
Physiologically Based Pharmacokinetic Models for Infliximab, Ipilimumab, and Nivolumab Developed with GastroPlus to Predict Hepatic Concentrations

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. PBPK Modeling Software
2.2. Infliximab PBPK Model Development
2.2.1. TNF-α Representation in Infliximab PBPK Model
2.2.2. PBPK Model Development for Infliximab
2.2.3. Accounting for the Development of Anti-Drug Antibodies (ADAs)
2.3. Ipilimumab PBPK Model Development
2.3.1. CTLA-4 Representation in Ipilimumab PBPK Model
2.3.2. PBPK Model Development for Ipilimumab
2.4. Nivolumab PBPK Model Development
2.4.1. PD-1 Representation in Nivolumab PBPK Model
2.4.2. PBPK Model Development for Nivolumab
2.5. Linear Correlation Model Used to Predict Liver Concentration from Plasma Concentration as Validation for the PBPK Models
3. Results
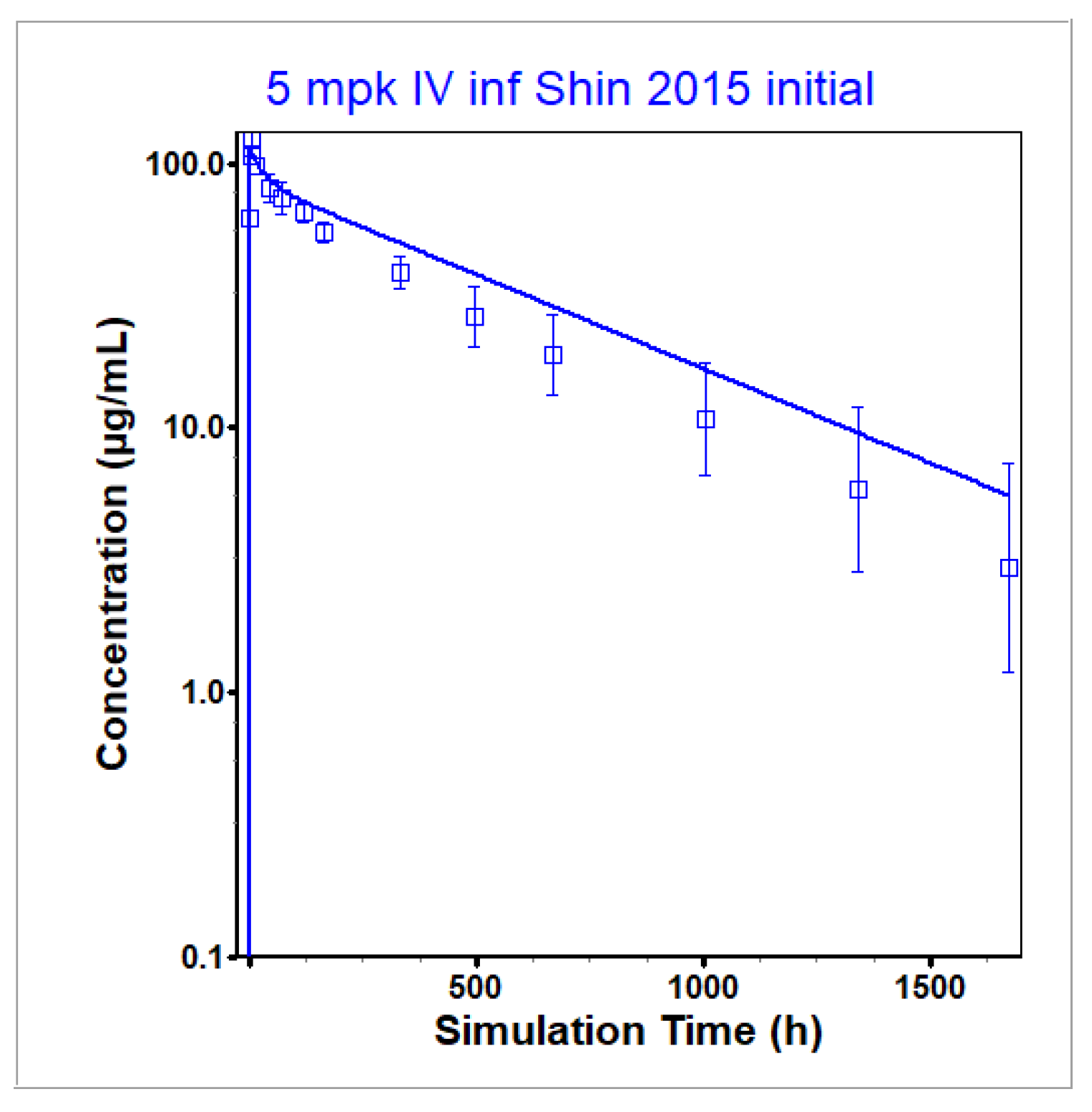
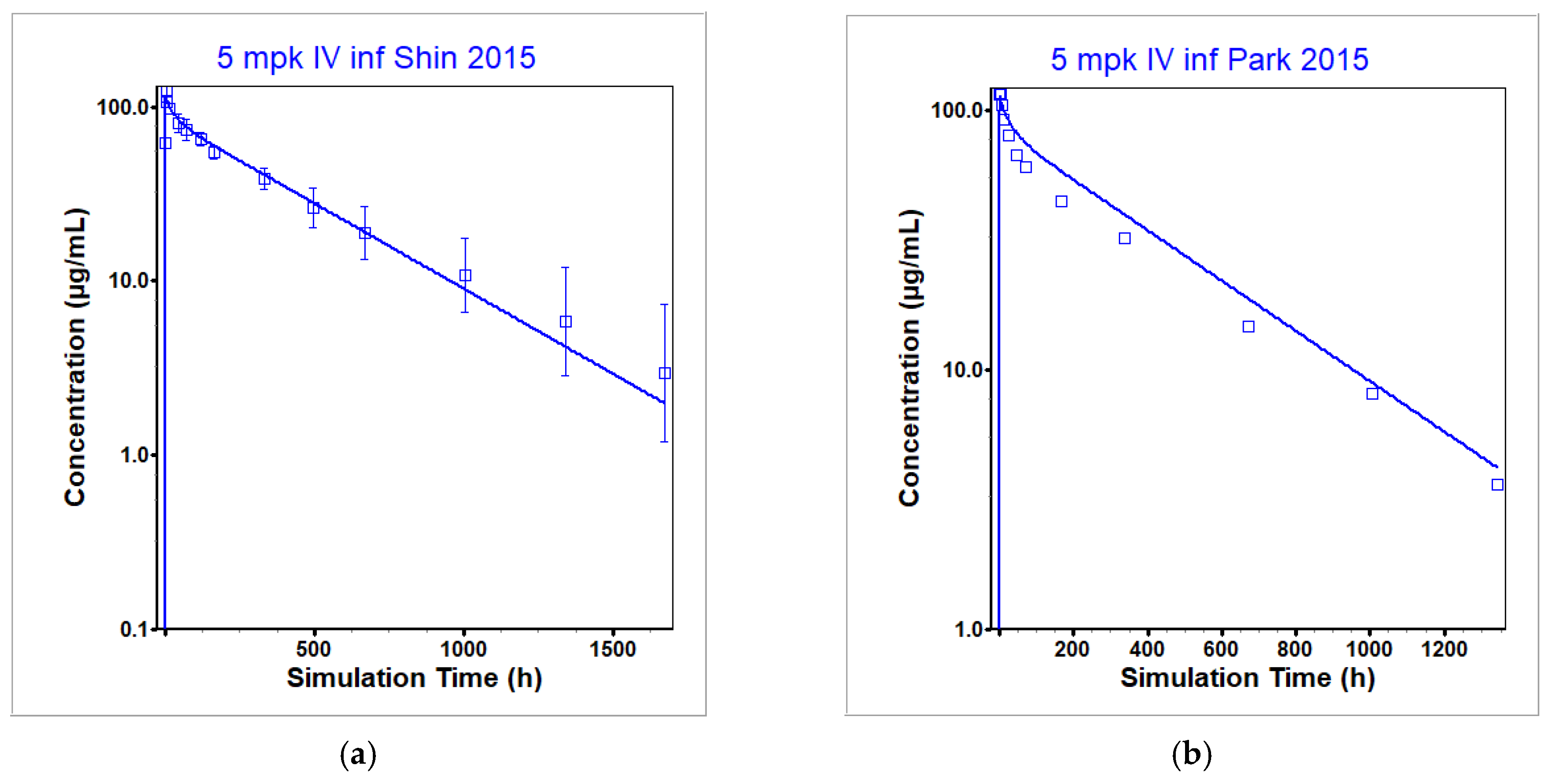
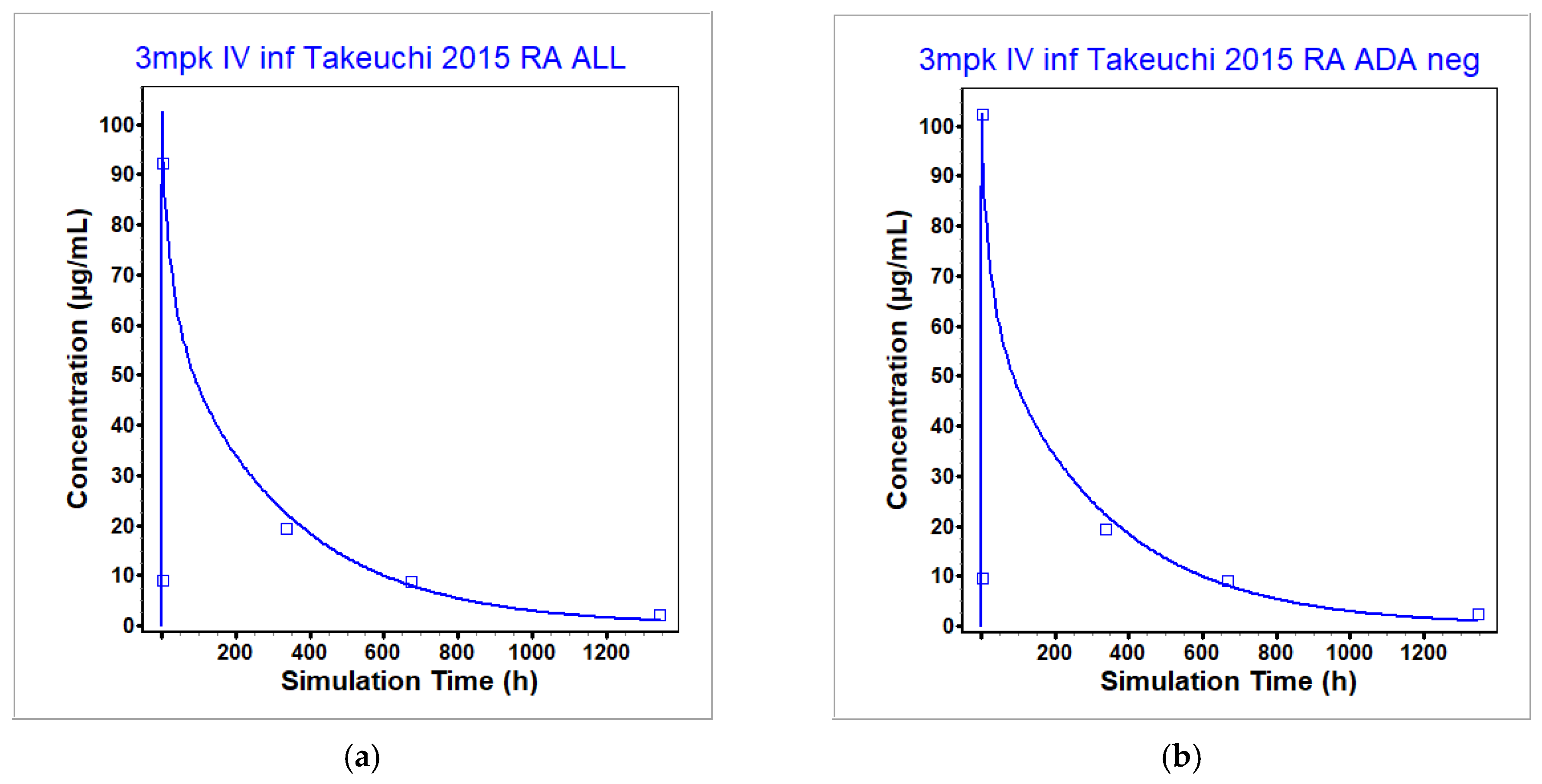
3.1. Infliximab PBPK Model
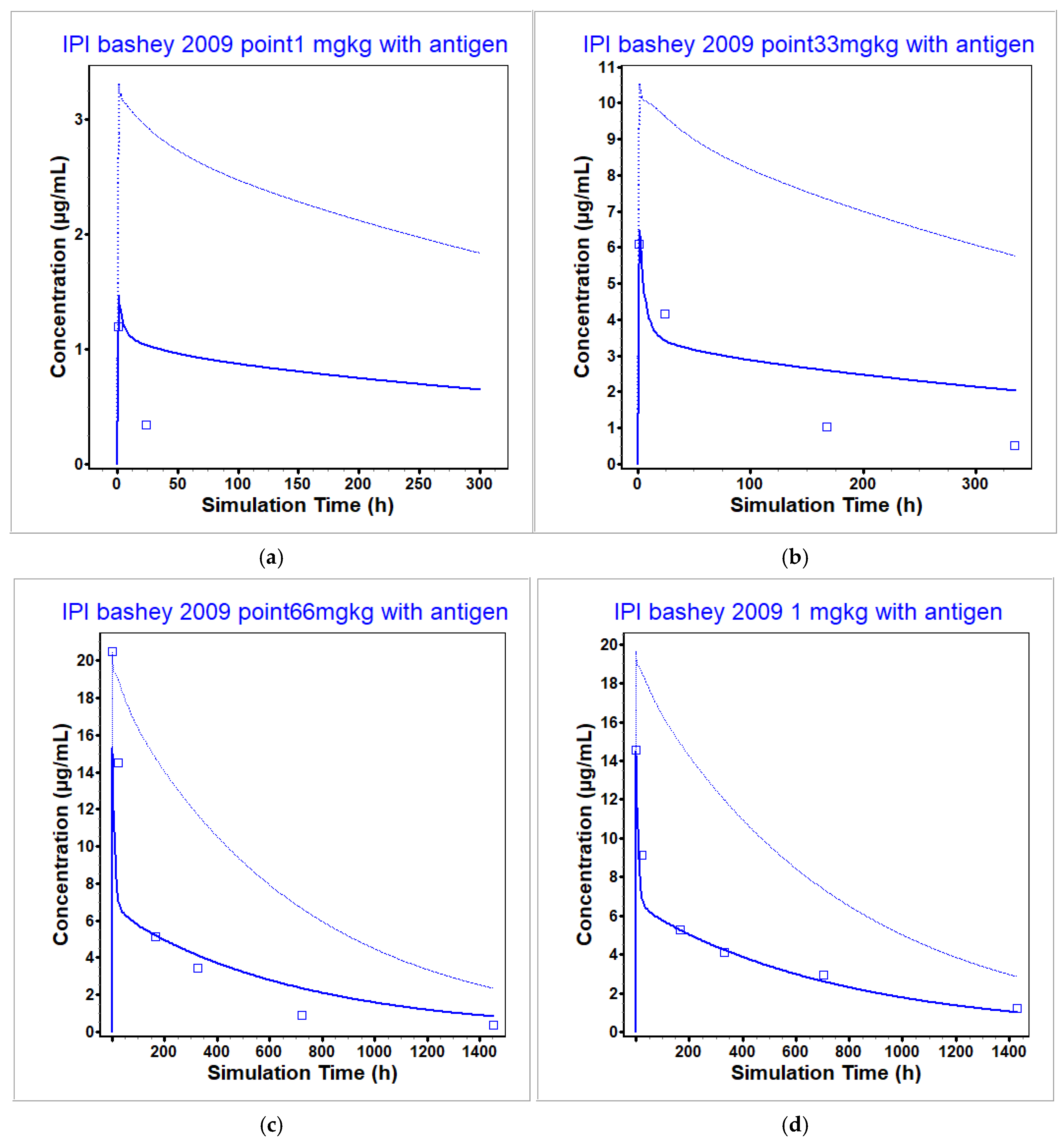
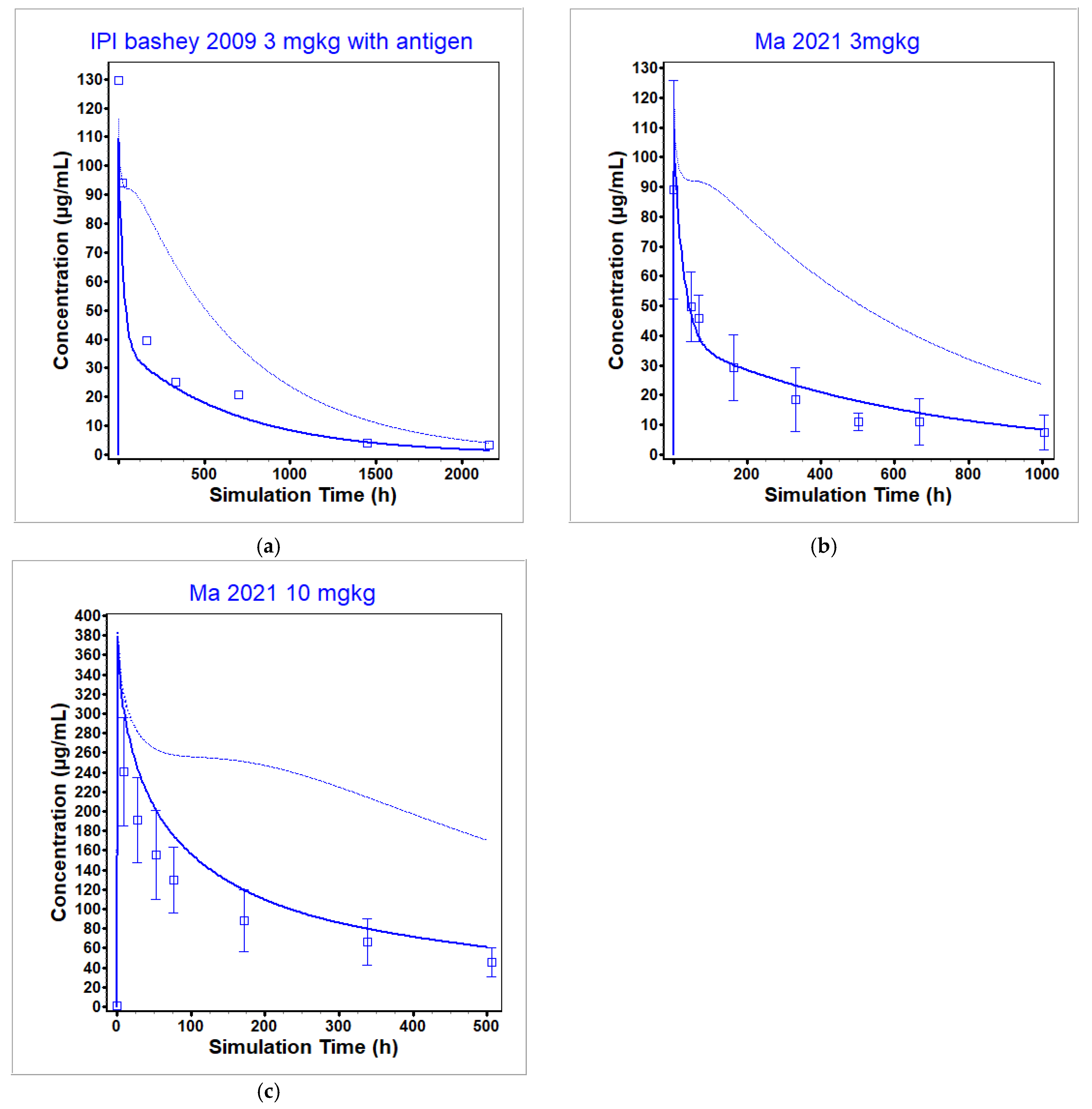
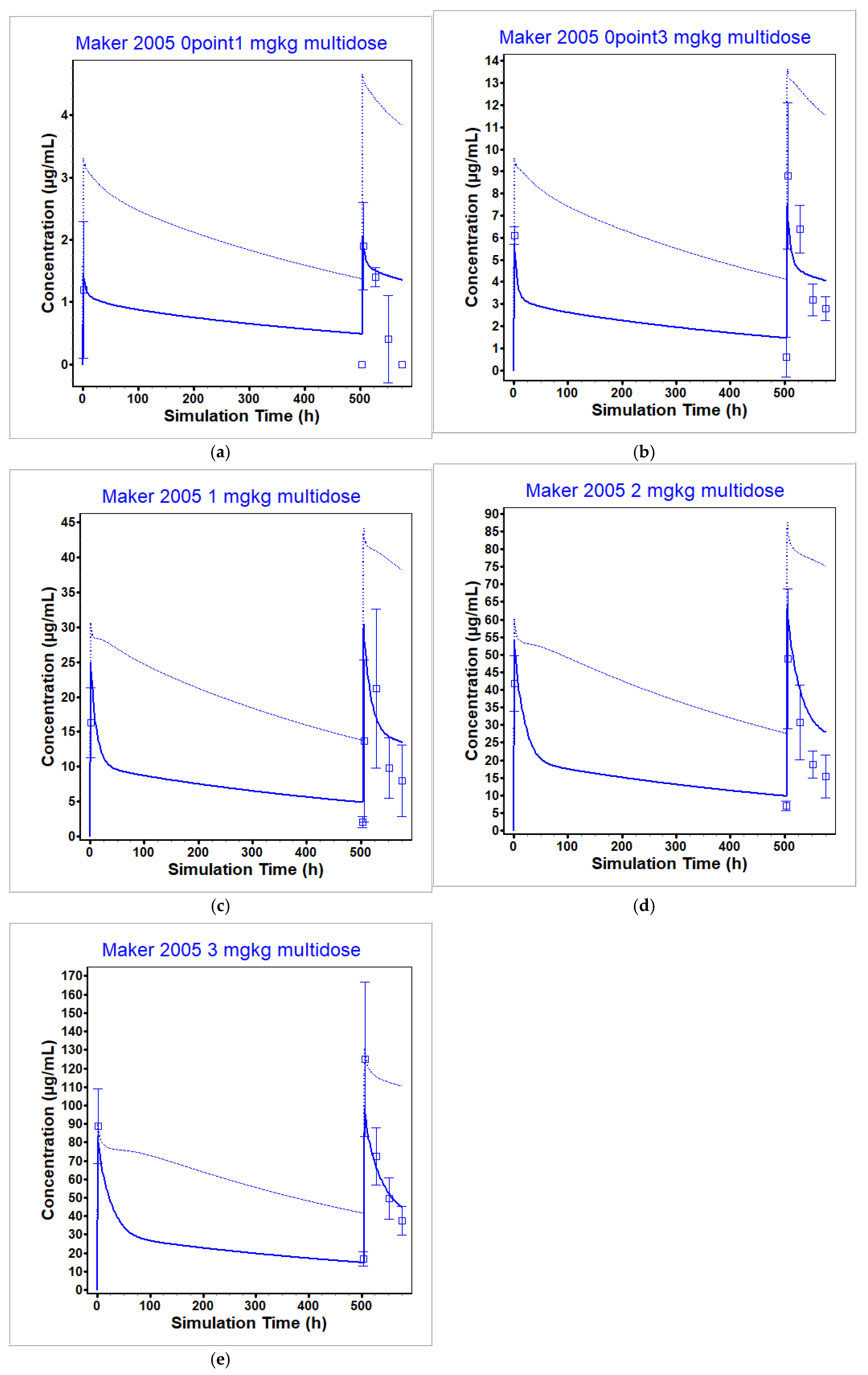
3.2. Ipilimumab PBPK Model
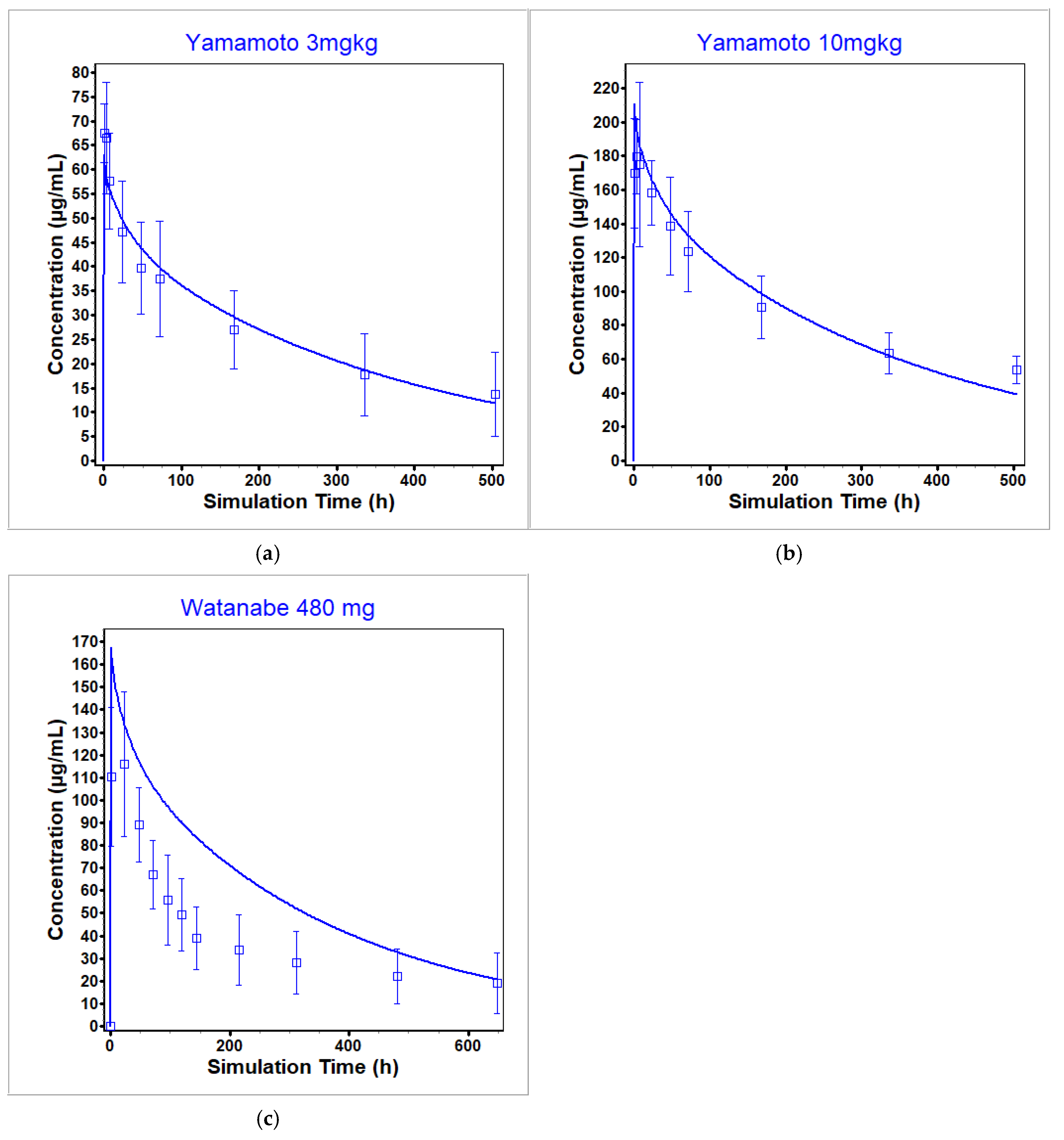
3.3. Nivolumab PBPK Model
3.4. Predicted Interstitial Liver Concentrations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Beaudoin, J.J.; Clemens, L.; Miedel, M.T.; Gough, A.; Zaidi, F.; Ramamoorthy, P.; Wong, K.E.; Sarangarajan, R.; Battista, C.; Shoda, L.K.M.; et al. The Combination of a Human Biomimetic Liver Microphysiology System with BIOLOGXsym, a Quantitative Systems Toxicology (QST) Modeling Platform for Macromolecules, Provides Mechanistic Understanding of Tocilizumab- and GGF2-Induced Liver Injury. Int. J. Mol. Sci. 2023, 24, 9692. [Google Scholar] [CrossRef] [PubMed]
- Howell, B.A.; Chauhan, A. A physiologically based pharmacokinetic (PBPK) model for predicting the efficacy of drug overdose treatment with liposomes in man. J. Pharm. Sci. 2010, 99, 3601–3619. [Google Scholar] [CrossRef] [PubMed]
- De Sutter, P.-J.; Gasthuys, E.; Vermeulen, A. Comparison of monoclonal antibody disposition predictions using different physiologically based pharmacokinetic modelling platforms. J. Pharmacokinet. Pharmacodyn. 2024, 51, 639–651. [Google Scholar] [CrossRef] [PubMed]
- GastroPlusTM User Manual for Version 9.9, Simulations Plus, Inc.: Lancaster, CA, USA, 2022.
- Shah, D.K.; Betts, A.M. Antibody biodistribution coefficients: Inferring tissue concentrations of monoclonal antibodies based on the plasma concentrations in several preclinical species and human. MAbs 2013, 5, 297–305. [Google Scholar] [CrossRef]
- Opdivo (Nivolumab) Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125554s058lbl.pdf (accessed on 9 December 2024).
- Remicade (Infliximab) Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/103772s5359lbl.pdf (accessed on 1 June 2023).
- Yervoy (Ipilimumab) Data Sheet. Available online: https://medsafe.govt.nz/profs/Datasheet/y/yervoyinj.pdf (accessed on 21 November 2023).
- Bashey, A.; Medina, B.; Corringham, S.; Pasek, M.; Carrier, E.; Vrooman, L.; Lowy, I.; Solomon, S.R.; Morris, L.E.; Holland, H.K.; et al. CTLA4 blockade with ipilimumab to treat relapse of malignancy after allogeneic hematopoietic cell transplantation. Blood 2009, 113, 1581–1588. [Google Scholar] [CrossRef]
- Pasquiers, B.; Benamara, S.; Felices, M.; Ternant, D.; Declèves, X.; Puszkiel, A. Translation of Monoclonal Antibodies Pharmacokinetics from Animal to Human Using Physiologically Based Modeling in Open Systems Pharmacology (OSP) Suite: A Retrospective Analysis of Bevacizumab. Pharmaceutics 2023, 15, 2129. [Google Scholar] [CrossRef]
- Hu, S.; Datta-Mannan, A.; D’Argenio, D.Z. Physiologically Based Modeling to Predict Monoclonal Antibody Pharmacokinetics in Humans from in vitro Physiochemical Properties. MAbs 2022, 14, 2056944. [Google Scholar] [CrossRef]
- Jones, H.M.; Zhang, Z.; Jasper, P.; Luo, H.; Avery, L.B.; King, L.E.; Neubert, H.; Barton, H.A.; Betts, A.M.; Webster, R. A Physiologically-Based Pharmacokinetic Model for the Prediction of Monoclonal Antibody Pharmacokinetics From In Vitro Data. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 738–747. [Google Scholar] [CrossRef]
- Lafourcade, C.; Sobo, K.; Kieffer-Jaquinod, S.; Garin, J.; van der Goot, F.G. Regulation of the V-ATPase along the endocytic pathway occurs through reversible subunit association and membrane localization. PLoS ONE 2008, 3, e2758. [Google Scholar] [CrossRef]
- Ober, R.J.; Martinez, C.; Vaccaro, C.; Zhou, J.; Ward, E.S. Visualizing the site and dynamics of IgG salvage by the MHC class I-related receptor, FcRn. J. Immunol. 2004, 172, 2021–2029. [Google Scholar] [CrossRef]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef]
- Antibody Isotypes & Subtypes. Absolute Antibody an Absolute Biotech Company. Available online: https://absoluteantibody.com/antibody-resources/antibody-overview/antibody-isotypes-subtypes/ (accessed on 25 June 2024).
- Lindena, J.; Küpper, W.; Trautschold, I. Catalytic enzyme activity concentration in thoracic duct, liver, and intestinal lymph of the dog, the rabbit, the rat and the mouse. Approach to a quantitative diagnostic enzymology, II. Communication. J. Clin. Chem. Clin. Biochem. 1986, 24, 19–33. [Google Scholar] [CrossRef]
- Stücker, O.; Pons-Himbert, C.; Laemmel, E. Towards a better understanding of lymph circulation. Phlebolymphology 2008, 15, 31–36. [Google Scholar]
- Chen, Y.; Balthasar, J.P. Evaluation of a catenary PBPK model for predicting the in vivo disposition of mAbs engineered for high-affinity binding to FcRn. AAPS J. 2012, 14, 850–859. [Google Scholar] [CrossRef]
- Urva, S.R.; Yang, V.C.; Balthasar, J.P. Physiologically based pharmacokinetic model for T84.66, A monoclonal anti-CEA antibody. J. Pharm. Sci. 2010, 99, 1582–1600. [Google Scholar] [CrossRef] [PubMed]
- Thilagar, S.; Theyagarajan, R.; Sudhakar, U.; Suresh, S.; Saketharaman, P.; Ahamed, N. Comparison of serum tumor necrosis factor-α levels in rheumatoid arthritis individuals with and without chronic periodontitis: A biochemical study. J. Indian Soc. Periodontol. 2018, 22, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Scallon, B.; Cai, A.; Solowski, N.; Rosenberg, A.; Song, X.-Y.; Shealy, D.; Wagner, C. Binding and functional comparisons of two types of tumor necrosis factor antagonists. J. Pharmacol. Exp. Ther. 2002, 301, 418–426. [Google Scholar] [CrossRef]
- Liang, S.; Dai, J.; Hou, S.; Su, L.; Zhang, D.; Guo, H.; Hu, S.; Wang, H.; Rao, Z.; Guo, Y.; et al. Structural basis for treating tumor necrosis factor α (TNFα)-associated diseases with the therapeutic antibody infliximab. J. Biol. Chem. 2013, 288, 13799–13807. [Google Scholar] [CrossRef]
- Clodi, M.; Vila, G.; Geyeregger, R.; Riedl, M.; Stulnig, T.M.; Struck, J.; Luger, T.A.; Luger, A. Oxytocin alleviates the neuroendocrine and cytokine response to bacterial endotoxin in healthy men. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E686–E691. [Google Scholar] [CrossRef]
- Mookerjee, R.P.; Stadlbauer, V.; Lidder, S.; Wright, G.A.K.; Hodges, S.J.; Davies, N.A.; Jalan, R. Neutrophil dysfunction in alcoholic hepatitis superimposed on cirrhosis is reversible and predicts the outcome. Hepatology 2007, 46, 831–840. [Google Scholar] [CrossRef]
- Song, M.Y.; Park, S.K.; Kim, C.S.; Yoo, T.H.; Kim, B.; Kim, M.S.; Kim, Y.S.; Kwag, W.J.; Lee, B.K.; Baek, K. Characterization of a novel anti-human TNF-alpha murine monoclonal antibody with high binding affinity and neutralizing activity. Exp. Mol. Med. 2008, 40, 35–42. [Google Scholar] [CrossRef]
- Ternant, D.; Le Tilly, O.; Picon, L.; Moussata, D.; Passot, C.; Bejan-Angoulvant, T.; Desvignes, C.; Mulleman, D.; Goupille, P.; Paintaud, G. Infliximab Efficacy May Be Linked to Full TNF-α Blockade in Peripheral Compartment-A Double Central-Peripheral Target-Mediated Drug Disposition (TMDD) Model. Pharmaceutics 2021, 13, 1821. [Google Scholar] [CrossRef]
- Shin, D.; Kim, Y.; Kim, Y.S.; Körnicke, T.; Fuhr, R. A Randomized, Phase I Pharmacokinetic Study Comparing SB2 and Infliximab Reference Product (Remicade®) in Healthy Subjects. BioDrugs 2015, 29, 381–388. [Google Scholar] [CrossRef]
- van Brummelen, E.M.J.; Ros, W.; Wolbink, G.; Beijnen, J.H.; Schellens, J.H.M. Antidrug Antibody Formation in Oncology: Clinical Relevance and Challenges. Oncologist 2016, 21, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Moots, R.J.; Xavier, R.M.; Mok, C.C.; Rahman, M.U.; Tsai, W.-C.; Al-Maini, M.H.; Pavelka, K.; Mahgoub, E.; Kotak, S.; Korth-Bradley, J.; et al. The impact of anti-drug antibodies on drug concentrations and clinical outcomes in rheumatoid arthritis patients treated with adalimumab, etanercept, or infliximab: Results from a multinational, real-world clinical practice, non-interventional study. PLoS ONE 2017, 12, e0175207. [Google Scholar] [CrossRef]
- Ungar, B.; Chowers, Y.; Yavzori, M.; Picard, O.; Fudim, E.; Har-Noy, O.; Kopylov, U.; Eliakim, R.; Ben-Horin, S.; ABIRISK Consortium. The temporal evolution of antidrug antibodies in patients with inflammatory bowel disease treated with infliximab. Gut 2014, 63, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Drake, C.G. Ipilimumab: An anti-CTLA-4 antibody for metastatic melanoma. Clin. Cancer Res. 2011, 17, 6958–6962. [Google Scholar] [CrossRef]
- Khailaie, S.; Rowshanravan, B.; Robert, P.A.; Waters, E.; Halliday, N.; Badillo Herrera, J.D.; Walker, L.S.K.; Sansom, D.M.; Meyer-Hermann, M. Characterization of CTLA4 Trafficking and Implications for Its Function. Biophys. J. 2018, 115, 1330–1343. [Google Scholar] [CrossRef]
- Simone, R.; Pesce, G.; Antola, P.; Rumbullaku, M.; Bagnasco, M.; Bizzaro, N.; Saverino, D. The soluble form of CTLA-4 from serum of patients with autoimmune diseases regulates T-cell responses. BioMed Res. Int. 2014, 2014, 215763. [Google Scholar] [CrossRef]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4, A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Valk, E.; Rudd, C.E.; Schneider, H. CTLA-4 trafficking and surface expression. Trends Immunol. 2008, 29, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Hebbar, M.; Jeannin, P.; Magistrelli, G.; Hatron, P.-Y.; Hachulla, E.; Devulder, B.; Bonnefoy, J.-Y.; Delneste, Y. Detection of circulating soluble CD28 in patients with systemic lupus erythematosus, primary Sjögren’s syndrome and systemic sclerosis. Clin. Exp. Immunol. 2004, 136, 388–392. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Chai, Y.; Qi, J.; Zhang, C.W.H.; Tong, Z.; Shi, Y.; Yan, J.; Tan, S.; Gao, G.F. Remarkably similar CTLA-4 binding properties of therapeutic ipilimumab and tremelimumab antibodies. Oncotarget 2017, 8, 67129–67139. [Google Scholar] [CrossRef] [PubMed]
- Pluim, D.; Ros, W.; Miedema, I.H.C.; Beijnen, J.H.; Schellens, J.H.M. Multiparameter Flow Cytometry Assay for Quantification of Immune Cell Subsets, PD-1 Expression Levels and PD-1 Receptor Occupancy by Nivolumab and Pembrolizumab. Cytometry Part A 2019, 95, 1053–1065. [Google Scholar] [CrossRef]
- Shchelokov, D.; Demin, O. Receptor occupancy assessment and interpretation in terms of quantitative systems pharmacology: Nivolumab case study. MAbs 2023, 15, 2156317. [Google Scholar] [CrossRef]
- Liu, C.; Yu, J.; Li, H.; Liu, J.; Xu, Y.; Song, P.; Liu, Q.; Zhao, H.; Xu, J.; Maher, V.E.; et al. Association of time-varying clearance of nivolumab with disease dynamics and its implications on exposure response analysis. Clin. Pharmacol. Ther. 2017, 101, 657–666. [Google Scholar] [CrossRef]
- Zhang, J.; Sanghavi, K.; Shen, J.; Zhao, X.; Feng, Y.; Statkevich, P.; Sheng, J.; Roy, A.; Zhu, L. Population Pharmacokinetics of Nivolumab in Combination With Ipilimumab in Patients With Advanced Malignancies. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 962–970. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, Y.-H.; Lee, J.H.; Kwon, H.J.; Lee, S.-H.; Park, D.I.; Kim, H.K.; Cheon, J.H.; Im, J.P.; Kim, Y.S.; et al. Post-marketing study of biosimilar infliximab (CT-P13) to evaluate its safety and efficacy in Korea. Expert Rev. Gastroenterol. Hepatol. 2015, 9 (Suppl. S1), 35–44. [Google Scholar] [CrossRef]
- Arora, T.; Padaki, R.; Liu, L.; Hamburger, A.E.; Ellison, A.R.; Stevens, S.R.; Louie, J.S.; Kohno, T. Differences in binding and effector functions between classes of TNF antagonists. Cytokine 2009, 45, 124–131. [Google Scholar] [CrossRef]
- Takeuchi, T.; Yamanaka, H.; Tanaka, Y.; Sakurai, T.; Saito, K.; Ohtsubo, H.; Lee, S.J.; Nambu, Y. Evaluation of the pharmacokinetic equivalence and 54-week efficacy and safety of CT-P13 and innovator infliximab in Japanese patients with rheumatoid arthritis. Mod. Rheumatol. 2015, 25, 817–824. [Google Scholar] [CrossRef]
- Maini, R.N.; Breedveld, F.C.; Kalden, J.R.; Smolen, J.S.; Davis, D.; Macfarlane, J.D.; Antoni, C.; Leeb, B.; Elliott, M.J.; Woody, J.N.; et al. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis Rheum. 1998, 41, 1552–1563. [Google Scholar] [CrossRef]
- Yoo, D.H.; Racewicz, A.; Brzezicki, J.; Yatsyshyn, R.; Arteaga, E.T.; Baranauskaite, A.; Abud-Mendoza, C.; Navarra, S.; Kadinov, V.; Sariego, I.G.; et al. A phase III randomized study to evaluate the efficacy and safety of CT-P13 compared with reference infliximab in patients with active rheumatoid arthritis: 54-week results from the PLANETRA study. Arthritis Res. Ther. 2016, 18, 82. [Google Scholar] [CrossRef] [PubMed]
- Svenson, M.; Geborek, P.; Saxne, T.; Bendtzen, K. Monitoring patients treated with anti-TNF-alpha biopharmaceuticals: Assessing serum infliximab and anti-infliximab antibodies. Rheumatology 2007, 46, 1828–1834. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Fang, W.; Zhao, H.; Bathena, S.P.; Tendolkar, A.; Sheng, J.; Zhang, L. A Phase I Dose Escalation Study of the Safety, Tolerability, and Pharmacokinetics of Ipilimumab in Chinese Patients with Select Advanced Solid Tumors. Oncologist 2021, 26, e549–e566. [Google Scholar] [CrossRef] [PubMed]
- Maker, A.V.; Phan, G.Q.; Attia, P.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Kammula, U.S.; Royal, R.E.; Haworth, L.R.; Levy, C.; et al. Tumor regression and autoimmunity in patients treated with cytotoxic T lymphocyte-associated antigen 4 blockade and interleukin 2, A phase I/II study. Ann. Surg. Oncol. 2005, 12, 1005–1016. [Google Scholar] [CrossRef]
- Yamamoto, N.; Nokihara, H.; Yamada, Y.; Shibata, T.; Tamura, Y.; Seki, Y.; Honda, K.; Tanabe, Y.; Wakui, H.; Tamura, T. Phase I study of Nivolumab, an anti-PD-1 antibody, in patients with malignant solid tumors. Investig. New Drugs 2017, 35, 207–216. [Google Scholar] [CrossRef]
- Watanabe, E.; Nishida, O.; Kakihana, Y.; Odani, M.; Okamura, T.; Harada, T.; Oda, S. Pharmacokinetics, Pharmacodynamics, and Safety of Nivolumab in Patients With Sepsis-Induced Immunosuppression: A Multicenter, Open-Label Phase 1/2 Study. Shock 2020, 53, 686–694. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Colston, E.; Yende, S.; Crouser, E.D.; Martin, G.S.; Albertson, T.; Bartz, R.R.; Brakenridge, S.C.; Delano, M.J.; Park, P.K.; et al. Immune checkpoint inhibition in sepsis: A Phase 1b randomized study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of nivolumab. Intensive Care Med. 2019, 45, 1360–1371. [Google Scholar] [CrossRef]
- Hurkmans, D.P.; Basak, E.A.; van Dijk, T.; Mercieca, D.; Schreurs, M.W.J.; Wijkhuijs, A.J.M.; Bins, S.; Hoop, E.O.; Debets, R.; Joerger, M.; et al. A prospective cohort study on the pharmacokinetics of nivolumab in metastatic non-small cell lung cancer, melanoma, and renal cell cancer patients. J. Immunother. Cancer 2019, 7, 192. [Google Scholar] [CrossRef]
- Chang, H.P.; Shakhnovich, V.; Frymoyer, A.; Funk, R.S.; Becker, M.L.; Park, K.T.; Shah, D.K. A population physiologically-based pharmacokinetic model to characterize antibody disposition in pediatrics and evaluation of the model using infliximab. Br. J. Clin. Pharmacol. 2022, 88, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Petric, Z.; Gonçalves, J.; Paixão, P. Infliximab in Inflammatory Bowel Disease: Leveraging Physiologically Based Pharmacokinetic Modeling in the Clinical Context. Biomedicines 2024, 12, 1974. [Google Scholar] [CrossRef] [PubMed]
- Malik, P.R.V.; Edginton, A.N. Physiologically-Based Pharmacokinetic Modeling vs. Allometric Scaling for the Prediction of Infliximab Pharmacokinetics in Pediatric Patients. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Tiegs, G.; Horst, A.K. TNF in the liver: Targeting a central player in inflammation. Semin. Immunopathol. 2022, 44, 445–459. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Parameter | Unit | Value | Source |
|---|---|---|---|
| Infliximab MW | g/mol | 149,100 | [7] |
| TNF-α MW | g/mol | 17,000 | [23] |
| TNF-α degradation | 1/day | 16.6 | Based on value used in BIOLOGXsym to promote stable dynamics |
| TNF-α NHV blood expression | µmol/g-tissue | 1.32 × 10−10 | [21,24,25] |
| TNF-α RA blood expression | µmol/g-tissue | 7.94 × 10−10 | [21] |
| Kon (antibody-antigen) | 1/µM/day | 8372.16 | [26] |
| Koff (antibody-antigen) | 1/day | 75.95 | [26] |
| Kint (internalization rate of the antibody-antigen complex) | 1/day | 0.089 | [27] |
| Model Parameter | Value | Source |
|---|---|---|
| Vascular reflection coefficient | 0.95 | Default |
| Lymphatic reflection coefficient | 0.2 | Default |
| Endosomal uptake rate (1/day) | 12 | Default |
| Recycle rate (1/day) | 12 | Default |
| Vascular rate fraction | 0.72 | Default |
| Kon, FcRn (pH 7.4) (1/µM/day) | 90 | Default |
| Kon, FcRn (pH 6.0) (1/µM/day) | 8000 | Default |
| Koff, FcRn (pH 7.4) (1/day) | 3 × 104 | Default |
| Koff, FcRn (pH 6.0) (1/day) | 800 | Optimized |
| Kdeg (1/day) | 1 × 104 | Default |
| Parameter | Unit | Value | Source |
|---|---|---|---|
| Ipilimumab MW | g/mol | 148,000 | [8] |
| CTLA-4 MW | g/mol | 25,000 | [32] |
| CTLA-4 decay rate | 1/day | 6.1235 | [33] |
| CTLA-4 blood concentration | µmol/g-tissue | 1.84 × 10−5 | Optimized |
| Kon (antigen-antibody) | 1/µM/day | 33,091.2 | [38] |
| Koff (antigen-antibody) | 1/day | 601.344 | [38] |
| Kint (internalization rate of the antibody-antigen complex) | 1/day | 0.04 | Optimized |
| Model Parameter | Value | Source |
|---|---|---|
| Vascular reflection coefficient | 0.95 | Default |
| Lymphatic reflection coefficient | 0.2 | Default |
| Endosomal uptake rate (1/day) | 12 | Default |
| Recycle rate (1/day) | 12 | Default |
| Vascular rate fraction | 0.72 | Default |
| Kon, FcRn (pH 7.4) (1/µM/day) | 90 | Default |
| Kon, FcRn (pH 6.0) (1/µM/day) | 8000 | Default |
| Koff, FcRn (pH 7.4) (1/day) | 3 × 104 | Default |
| Koff, FcRn (pH 6.0) (1/day) | 500 | Default |
| Kdeg (1/day) | 6000 | Optimized |
| Parameter | Unit | Value | Source |
|---|---|---|---|
| Nivolumab MW | g/mol | 143,600 | [10] |
| PD-1 MW | g/mol | 29,289.06 | [39] |
| PD-1 decay rate | 1/day | 0.33648 | [40] |
| PD-1 blood concentration | µmol/g-tissue | 1.04 × 10−9 | [39] |
| Kon (antigen-antibody) | 1/µM/day | 21,685 | [6,40] |
| Koff (antigen-antibody) | 1/day | 66.3552 | [6,40] |
| Kint (internalization rate of the antibody-antigen complex) | 1/day | 0.4474 | Calculated using [39,40] |
| Model Parameter | Value | Source |
|---|---|---|
| Vascular reflection coefficient | 0.95 | Default |
| Lymphatic reflection coefficient | 0.2 | Default |
| Endosomal uptake rate (1/day) | 12 | Default |
| Recycle rate (1/day) | 12 | Default |
| Vascular rate fraction | 0.72 | Default |
| Kon, FcRn (pH 7.4) (1/µM/day) | 90 | Default |
| Kon, FcRn (pH 6.0) (1/µM/day) | 8000 | Default |
| Koff, FcRn (pH 7.4) (1/day) | 3 × 104 | Default |
| Koff, FcRn (pH 6.0) (1/day) | 500 | Default |
| Kdeg (1/day) | 20,000 | Optimized (single dose) |
| Kdeg (1/day) | 11,000 | Optimized (multidose) |
| Study | Health Status | ADA Positive or Negative | Dose [mg/kg] | PK Parameter | Obs | Sim | Sim/Obs |
|---|---|---|---|---|---|---|---|
| Shin 2015 [28] | Healthy | Both | 5 | Cmax [µg/mL] | 124.1 | 117.3 | 0.95 |
| AUC0-t [µg·h/mL] | 38,550 | 38,070 | 0.99 | ||||
| AUC0-inf [µg·h/mL] | 39,990 | 38,960 | 0.97 | ||||
| Park 2015 [43] | Healthy | NA | 5 | Cmax [µg/mL] | 116.7 | 114.9 | 0.98 |
| AUC0-t [µg·h/mL] | 30,820 | 36,720 | 1.19 | ||||
| AUC0-inf [µg·h/mL] | 32,320 | 38,600 | 1.19 | ||||
| Takeuchi 2015 [45] | RA | Both | 3 | Cmax [µg/mL] | 108 | 102.4 | 0.95 |
| AUC0-t [µg·h/mL] | 21,900 | 21,020 | 0.96 | ||||
| Takeuchi 2015 [45] | RA | Negative | 3 | Cmax [µg/mL] | 111 | 102.4 | 0.92 |
| AUC0-t [µg·h/mL] | 24,700 | 21,020 | 0.85 | ||||
| Shin 2015 [28] | Healthy | Positive | 5 | Cmax [µg/mL] | 124.1 | 117.3 | 0.95 |
| AUC0-t [µg·h/mL] | 31,870 | 32,030 | 1.01 | ||||
| AUC0-inf [µg·h/mL] | 32,620 | 32,370 | 0.99 |
| Source | Dose [mg/kg] | Obs Cmax [µg/mL] | Sim Cmax [µg/mL] | Sim/Obs | Obs AUC0-t [µg·h/mL] | Sim AUC0-t [µg·h/mL] | Sim/Obs |
|---|---|---|---|---|---|---|---|
| Bashey 2009 [9] | 0.1 | 1.195 | 1.38 | 1.15 | NA 1 | 25.14 | NA |
| Bashey 2009 [9] | 0.33 | 6.094 | 6.47 | 1.06 | 626.2 | 900.58 | 1.44 |
| Bashey 2009 [9] | 0.66 | 20.48 | 15.3 | 0.75 | 3766.7 | 4104.44 | 1.09 |
| Bashey 2009 [9] | 1 | 14.56 | 14.51 | 1.00 | 4871.1 | 4288.13 | 0.88 |
| Bashey 2009 [9] | 3 | 129.5 | 109.61 | 0.85 | 37,790 | 25,991.08 | 0.69 |
| Ma 2021 [49] | 3 | 88.96 | 109.61 | 1.23 | 19,370 | 25,991.08 | 1.34 |
| Ma 2021 [49] | 10 | 241 | 376.13 | 1.56 | 45,460 | 58,740.24 | 1.29 |
| Source | Dose [mg/kg] | Obs Cmax [µg/mL] | Sim Cmax [µg/mL] | Sim/Obs | Obs AUC0-t [µg·h/mL] | Sim AUC0-t [µg·h/mL] | Sim/Obs |
|---|---|---|---|---|---|---|---|
| Yamamoto 2017 [51] | 1 | 22.9 | 21.0 | 0.92 | 4980 | 4390 | 0.88 |
| Yamamoto 2017 [51] | 3 | 67.4 | 63.1 | 0.94 | 1.28 × 104 | 1.32 × 104 | 1.03 |
| Yamamoto 2017 [51] | 10 | 180 | 211 | 1.17 | 4.37 × 104 | 4.39 × 104 | 1.00 |
| Watanabe 2020 [52] | 480 mg | 116 | 167 | 1.44 | 2.41 × 104 | 3.83 × 104 | 1.59 |
| Hotchkiss 2019 [54] | 480 mg | 82.8 | 102 | 1.23 | 2.1 × 104 | 3.08 × 104 | 1.47 |
| Brahmer 2010 [53] | 1 | 38.1 | 19.1 | 0.5 | 1.03 × 104 | 5550 | 0.54 |
| Brahmer 2010 [53] | 3 | 68.4 | 57.4 | 0.84 | 1.93 × 104 | 1.42 × 104 | 0.74 |
| Brahmer 2010 [53] | 10 | 216 | 191 | 0.88 | 9.94 × 104 | 5.69 × 104 | 0.57 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallejo, C.; Meaney, C.; Clemens, L.; Yang, K.; Lukacova, V.; Zhou, H. Physiologically Based Pharmacokinetic Models for Infliximab, Ipilimumab, and Nivolumab Developed with GastroPlus to Predict Hepatic Concentrations. Pharmaceutics 2025, 17, 372. https://doi.org/10.3390/pharmaceutics17030372
Vallejo C, Meaney C, Clemens L, Yang K, Lukacova V, Zhou H. Physiologically Based Pharmacokinetic Models for Infliximab, Ipilimumab, and Nivolumab Developed with GastroPlus to Predict Hepatic Concentrations. Pharmaceutics. 2025; 17(3):372. https://doi.org/10.3390/pharmaceutics17030372
Chicago/Turabian StyleVallejo, Celeste, Cameron Meaney, Lara Clemens, Kyunghee Yang, Viera Lukacova, and Haiying Zhou. 2025. "Physiologically Based Pharmacokinetic Models for Infliximab, Ipilimumab, and Nivolumab Developed with GastroPlus to Predict Hepatic Concentrations" Pharmaceutics 17, no. 3: 372. https://doi.org/10.3390/pharmaceutics17030372
APA StyleVallejo, C., Meaney, C., Clemens, L., Yang, K., Lukacova, V., & Zhou, H. (2025). Physiologically Based Pharmacokinetic Models for Infliximab, Ipilimumab, and Nivolumab Developed with GastroPlus to Predict Hepatic Concentrations. Pharmaceutics, 17(3), 372. https://doi.org/10.3390/pharmaceutics17030372