; 17(3):380. https://doi.org/10.3390/pharmaceutics17030380
Chicago/Turabian StyleJabbar, Alhasan A., Israa Al-Ani, Ramadan I. Al-Shdefat, Nadia Ghazal, Anwar Jaffal, and Mohamed H. Fayed. 2025. "Design of Experiment Approach for Enhancing the Dissolution Profile and Robustness of Loratadine Tablet Using D-α-Tocopheryl Polyethylene Glycol 1000 Succinate" Pharmaceutics 17, no. 3: 380. https://doi.org/10.3390/pharmaceutics17030380
APA StyleJabbar, A. A., Al-Ani, I., Al-Shdefat, R. I., Ghazal, N., Jaffal, A., & Fayed, M. H. (2025). Design of Experiment Approach for Enhancing the Dissolution Profile and Robustness of Loratadine Tablet Using D-α-Tocopheryl Polyethylene Glycol 1000 Succinate. Pharmaceutics, 17(3), 380. https://doi.org/10.3390/pharmaceutics17030380